

Abstract
Protein Z-dependent protease inhibitor (ZPI) is a plasma serpin which can rapidly inactivate factor Xa (fXa) in the presence of protein Z (PZ), negatively charged phospholipids and Ca2+. To investigate the mechanism by which ZPI inactivates fXa, we expressed the serpin in mammalian cells and characterized its reactivity with both wild-type and selected mutants of fXa which 1) contained substitutions in the autolysis loop and the heparin-binding exosite, 2) lacked the first EGF-like domain (fXa-des-EGF-1), or 3) contained the Gla domain of protein C (fXa/PC-Gla). Inhibition studies in both the presence and absence of PZ revealed that Arg-143, Lys-147 and Arg-154 of the autolysis loop and Lys-96, Lys-169 and Lys-236 of the heparin-binding exosite are required for recognition of ZPI, with Arg-143 being essential for the interaction. Similar studies with fXa-des-EGF-1 and fXa/PC-Gla suggested that protein-protein interaction with either the Gla or the EGF-1 domain may not play a dominant role in the PZ-dependent recognition of fXa by the serpin on phospholipid vesicles. Further studies showed that an inactive Ser-195 to Ala mutant of fXa effectively competes with wild-type fXa for binding to the non-serpin inhibitors, tissue factor pathway inhibitor and recombinant tick anticoagulant peptide, but does not compete for binding to ZPI. This suggests that the catalytic residue of fXa is required for interaction with ZPI.
Factor Xa (fXa)1 is a vitamin-K dependent serine protease that is responsible for generation of thrombin from prothrombin in the coagulation cascade (1-4). It circulates in plasma as a light and heavy chain molecule held together by a disulfide bond (5). The N-terminal light chain contains the non-catalytic γ-carboxyglutamic acid (Gla) and two epidermal growth factor-like (EGF) domains, while the C-terminal heavy chain contains the trypsin-like catalytic domain of the molecule (2,5). The proteolytic activity of fXa in plasma is regulated by three physiological inhibitors, tissue factor pathway inhibitor (TFPI) (6), antithrombin (AT) (7,8), and protein Z (PZ)-dependent protease inhibitor (ZPI) (9-12). TFPI is a multi-domain Kunitz-type inhibitor that regulates the coagulation cascade by the fXa-dependent inhibition of the factor VIIa-tissue factor complex during the initial stages of the clotting cascade (6). It functions by binding to the active-site pocket of fXa by the second Kunitz domain, and thereafter binding tightly to the active-site of the factor VIIa-tissue factor complex via the first Kunitz domain, thereby rendering both proteases inactive in a quaternary complex (6). AT, on the other hand, is a serine protease inhibitor (serpin) that regulates the proteolytic activity of fXa and other coagulation proteases by covalently binding to the active-site through an exposed reactive center loop, and undergoing a conformational change which leads to entrapment of the target protease in inactive and SDS-stable complex with the serpin (13-15). ZPI is also a serpin, and there is some evidence that it interacts with the active-site pocket of fXa by a similar covalent mechanism (11). However, ZPI does not form an SDS-stable complex with fXa, suggesting that, unlike the AT-fXa complex, the ZPI-fXa complex has a higher dissociation constant (11). This has been previously documented by the reversibility of the ZPI-fXa reaction and the observation that the activity of fXa is partially recovered following the incubation of the ZPI-fXa complex with EDTA at 37 °C (11).
Both AT and ZPI require cofactors for their effective interaction with fXa. Thus, the AT interaction with the heparin-like glycosaminoglycans on the surface of the endothelium (16), and the ZPI complex formation with PZ on membrane phospholipids in the presence of Ca2+ (9) is required for the ability of both serpins to effectively interact with fXa. In the case of AT, the mechanism of the cofactor function of heparin in promoting the serpin inhibition of fXa has been extensively studied (14,17). It has been demonstrated that the binding of a unique pentasaccharide fragment of heparin to a basic helical structure of AT induces a conformational change in the serpin that leads to a better recognition and a high affinity interaction of the serpin with fXa (18). Moreover, previous studies have indicated that the basic residues of the autolysis loop (143-154 in chymotrypsin numbering (19)) play a key role in recognition of the heparin-activated conformation of AT by fXa (20). In addition to promotion of the AT-fXa reaction by this allosteric mechanism, full-length heparins are also known to bind to a basic exosite on fXa in the presence of Ca2+, thereby bridging both the serpin and the protease in one complex and further contributing to the promotion of the reaction by a template mechanism (21,22). Unlike AT, it is not known how ZPI interacts with fXa and unlike the cofactor function of heparin, it is not known how PZ promotes the inhibition of fXa by ZPI on membrane surfaces in the presence of Ca2+. PZ is a vitamin K-dependent plasma protein with a domain organization identical to that of fXa, and thus capable of interacting with negatively charged phospholipid membranes via its Gla domain, however, it merely functions as a cofactor in the reaction and lacks any catalytic function (23). It is not known whether the interaction of fXa with PZ on the membrane surface is required for the protease recognition of ZPI.
To investigate these questions, we expressed ZPI in mammalian cells and following purification, characterized its properties in inhibition reactions with wild-type and selected mutants of fXa containing substitutions in several exposed surface loops (exosites) on the catalytic domain, known to be critical for the macromolecular substrate and inhibitor specificity of fXa and other coagulation enzymes. Moreover, the role of the non-catalytic domain of fXa for the ZPI-PZ interaction was examined using two mutants of fXa from one of which the first EGF-like domain was deleted (fXa-des-EGF-1). In the other mutant, the Gla domain of fXa was substituted with the corresponding domain of protein C (fXa/PCGla). Recombinant ZPI, similar to plasma ZPI, rapidly inactivated fXa in the presence of PZ with a second-order inhibition rate constant of 6.2 × 105 M-1 s-1. A similar high reactivity for ZPI was observed with fXa mutants of the 39, 60 and 70-80 loops. However, the reactivity of ZPI with the autolysis loop mutant of fXa was markedly impaired and Arg-143 was identified to be essential for interaction with the serpin both in the absence and presence of PZ. Similarly, three Ala substitution mutants of heparin-binding exosite of fXa (K96A, K169A and K236A) exhibited diminished reactivity with ZPI both in the absence and presence of PZ. A near normal cofactor effect for PZ with both fXa-des-EGF-1 and fXa/PC-Gla suggested that neither the Gla nor the EGF-1 domain plays a dominant role in the PZ-dependent recognition of fXa by the serpin on phospholipid vesicles. Finally, an inactive Ser-195 to Ala mutant of fXa effectively competed with wild-type fXa for binding to TFPI and recombinant tick anticoagulant peptide (rTAP), but failed to compete for binding to ZPI. These results suggest that, similar to other serine protease-serpin reactions, the catalytic Ser-195 is required for a high affinity interaction of fXa with ZPI.
