

Abstract
Caldesmon interacts with the NH2-terminal region of actin. It is now shown in airfuge centrifugation experiments that modification of the penultimate cysteine residue of actin significantly weakens its binding to caldesmon both in the presence and absence of tropomyosin. Furthermore, as revealed by fluorescence measurements, caldesmon increases the exposure of the COOH-terminal region of actin to the solvent. This effect of caldesmon, like its inhibitory effect on actomyosin ATPase activity, is enhanced in the presence of tropomyosin. Proteolytic removal of the last three COOH-terminal residues of actin, containing the modified cysteine residue, restores the normal binding between caldesmon and actin. These results establish a correlation between the binding of caldesmon to actin and the conformation of the COOH-terminal region of actin and suggest an indirect rather than direct interaction between caldesmon and this part of actin.
Caldesmon is an actin-binding protein associated with thin filaments of smooth muscle (Sobue et al., 1981; Marston and Lehman, 1985) and nonmuscle cells (Owada et al, 1984). Caldesmon is a possible component of the contractile regulatory apparatus in these cells since it inhibits superprecipitation of actomyosin (Sobue et al, 1981) and the actin-activated ATPase activity of myosin (Dabrowska et al, 1985; Smith and Marston, 1985; Ngai and Walsh, 1985; Moody et al, 1985; Sobue et al, 1985) and its subfragments (Lash et al, 1986; Chalovich et al, 1987). However, defining the precise role of caldesmon in cells requires an understanding of its interactions with several proteins.
The amino-terminal region of caldesmon binds to myosin (Hemric and Chalovich, 1988; Ikebe and Reardon, 1988). This interaction is moderately strong and it saturates with the formation of a 1:1 complex in the presence of ATP (Hemric and Chalovich, 1990). The binding of caldesmon to myosin is predominantly through the S2 region of myosin (Ikebe and Reardon, 1988; Hemric and Chalovich, 1990). The NH2-terminal region of caldesmon has also been reported to contain a calmodulin-binding site (Wang et al, 1989). The center region of caldesmon is thought to serve a structural role as it has no known activity, exists primarily as an extended helix (Wang et al., 1991), and is absent in some nonmuscle caldesmons (Ball and Kovala, 1988; Bryan and Lee, 1991; Hayashi et al., 1991). The COOH-terminal 40-kDa region of caldesmon binds to actin and calmodulin (Szpacenko and Dabrowska, 1986; Fujii et al, 1987; Yazawa et al, 1987) and reversibly inhibits actin-activated ATP hydrolysis of myosin (Dabrowska et al, 1986; Fujii et al, 1987). Several laboratories are working to further define the actin and calmodulin-binding regions in the COOH-terminal domain of caldesmon (see Wang et al, 1991; and Hayashi et al, 1991; and references therein).
Several attempts have been made to localize the caldesmon-binding sites on actin. These studies have been confined to NH2-terminal sequences 1–7 and 18–28 of actin which are implicated in other macromolecular interactions. Segment 1–7 is involved in the binding of both troponin I (Garbarek and Gergely, 1987; Levine et al, 1988) and myosin-ATP (DasGupta and Reisler, 1989; Bertrand et al, 1989), and to a lesser extent myosin in rigor conditions (Mejean et al, 1987, Miller et al, 1987). Cross-linking experiments (Bartegi et al, 1990), nmr spectroscopy (Patchell et al, 1989), and competition studies with antibodies directed against residues 1–7 of actin (Adams et al, 1990) have shown that the same segment of actin is involved also in the binding of caldesmon. This deduced overlap between caldesmon and myosin-ATP sites on actin is reasonable since caldesmon readily displaces myosin S1-ATP from actin (Chalovich et al, 1987; Hemric et al, 1988). Region 18–28 of actin appears to be important in binding to both troponin I (Levine et al, 1988) and myosin in rigor (Mejean et al, 1987). Antibodies directed against this region have little effect on the binding of caldesmon to actin but do have somewhat greater impact on the binding of the 20-kDa fragments of caldesmon to actin (Adams et al, 1990).
The COOH-terminal region of actin and its possible role in the binding of caldesmon merit attention for at least three reasons. First, the atomic resolution structure of actin suggests that its COOH-terminus is in close proximity with the amino-terminal region (Kabsch et al, 1990). Second, myosin heads which compete with caldesmon for the binding to actin interact via their light chains with the COOH-terminal segment of actin (Sutoh, 1982). Third, the COOH-terminal region of actin has been implicated in interactions with other proteins such as profilin (Malm et al, 1980), gelsolin (Boyer et al, 1987; Sutoh and Yin, 1989; Doi et al, 1991), and depactin (Sutoh and Mabuchi, 1984).
We now show that the binding of caldesmon to actin is influenced by changes in the COOH-terminal region of actin. The binding of caldesmon to pyrene labeled- and unmodified actin was measured by a co-sedimentation method. Modification of Cys-374 caused a 3–4-fold decrease of the binding constant in the presence and absence of tropomyosin. The binding of caldesmon to pyrene-labeled actin was also monitored by fluorescence titration measurements. The fluorescence decreased by up to 15% as the molar ratio of caldesmon to actin was increased to 0.4:1. Similar titrations in the presence of tropomyosin revealed an enhanced fluorescence loss, up to 25%. Titrations with nitromethane revealed a caldesmon-induced increase in the accessibility of the pyrene probe. Interestingly, proteolytic removal of the last several COOH-terminal residues of actin abolished the negative effect of Cys-374 labeling on the binding of caldesmon. Therefore, while the COOH-terminus of actin is not required for the binding of caldesmon to actin, it is involved indirectly in that interaction.