Experimental Procedures
Construction, Mutagenesis and Expression of Recombinant Proteins- ZPI cDNA (obtained from Open Biosystems, Huntsville, AL) was sub-cloned into the StuI and XbaI restriction enzyme sites of the RSV-PL4 expression/purification vector system and expressed in human embryonic kidney cells (HEK-293) as described (24). This vector contains the sequence of the transferrin signal peptide for secretion, the sequence of a 12-residue epitope for a Ca2+-dependent monoclonal antibody, HPC4, for purification and a neomycin gene for the G418 resistant selection in mammalian cells (24). Following transfer of the vector into HEK-293 cells, several G418 resistant clones were selected and examined for ZPI expression by Western-blot analysis using the HPC4 monoclonal antibody. A clone positive for ZPI expression was identified, expanded, and 20 liters of cell culture supernatant were collected, concentrated and purified by immunoaffinity chromatography using the HPC4 antibody linked to Affi-gel 10 (Bio-Rad) as described (24). The HPC4 eluate was further chromatographed on a size exclusion column using Superdex 200 (Amersham Biosciences Corp). The ZPI concentration was calculated from the absorbance at 280 nm using a molar absorption coefficient of 31525 M-1 cm-1, calculated based on the equation ε(280) = (#Trp × 5500) + (#Tyr × 1490) + (#cystine × 125) as described (25). The fX mutant in which the exon coding for the Gla domain (Ala-1 to Asn-38) of fX was exchanged with the corresponding domain of protein C was prepared by PCR methods and expressed in the same expression system as described for wild-type fX (20). The expression and purification of the fX mutant lacking the N-terminal EGF-like domain (fX-des-EGF-1) (26), 39 loop mutants (E36Q, E37Q and E39Q) (27), 60 loop mutants (K62E and R63E) (27), 70-80 loop mutants (R71A, E74A, E76A, and E77A) (28), the autolysis loop mutants (R143A, K147A, R150A, and R154A) (20), and the heparin-binding exosite mutants (R93A, K96A, R165A, K169A, K236A, and R240A) (29,30) have been described. The inactive Ser195 → Ala (S195A) substitution mutant of fX was expressed in the same expression/purification vector system (20). The accuracy of all constructs was confirmed by DNA sequencing prior to their expression in mammalian cells. All fX mutants containing the fully γ-carboxylated proteins were purified to homogeneity (28), activated by the fX activating enzyme from Russell's viper venom (RVV-X), purified on heparin-Sepharose column and active-site titrated with known concentrations of AT as described (20).
Tissue factor pathway inhibitor (TFPI) was from Monsanto Chemical Co. (St. Louis, MO). Recombinant tick anticoagulant peptide (rTAP) was a generous gift from Dr. G. Vlasuk (Corvas International Inc., San Diego, CA), and human plasma-derived ZPI (pZPI) was a generous gift from Dr. Mary Jo Heeb (Scripps Research Institute). Protein Z (PZ) and RVV-X were purchased from Haematologic Technologies Inc. (Essex Junction, VT). Phospholipid vesicles containing 80% phosphatidylcholine and 20% phosphatidylserine (PC/PS) were prepared as described (31). The chromogenic substrates, Spectrozyme FXa (SpFXa, MeO-CO-Dcyclohexylglycyl-Gly-Arg-p-nitroaniline-dihydrochloride) was purchased from American Diagnostica (Greenwich, CT), and S2765 (N-α-Benzyloxycarbonyl-D-Arg-Gly-Arg-p-nitroaniline-dihydrochloride) was purchased from Kabi Pharmacia/Chromogenix (Franklin, OH).
Inhibition Assays- The time course and concentration dependence of fXa inhibition by ZPI was studied both in the absence and presence of PZ on PC/PS vesicles under pseudo-first-order conditions as described (21,22). In the absence of PZ, fXa derivatives (1 nM) were incubated with ZPI (100-250 nM) on PC/PS vesicles (25 μM) in 0.1 M NaCl, 0.02 M TrisHCl (pH 7.5), 0.1% polyethylene glycol 8000 (PEG 8000), 0.1 mg/mL bovine serum albumin (BSA) and 5 mM Ca2+ (TBS/Ca2+) for 30-120 min in 50 μL volumes in 96-well polystyrene plates at room temperature. The presence of BSA in the reaction did not influence the reactivity of the serpin with the protease. In the presence of PZ, the reaction conditions were the same except that all proteases (0.5 nM) were incubated with ZPI (10-80 nM) in complex with a saturating concentration of PZ (50-150 nM) for 15 sec to 50 min on PC/PS vesicles (25 μM) in the same TBS buffer. The inactivation reactions were stopped by addition of 50 μL S2765 (0.25 mM final) in TBS containing 50 mM EDTA and the remaining enzyme activity was measured with a Vmax Kinetics Microplate Reader (Molecular Devices, Menlo Park, CA) at 405 nm. The cofactor concentration dependence of PZ-mediated fXa inhibition by ZPI indicated that PZ concentrations are saturating under these conditions. The observed pseudo-first-order (kobs) rate constants were calculated from a first-order rate equation and the second-order rate constants (k2) were calculated from the slope of linear plot of kobs values versus ZPI concentrations as described (21,22).
Competitive Inhibition Studies- The competitive effect of the inactive S195A mutant of fXa on the wild-type fXa inhibition by the ZPI-PZ complex, and the non-serpin inhibitors TFPI and rTAP was studied using the same amidolytic activity assay described above. In this case, the inhibition of fXa (1 nM) on PC/PS vesicles (25 μM) by each inhibitor (5 nM TFPI and rTAP, and 10 nM ZPI) was monitored in the presence of increasing concentrations of fXa S195A (0-200 nM) in TBS/Ca2+. Following 10 min incubation at room temperature, 50 μL SpFXa (0.5 mM in EDTA) was added to each well and the remaining activity of fXa was measured as described above.
Results
Expression and Purification of Recombinant Proteins- Human recombinant ZPI (rZPI) was expressed in HEK-293 cells using the RSV-PL4 expression/purification vector system as described (24). rZPI expressed by this system contains a 12-residue epitope for the Ca2+-dependent monoclonal antibody HPC4 at its N-terminal domain for easy purification. Following passage of the rZPI supernatants through the immobilized HPC4 antibody, the substitution of Ca2+ with EDTA in the wash buffer was sufficient to elute the serpin from the antibody column (24). The HPC4 eluate was further purified by size exclusion chromatography and its concentration was determined as described under “Experimental Procedures”. We have previously utilized this expression/purification vector system for expression of other serpins including AT (32) and protein C inhibitor (33). Both serpins expressed in this system, have exhibited identical activities as their wild-type counterparts both in the absence and presence of related cofactors, suggesting that the N-terminal HPC4 epitope does not interfere with the inhibitory properties of either serpin (32,33). This is also true for the ZPI inhibition of fXa since as shown in Table 1, rZPI inhibited fXa with k2 values which were comparable to those of pZPI in both the absence and presence of PZ. SDS-PAGE analysis under non-reducing conditions suggested that rZPI migrates as a single band with an expected molecular mass of ∼72 kDa (Fig. 1, lane 1), as reported previously for this protein (9,11). The results further suggested that fXa does not cleave ZPI either in the absence or presence of PZ, however, it does not also form an SDS-stable complex with the serpin under both conditions (Fig. 1, lanes 4 and 5). This is consistent with previous results in the literature (11). All of the fXa derivatives were also expressed in a similar HPC4-based expression/purification vector system and the fully γ-carboxylated proteins were isolated as described (20,28). In the case of the fX expression system, however, the construct has been prepared such that the HPC4 epitope replaces the first 12-residues of the zymogen activation peptide on the heavy chain of the molecule, and since the activation peptide of fX is cleaved off during activation by RVV-X, the N-terminal HPC4 epitope is removed along with the activation peptide from all fXa derivatives. Thus, with the exception of specific mutations, all fXa derivatives have identical amino acid sequences as the plasma-derived fXa. The characterization of all fXa mutants with the exception of fXa/PC-Gla has been described previously (20,27-30) As expected, the fXa/PCGla mutant also exhibited normal amidolytic activity and reactivity with AT (data not shown).
Table 1.
Second-order rate (k2) constants for the ZPI inhibition of fXa derivatives in the absence and presence of PZ on PC/PS vesicles.
| k2 (-PZ) (M-1 s-1) | k2 (+PZ) (M-1 s-1) | Fold Enhancement +PZ/-PZ | |
|---|---|---|---|
| fXa (pZPI) | (2.3 ± 0.1) x 103 | (6.1 ± 0.4) x 105 | 265 ± 29 |
| fXa (rZPI) | (1.6 ± 0.1) x 103 | (6.2 ± 0.5) x 105 | 388 ± 55 |
| fXa-des-EGF-1 | (1.3 ± 0.1) x 103 | (4.2 ± 0.7) x 105 | 323 ± 79 |
| fXa/PC-Gla | (7.5 ± 0.4) x 102 | (1.2 ± 0.1) x 105 | 160 ± 22 |
| R143A | ND | (1.2 ± 0.3) x 102 | ND |
| K147A | (2.1 ± 0.4) x 102 | (1.5 ± 0.1) x 105 | 714 ± 184 |
| R150A | (6.4 ± 0.2) x 102 | (8.5 ± 0.4) x 105 | 1328 ± 104 |
| R154A | (5.0 ± 0.3) x 102 | (3.3 ± 0.5) x 104 | 66 ± 14 |
| R93A | (1.0 ± 0.1) x 103 | (4.9 ± 0.1) x 105 | 490 ± 59 |
| K96A | (8.8 ± 0.3) x 102 | (1.0 ± 0.1) x 105 | 114 ± 15 |
| R165A | (1.3 ± 0.1) x 103 | (5.5 ± 0.4) x 105 | 423 ± 63 |
| K169A | (6.6 ± 0.2) x 102 | (6.6 ± 0.5) x 104 | 100 ± 11 |
| K236A | (9.3 ± 0.5) x 102 | (6.1 ± 0.3) x 104 | 66 ± 7 |
| R240A | (5.9 ± 0.1) x 102 | (8.6 ± 0.1) x 105 | 1458 ± 42 |
The second-order rate (k2) constants in the absence of PZ were determined from the remaining activities of fXa derivatives (1 nM each) after incubation with ZPI (100-250 nM) on PC/PS vesicles (25 μM) for 30-120 min in TBS/Ca2+ at room temperature by an amidolytic activity assay described under "Experimental Procedures". The values in the presence of PZ was determined by the same methods except that the activation of fXa derivatives (0.5 nM) by ZPI (10-80 nM) was monitored in the presence of PZ (50-150 nM) for 15 sec to 50 min under the same experimental conditions. In the case R143A, the k2 value was determined by incubation of 1 nM mutant protease with 200 nM ZPI in the presence of 500 nM PZ for 50 min. All values are averages of at least 3 independent measurements ± S.D. ND, no reactivity for R143A was detected after incubation of the protease with 250 nM ZPI for 120 min.
Figure 1.
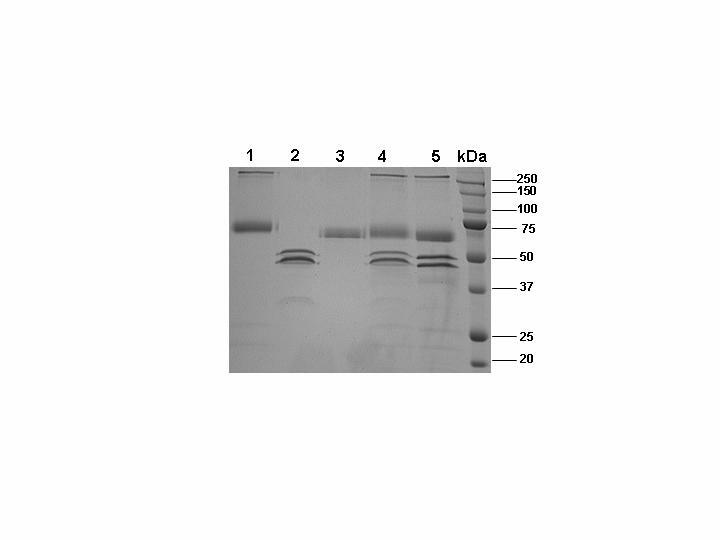
SDS-PAGE analysis of ZPI reaction with fXa in both the absence and presence of PZ in TBS containing 25 μM PC/PS and 5 mM Ca2+ vesicles. Under non-reducing conditions lane 1, rZPI; lane 2, fXa; lane 3, PZ; lane 4, equimolar concentrations of fXa and ZPI; lane 5, equimolar concentrations of fXa, ZPI and PZ. Lane 6 is molecular mass standards in kDa. The two closely migrating bands in fXa lanes are α and β forms of the protease.
FXa Inhibition by ZPI- The extent of the reactivity of fXa with ZPI was evaluated both in the absence and presence of PZ on PC/PS vesicles and in TBS/Ca2+ under pseudo first-order conditions as described under “Experimental Procedures”. Both time course and concentration dependence of the inhibition reactions indicated that the inhibitory activity of ZPI toward fXa is not complete, but rather reaches to a maximum of 80% inhibition and thereafter no further decline in the amidolytic activity of fXa is observed. Previously, a similar 20% remaining activity for fXa in the presence of high concentrations of ZPI has been observed in a similar amidolytic activity assay (9). The reason for the incomplete inhibitory activity of ZPI toward fXa in the amidolytic activity assays is not known. One possibility is that since ZPI exhibits reversibility in reaction with fXa (11), the addition of the chromogenic substrate in EDTA and subsequent dilution of the reaction leads to the dissociation of active fXa from the inhibitory complex (11). Alternative possibilities including differential reactivity for α and β forms of fXa with the ZPI-PZ complex have been postulated in the past (9). However, the exact cause of an incomplete inhibition of fXa by the ZPI-PZ complex, as measured by an amidolytic activity assay remains an open question. To minimize the effect of an incomplete inhibition of fXa by ZPI, all rate constants were derived from inhibition reactions in which ~40-60% of the enzyme activity was inhibited. The second-order inhibition rate constants were calculated from the slopes of linear plots of kobs values as a function of increasing concentrations of ZPI in complex with a saturating concentration of PZ (Fig. 2). The values presented in Table 1 indicated that, rZPI similar to pZPI, inhibits fXa with k2 values of 1.6 × 103 M-1 s-1 in the absence and 6.2 × 105 M-1 s-1 in the presence of PZ on PC/PS vesicles. The cofactor effect of PZ enhanced the reactivity of rZPI with fXa 338-fold (Table 1). Similar reactivities of pZPI and rZPI with fXa both in the absence and presence of PZ suggest that the N-terminus of the serpin does not interact with either the cofactor or the protease in the inhibitory complex.
Figure 2.
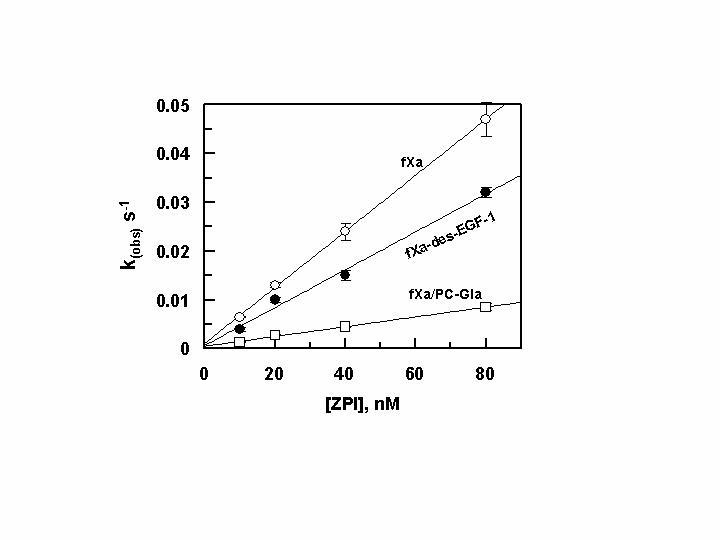
Dependence of kobs values on ZPI concentration with fXa derivatives in the presence of PZ. The kobs values for the ZPI inhibition of fXa (○), fXa-des-EGF-1 ([unk]) and fXa/PC-Gla (…) (0.5 nM each) were determined in the presence of PZ (50-150 nM) and varying concentration of ZPI (10-80 nM) on PC/PS vesicles (25 μM) in TBS containing 0.1 mg/mL BSA, 0.1% PEG 8000 and 5 mM Ca2+ at room temperature by an amidolytic activity assay described under “Experimental Procedures”. The k2 values were calculated from the slopes of the straight lines and presented in Table 1. Data are derived from averages of at least 3 independent measurements.
Role of the Gla and EGF-1 domains in the PZ-dependent Interaction of FXa with ZPI-Similar to fXa, PZ contains a Gla domain that can interact with the negatively charged membrane surfaces. To investigate the possibility that the interaction of the two proteins via their Gla and/or EGF-1 domains on or near the membrane surface contributes to the high affinity of the fXa-PZ-ZPI interaction, k2 values for the ZPI inhibition of fXa-des-EGF-1 and fXa/PC-Gla derivatives were determined. As shown in Fig. 2 and Table 1, the reactivity of the fXa mutant lacking the EGF-1 domain with the ZPI-PZ complex was comparable to that of wild-type fXa, suggesting that the N-terminal EFG domain does not interact with either PZ or ZPI in the inhibitory complex. The reactivity of ZPI with fXa mutant containing the Gla domain of protein C was impaired ∼2-3-fold in the absence and ∼5-fold in the presence of PZ (Fig. 2, Table 1), thus leading to ∼2-fold impairment in the cofactor function of PZ in the reaction. These results suggest that the specific interaction of the Gla domain of fXa with PZ/ZPI on PC/PS vesicles makes some contribution to protease inhibition by the serpin.
Role of the Autolysis loop in the FXaZPI Interaction- The catalytic groove of fXa and other coagulation proteases is surrounded by several surface loops including 39 loop, autolysis loop (residues 143-154), and 60 loop, all of which contain variant residues with important roles in determining the substrate and inhibitor specificity of these proteases (34,35). To determine whether any one of the variant residues of these loops constitute a recognition site for ZPI, the ability of the serpin to inhibit selected loop mutants of fXa was evaluated as a function of increasing concentrations of ZPI in the presence of a saturating concentration of PZ. Among the three loops mentioned above, only the basic residues of the autolysis loop were important for the recognition of the serpin (Fig. 3). Thus, the reactivity of the three mutants R143A, K147A, and R154A with ZPI was impaired to varying degrees, with the first mutant showing a dramatic effect in the reaction (Fig. 3A). No reactivity for R143A with ZPI could be detected in the presence of PZ under experimental conditions described under legend of Fig. 3. However, if incubated for a longer time (50 min) in the presence of a higher concentration of ZPI (200 nM), the R143A mutant was inhibited by the serpin with k2 = 1.2 × 102 M-1 s-1 in the presence of PZ (500 nM). No reactivity for R143A with ZPI (250 nM) in the absence of PZ was observed even if the incubation time was increased to 120 min. These results suggest that Arg-143 is an important cofactor independent recognition site for ZPI on the protease. The reactivities of both R150A and R154A with ZPI were impaired ∼2-3-fold in the absence of the cofactor (Table 1). Interestingly, while the reactivity of R154A with ZPI was impaired ∼20-fold in the presence of PZ, it was slightly improved with the R150A mutant, thus leading to a much higher cofactor effect of 1328-fold for the PZ acceleration of the ZPI inhibition of this mutant (Table 1). Unlike the autolysis loop mutants, E36Q, E37Q and E39Q mutants of 39 loop, and the K62E and R63E mutants of 60 loop did not exhibit significant changes in their reactivities with ZPI either in the absence or presence of PZ (data not shown). It is worth noting that we have previously demonstrated that all autolysis loop mutants of fXa have normal amidolytic and proteolytic activities (20).
Figure 3.
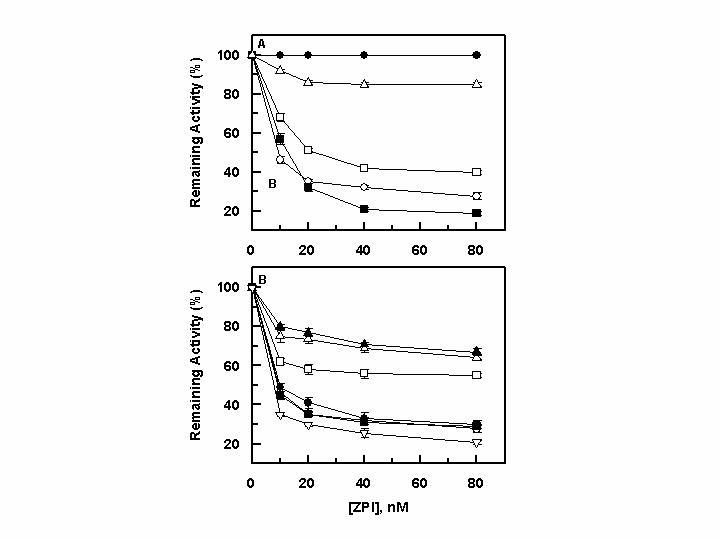
Concentration-dependence of inhibition of the autolysis loop mutants of fXa by ZPI in the presence of PZ. A, the inhibition of fXa derivatives: wild-type fXa (○), R143A ([unk]), K147A (…), R150A („), and R154A (∆) (0.5 nM each) was monitored by increasing concentrations of ZPI (x-axis) in the presence of PZ (50-150 nM) on PC/PS vesicles (25 μM) in TBS containing 0.1 mg/mL BSA, 0.1% PEG 8000 and 5 mM Ca2+ at room temperature for 1 min by an amidolytic activity assay described under “Experimental Procedures”. B, the same as A except that the inhibition reactions on PC/PS vesicles were monitored for the heparin-binding exosite mutants of fXa. The symbols in panel B are: wild-type fXa (○ ), R93A ([unk]), K96A (†), R165A („), K169A (U), K236 A (S) and R240A (V). Data in both panels are derived from averages of 3 independent measurements.
Role of the Ca2+-binding 70-80 loop and Basic Heparin-binding Exosite in the FXa-ZPI Interaction- The reactivity of 70-80 loop mutants of fXa (R71A, E74A, E76A, and E77A) with ZPI either in the absence or presence of PZ was not significantly affected, suggesting that this loop does not provide a recognition site for interaction with the serpin (data not shown). On the other hand, the reactivity of three basic heparin-binding exosite mutants of fXa with ZPI was slightly or moderately impaired. Thus, while the R93A and R165A mutants exhibited near normal reactivity with ZPI, the reactivity of K96A, K169, and K236A with ZPI was impaired ∼2-3-fold in the absence and ∼6-10-fold in the presence of PZ (Fig. 3B, Table 1). The reactivity of R240A with ZPI was impaired 3-fold in the absence of PZ, but it was slightly improved in the presence of the cofactor. Similar to inhibition of R150A, the cofactor effect of PZ in accelerating the ZPI inhibition of R240A was considerably improved, suggesting that Arg-240 makes an inhibitory interaction with the serpin in the absence of the cofactor. Nevertheless, the cofactor function of PZ overcomes the inhibitory interaction of Arg-240 with ZPI (Table 1).
It should be noted that the heparin-binding exosite mutants of fXa, most likely, folded properly since they have normal amidolytic activities and reactivities with AT both in the absence and presence of the pentasaccharide fragment of the high affinity heparin (30).
Role of the Catalytic Ser-195 in the PZ-dependent Interaction of FXa with ZPI- The reactivity of nearly all serpins with their target serine proteases is irreversible, because the attack of the P1 recognition site of the serpin by Ser-195 results in the covalent acylation of the active-site residue of the protease. This accounts for the kinetic stability of the protease-serpin complexes (13). In contrast to serpins, the Kunitz-type inhibitors bind tightly to the active-site pocket of their target proteases by a reversible mechanism which is independent of the catalytic residue Ser-195 (13). In light of the previous observation that the ZPI-fXa complex is reversible (11), we decided to investigate the role of Ser-195 of fXa in this reaction. As shown in Fig. 4, increasing concentrations of the S195A mutant of fXa effectively competed with wild-type fXa for binding to both TFPI and rTAP, suggesting no major role for Ser-195 in interaction with the non-serpin inhibitors. On the other hand, a 200-fold molar excess of the inactive fXa mutant failed to compete with fXa for interaction with ZPI, suggesting that fXa reacts with ZPI by a reaction mechanism that is similar to that of other serpins.
Figure 4.
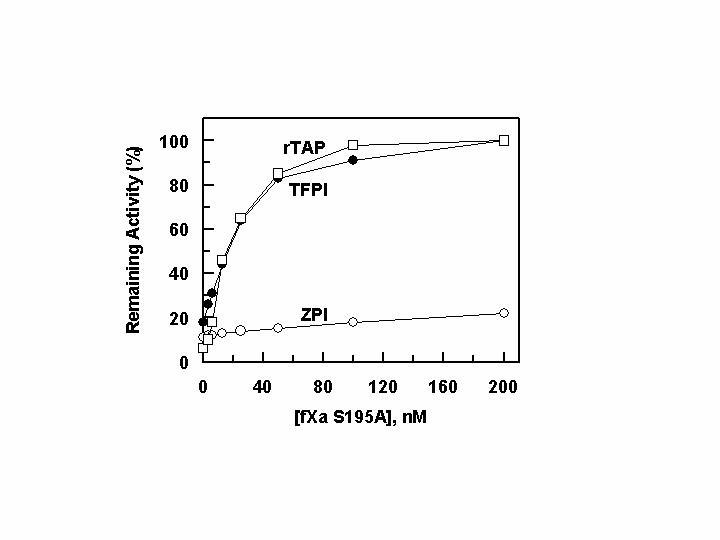
The competitive effect of inactive S195A fXa on the inhibition of wild-type fXa by ZPI, TFPI and rTAP. One nM fXa was incubated with each inhibitor: 10 nM ZPI in the presence of PZ (50 nM) (○), 5 nM TFPI ([unk]), and 5 nM rTAP (†) on PC/PS vesicles (25 μM) in TBS containing 0.1 mg/mL BSA, 0.1% PEG 8000 and 5 mM Ca2+ at room temperature for 10 min followed by monitoring the amidolytic activity of fXa as described under “Experimental Procedures”.
Discussion
We have expressed ZPI in mammalian cells and characterized its reactivity with several derivatives of fXa and demonstrated that, in addition to interaction with the active-site pocket, extended interaction of the serpin with two basic sites of fXa is essential for its high affinity interaction with the protease. This is derived from the observation that the reactivity of the autolysis loop mutants and heparin-binding exosite mutants of fXa with ZPI was altered. Among the autolysis loop mutants, R143A exhibited the highest degree of impairment in reaction with ZPI, suggesting that Arg-143 is a crucial recognition site on the protease for interaction with the serpin. It appears that Arg-143 directly interacts with ZPI since no reactivity for the mutant was observed in the absence of PZ, but the cofactor enhanced the reaction, yielding a k2 of 1.2 × 102 M-1 s-1 for the ZPI inhibition of the mutant (Table 1). Our previous results have indicated that the R143A mutant of fXa has a normal reactivity with the other two target inhibitors AT and TFPI, nevertheless, the same mutant also exhibits more than two orders of magnitude elevated dissociation constant in interaction with rTAP (36). In addition to Arg-143, the other three basic residues of fXa are also critical for high affinity interaction of the protease with ZPI. This is derived from the observation that the ZPI reactivities of all three mutants were impaired ∼3-10-fold (Table 1). Nevertheless, the extent of the rate accelerating effect of PZ was improved for both K147A and R150A mutants, suggesting that the interaction of either residue with ZPI is independent of the cofactor. However, there was an ∼10-fold impairment in k2 of R154A inhibition by ZPI in the presence of PZ and the extent of the cofactor function of PZ (66-fold) was also impaired ∼5-fold, possibly suggesting that Arg-154 makes a cofactor dependent interaction with the serpin. Similar to ZPI, the basic residues of the autolysis loop are also involved in fXa interaction with AT in the absence and presence of the heparin cofactors, with Arg-150 being required for the fXa recognition of the heparin-activated conformation of the serpin (20,37). It is interesting to note that the other two proteases that also are known to be possible targets for regulation by ZPI are factors IXa and XIa in both of which the basic residues of the autolysis loop have been conserved. Thus, the autolysis loop of both proteases may be involved in interaction with ZPI.
In addition to the autolysis loop, several heparin-binding exosite mutants of fXa (K96A, K169A, and K236A) also exhibited impaired reactivity with ZPI, as evidenced by an ∼5-10-fold lower k2 values for these mutants with the serpin in the presence of PZ. Nevertheless, the reactivity of all three mutants with ZPI was also impaired in the absence of the cofactor, though to a lower extent of ∼2-3-fold (Table 1). The reactivity of R240A with ZPI was also impaired ∼3-fold in the absence of PZ, but the mutant reacted with the serpin with an improved rate in the presence of PZ, leading to a considerably higher cofactor effect of 1458-fold for PZ in the reaction. The presence of overlapping binding sites for heparin and ZPI on fXa may provide a possible explanation for the previous observation that the reactivity of the serpin with fXa is not significantly influenced by the polysaccharide (9,11). Previous results have indicated that certain basic residues of the heparin-binding exosite of fXa may also interact with factor Va in the prothrombinase complex (30). It is interesting to note that Arg-165, which is the most important residue for fXa interaction with factor Va in the prothrombinase complex (29,38), makes no contact with ZPI since its substitution with Ala had no effect on the reactivity of the mutant with the serpin either in the absence or presence of PZ. The lack of requirement for ZPI interaction with Arg-165 provides a possible explanation for the ability of the serpin to inhibit fXa in the prothrombinase complex, as has been reported in the past (11).
Similar to fXa, PZ is a vitamin K-dependent plasma protein that interacts with negatively charged membrane surfaces in the presence of Ca2+ (23). Although it has been established that PZ interacts with ZPI with a high affinity (11), it is not known whether PZ makes any interaction with fXa on the membrane surface. To determine whether ZPI/PZ complex interacts with either the Gla or the membrane proximal N-terminal EGF domain of fXa, the reactivity of fXa/PC-Gla and fXades-EGF-1 with ZPI was examined both in the absence and presence PZ. The former mutant reacted with the serpin with ∼2-fold lower k2 value in the absence and ∼5-fold slower k2 value in the presence of PZ. The extent of the cofactor effect of PZ in promoting the ZPI inhibition of the Gla mutant was decreased ∼2-fold, suggesting that protein-protein interactions via the Gla domains may play a small role in the protease recognition by the ZPI-PZ complex. On the other hand, ZPI inhibited fXa-des-EGF-1 with similar k2 values both in the absence and presence of PZ, ruling out any functionally critical interaction for the first EGF domain of fXa with either ZPI or PZ in the inhibitory complex. Based on these mutagenesis data, we developed a schematic model for the fXa-ZPI-PZ interaction on PC/PS vesicles which is presented in Fig. 5. It appears that PZ via its Gla domain places ZPI on the negatively charged membrane surface, where fXa is bound via its own Gla domain. The interaction of the serpin with the autolysis loop of fXa then leads to docking of the reactive site loop of the serpin into the active-site pocket of the protease. The ZPI-PZ complex makes additional stabilizing interactions with the basic residues of the heparin-binding exosite, in particular with Lys-236 of fXa located at the C-terminal helix of the protein (35). Thus, apart from the active-site, the basic residues of the autolysis loop (in particular Arg-143) provide most of the binding energy of the protease interaction with the ZPI-PZ complex. It should be noted that the specific interaction of the Gla domain of PZ with the Gla domain of fXa may also make some contribution to the recognition mechanism, since the cofactor function of PZ with the fXa/PC-Gla mutant was impaired∼2-fold. The binding of the Gla domain of fXa (and other coagulation proteases) to PC/PS vesicles positions the active-site pocket of the protease some 60-70 Å above the membrane surface (39). Thus, the EGF-1 domain of fXa may function as a spacer to maintain the active-site pocket of the protease far above the membrane surface. This has been evidenced by the observation that the catalytic efficiency of fXa-des-EGF-1 toward prothrombin in the prothrombinase complex has been markedly impaired (26). The normal reactivity of the EGF-1 deletion mutant of fXa with the ZPI-PZ complex was surprising, and suggests that the cofactor-inhibitor complex has sufficient flexibility to interact with the mutant protease on PC/PS vesicles. Nevertheless, mapping the ZPI interactive sites on PZ will be required before one can speculate about the exact topographical orientation of the fXa-ZPIPZ complex at the membrane surface.
Figure 5.
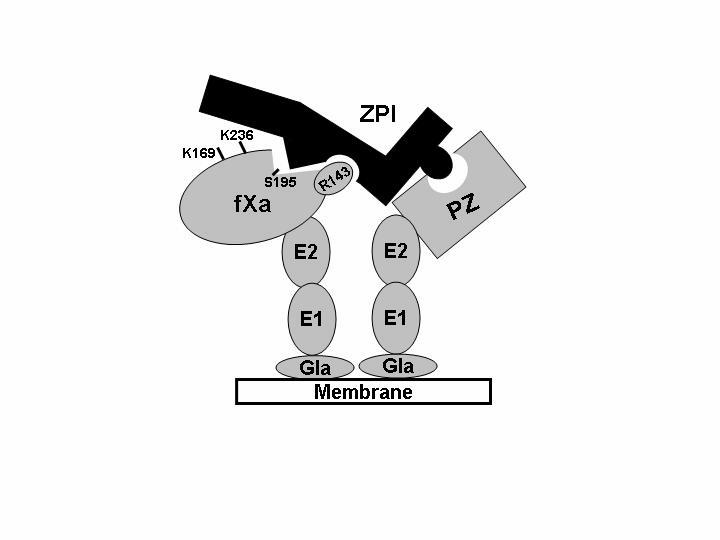
Schematic representation of fXa interaction with the ZPI-PZ complex on the negatively charged membranes. Protein Z interaction with a negatively charged membrane surface via the N-terminal Gla-domain condensates ZPI in the vicinity of fXa bound to the same surface. The ZPI recognition of the autolysis loop of fXa (in particular Arg-143) then leads to docking of the reactive site loop of the serpin into the active-site pocket of protease (marked by Ser-195). In this model, the complex is further stabilized by additional ionic interactions between basic residues of the heparin-binding exosite (particularly Lys-236) of the protease and a complementary site of the serpin. In addition to Lys-236, several other basic residues of this exosite (only Lys-169 shown in the model) also contribute to the interaction (see text for more details). E1, first EGF domain; E2, second EGF domain.
Previous results have indicated that the high affinity interaction of serine proteases with serpins requires an intact active-site pocket where Ser-195 establishes a covalent bond with the P1 residue of the serpin to form an irreversible, SDS-stable enzyme-inhibitor complex (13). However, this is not true with the non-serpin family of inhibitors (such as TFPI), which do not require Ser-195 for their high affinity interaction with the proteases, thus forming reversible and SDS-dissociable enzyme-inhibitor complexes (13). Since unlike the AT-fXa complex, the ZPI-fXa complex is known to be reversible, dissociating upon SDS-gel electrophoresis (11), we investigated the role of Ser-195 of fXa in its interaction with ZPI and compared it with that of the non-serpin inhibitors TFPI and rTAP. The inactive S195A mutant of fXa effectively bound to both TFPI and rTAP, as evidenced by the ability of the mutant to displace both inhibitors from the active-site pocket of fXa, thus restoring all amidolytic activity of the protease. On the other hand, a 200-fold molar excess of S195A fXa failed to compete with fXa for binding to ZPI, which suggests that the catalytic residue of fXa is required for its high affinity interaction with the serpin. Thus, the mechanism of the ZPI-fXa reaction is similar to that of other serpins. The results with the active-site mutant of fXa further support the hypothesis that the physical interaction of PZ with fXa may not be essential for the high affinity interaction of ZPI with the protease, because if the PZ interaction with both ZPI and fXa was required for the reaction, it was expected that the resulting non-productive complexes of the inactive S195A and PZ would have diminished or eliminated the inhibitory function of the serpin.
Acknowledgements
We would like to thank Drs. Mary Jo Heeb and Katia Cabral of The Scripps Research Institute for their generous gift of plasma-derived ZPI, and Andrew Deffenbaugh of The Department of Biochemistry of St. Louis University for his technical assistance with the size exclusion column chromatography of recombinant ZPI on Superdex 200. The research discussed herein was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL 62565 and HL 68571 to ARR).
Footnotes
1. Abbreviations- PZ, protein Z; ZPI, protein Z-dependent protease inhibitor; pZPI, plasma-derived ZPI; rZPI, recombinant ZPI; TFPI, tissue factor pathway inhibitor; rTAP, recombinant tick anticoagulant peptide; fX, factor X; fXa, activated factor X; AT, antithrombin; Gla, γ-carboxyglutamic acid; EGF, epidermal growth factor; fXa-des-EGF-1, fXa mutant in which the first EGF domain has been deleted by recombinant DNA methods; fXa/PC-Gla, fXa mutant in which the Gla domain has been replaced with the corresponding domain of protein C; RVV-X, factor X activating enzyme from Russell's viper venom; SpFXa, Spectrozyme factor Xa; PEG, polyethylene glycol; BSA, bovine serum albumin.
References
- 1.Nesheim ME, Kettner C, Shaw E, Mann KG. J. Biol. Chem. 1981;256:6537–6540. [PubMed] [Google Scholar]
- 2.Furie B, Furie BC. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 3.Davie EW, Fujikawa K, Kisiel W. Biochemistry. 1991;30:10363–10370. doi: 10.1021/bi00107a001. [DOI] [PubMed] [Google Scholar]
- 4.Jackson CM, Nemerson Y. Ann. Rev. Biochem. 1980;49:765–811. doi: 10.1146/annurev.bi.49.070180.004001. [DOI] [PubMed] [Google Scholar]
- 5.Stenflo J. Blood. 1991;78:1637–1651. [PubMed] [Google Scholar]
- 6.Broze GJ, Jr., Girard TJ, Novotny WF. Biochemistry. 1990;29:7539–7546. doi: 10.1021/bi00485a001. [DOI] [PubMed] [Google Scholar]
- 7.Damus PS, Hicks M, Rosenberg RD. Nature. 1973;246:355–357. doi: 10.1038/246355a0. [DOI] [PubMed] [Google Scholar]
- 8.Olson ST, Björk I. In: Thrombin: Structure and Function. Berliner LJ, editor. Plenum Press; New York: 1992. pp. 159–217. [Google Scholar]
- 9.Han X, Fiehler R, Broze GJ., Jr. Proc. Natl. Acad. Sci. (USA) 1998;95:9250–9255. doi: 10.1073/pnas.95.16.9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han X, Huang Z-F, Fiehler R, Broze GJ., Jr. Biochemistry. 1999;38:11073–11078. doi: 10.1021/bi990641a. [DOI] [PubMed] [Google Scholar]
- 11.Han X, Fiehler R, Broze GJ., Jr. Blood. 2000;96:3049–3055. [PubMed] [Google Scholar]
- 12.Yin Z-F, Huang Z-F, Cui J, Fiehler R, Lasky N, Ginsburg D, Broze GJ., Jr. Proc. Natl. Acad. Sci. (USA) 2000;97:6734–6738. doi: 10.1073/pnas.120081897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson ST, Bock PE, Kvassman J, Shore JD, Lawrence DA, Ginsburg D, Björk I. J. Biol. Chem. 1995;270:30007–30017. doi: 10.1074/jbc.270.50.30007. [DOI] [PubMed] [Google Scholar]
- 14.Jin L, Abrahams J, Skinner R, Petitou M, Pike RN, Carrell RW. Proc. Natl. Acad. Sci. (USA) 1997;94:14683–14688. doi: 10.1073/pnas.94.26.14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gettins PGW, Fan B, Crews BC, Turko IV, Olson ST, Streusand VJ. Biochemistry. 1993;32:8385–8389. doi: 10.1021/bi00084a001. [DOI] [PubMed] [Google Scholar]
- 16.de Agostini AI, Watkins SC, Slayter HS, Youssoufian H, Rosenberg RD. J. Cell Biol. 1990;111:1293–1304. doi: 10.1083/jcb.111.3.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J. J. Biol. Chem. 1992;267:12528–12538. [PubMed] [Google Scholar]
- 18.Belzar KJ, Zhou A, Carrell RW, Gettins PGW, Huntington JA. J. Biol. Chem. 2002;277:8551–8558. doi: 10.1074/jbc.M110807200. [DOI] [PubMed] [Google Scholar]
- 19.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manithody C, Yang L, Rezaie AR. Biochemistry. 2002;41:6780–6788. doi: 10.1021/bi0255367. [DOI] [PubMed] [Google Scholar]
- 21.Rezaie AR. J. Biol. Chem. 1998;273:16824–16827. doi: 10.1074/jbc.273.27.16824. [DOI] [PubMed] [Google Scholar]
- 22.Rezaie AR, Olson ST. Biochemistry. 2000;39:12083–12090. doi: 10.1021/bi0011126. [DOI] [PubMed] [Google Scholar]
- 23.Sejima H, Hayashi T, Deyashiki Y, Nishioka J, Suzuki K. Biochem. Biophys. Res. Commun. 1990;171:661–668. doi: 10.1016/0006-291x(90)91197-z. [DOI] [PubMed] [Google Scholar]
- 24.Rezaie AR, Esmon CT. J. Biol. Chem. 1992;267:26104–26109. [PubMed] [Google Scholar]
- 25.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kittur FS, Manithody C, Rezaie AR. J. Biol. Chem. 2004;279:24189–24196. doi: 10.1074/jbc.M402302200. [DOI] [PubMed] [Google Scholar]
- 27.Rezaie AR, Yang L, Manithody C. Biochemistry. 2004;43:2898–2905. doi: 10.1021/bi036145a. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Manithody C, Yang L, Rezaie AR. Protein Sci. 2004;13:431–442. doi: 10.1110/ps.03406904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rezaie AR, Kittur FS. J. Biol. Chem. 2004;279:48262–48269. doi: 10.1074/jbc.M409964200. [DOI] [PubMed] [Google Scholar]
- 30.Rezaie AR. J. Biol. Chem. 2000;275:3320–3327. doi: 10.1074/jbc.275.5.3320. [DOI] [PubMed] [Google Scholar]
- 31.Smirnov MD, Esmon CT. J. Biol. Chem. 1994;269:816–819. [PubMed] [Google Scholar]
- 32.Rezaie AR, Yang L. Biochem. Biophys. Acta. 2001;1528:167–176. doi: 10.1016/s0304-4165(01)00189-1. [DOI] [PubMed] [Google Scholar]
- 33.Yang L, Manithody C, Rezaie AR. Biochemistry. 2002;41:6149–6157. doi: 10.1021/bi015899r. [DOI] [PubMed] [Google Scholar]
- 34.Furie B, Bing DH, Feldman RJ, Robison DJ, Burnier JF, Furie BC. J. Biol. Chem. 1982;257:3875–3882. [PubMed] [Google Scholar]
- 35.Padmanabhan K, Padmanabhan KP, Tulinsky A, Park CH, Bode W, Huber R, Blankenship DT, Cardin AD, Kisiel W. J. Mol. Biol. 1993;232:947–966. doi: 10.1006/jmbi.1993.1441. [DOI] [PubMed] [Google Scholar]
- 36.Rezaie AR. Biochemistry. 2004;43:3368–3375. doi: 10.1021/bi036177y. [DOI] [PubMed] [Google Scholar]
- 37.Yang L, Manithody C, Rezaie AR. Blood. 2004;104:1753–1759. doi: 10.1182/blood-2004-03-1092. [DOI] [PubMed] [Google Scholar]
- 38.Rudolph AE, Porche-Sorbet R, Miletich JP. J. Biol. Chem. 2001;276:5123–5128. doi: 10.1074/jbc.M006961200. [DOI] [PubMed] [Google Scholar]
- 39.Husten EJ, Esmon CT, Johnson AE. J. Biol. Chem. 1987;262:12953–12962. [PubMed] [Google Scholar]