MATERIALS AND METHODS
Reagents
Trypsin, soybean trypsin inhibitor, ATP, N,N-dimethylformamide, Folin and Ciocalteu’s phenol reagent, and β-mercapto-ethanol were from Sigma. Bovine serum albumin was obtained from United States Biochemical Corp. (Cleveland, OH). Dithiothreitol (DTT)1 was purchased from Schwarz/Mann Biotech (Cleveland, OH). N-(l-Pyrenyl) iodoacetamide, 5-(iodoacetamido)fluorescein, and 5-(iodoacetamidoethyl) aminonaphthalene-1-sulfonic acid (1,5-IAEDANS) were obtained from Molecular Probes (Junction City, OR). The Bradford protein assay was purchased from Bio-Rad. Nitromethane was purchased from Matheson Coleman and Bell (Los Angeles, CA). Distilled and Millipore-filtered water and analytical grade reagents were used in all experiments.
Preparations of Proteins
Rabbit skeletal muscle actin was prepared in G-actin buffer (0.5 mm β-mercaptoethanol, 0.2 mm ATP, 0.2 mm CaCl2, and 5 mm Tris, pH 7.6) by the procedure of Spudich and Watt (1971). Smooth muscle actin was isolated from turkey gizzards as described by Strzelecka-Golaszewska et al. 1980 and prepared in G-actin buffer identical to that used in skeletal muscle actin. Tropomyosin was isolated from turkey gizzards (Bretscher, 1984). Caldesmon was purified from turkey gizzards by two methods as described by Velaz et al. 1989. Caldesmon was concentrated by ultrafiltration on a YM-2 membrane (Amicon), frozen in dry ice/ acetone and shipped to UCLA in a buffer consisting of 100 mm NaCl, 10 mm imidazole, 1 mm DTT, pH 7.4. Caldesmon concentrations were determined by the Lowry assay (1951) using bovine serum albumin as a standard. The molecular weight of caldesmon used in this work (87,000) is the value assigned by cDNA sequencing of this protein (Bryan et al., 1989).
Labeling of Proteins
Actin was labeled at Cys-374 with N-(1-pyrenyl)iodoacetamide following the method established by Cooper et al. 1983. The extent of labeling was measured by using a molar extinction coefficient of E344 = 2.2 × 104 M−1 cm−1 for the protein-dye complex (Kouyama and Mihashi, 1981). The labeling stoichiometry ranged between 0.80 and 1.00 probe/actin. 5-(Iodoacetamido) fluorescein-actin modified at Cys-374 was obtained by incubating F-actin with a 20-fold molar excess of 5-(iodoacetamido) fluorescein for 19 h at room temperature (Lin and Dowben, 1983). The excess reagent was removed on two Penefsky columns (1977) and by subsequent dialysis in G buffer. The labeling efficiency was determined using a molar extinction coefficient of E492 = 60,000 M−1 cm−1 and ranged between 0.60 and 0.80 dye/actin. 1,5-IAEDANS modified actin was prepared by a procedure identical to that described for fluorescein labeling except the reaction time was decreased to 3 h. The molar extinction coefficient used for labeled protein was E337 = 6300 M−1 cm−1. The modified protein contained between 0.60 and 0.70 dye/actin. Caldesmon was labeled with [14C] iodoacetamide as described by Velaz et al. 1989. The concentrations of modified actin and caldesmon were measured using the Bradford assay (1976) or the Lowry assay (1951), respectively.
Airfuge Binding Experiments
Unmodified and labeled G-actin (4.0 μm) were polymerized with 40 mm NaCl and 2 mm MgCl2 in G buffer and subsequently incubated with 1.5 μ m caldesmon. The protein mixtures were centrifuged at 160,000 × g for 25 min in an air-driven ultracentrifuge (Beckman Instruments). Caldesmon solutions were prespun under identical conditions prior to these experiments in order to remove aggregates. The pelleted proteins were resolubilized with G buffer. Supernatant and resolubilized pellet fractions were denatured and run on either 10 or 15% polyacrylamide gels (Laemmli, 1970). Coomassie Blue R-stained protein bands were scanned with a Biomed Instruments (Fullerton, CA) scanning densitometer interfaced to a DTK computer. The densitometric traces of the scanned protein bands were analyzed to determine the molar ratios of caldesmon bound to actin. Molar ratios of bound proteins were calculated using molar stain ratios obtained from appropriate calibration gels.
Determination of Binding Constants of Caldesmon to Actin
Binding constants of caldesmon to unmodified and pyrene-labeled actin were determined by measuring the binding of [14C] iodoacetamide-labeled caldesmon to actin. The binding was measured by the depletion of radioactivity from the supernatants following sedimentation of the reaction mixtures at 160,000 × g for 25 min. Binding data were corrected for the amount of caldesmon which sedimented in the absence of actin and for the amount of caldesmon which did not sediment at high actin concentrations. The treatment of data is as described by Velaz et al. 1989. All binding experiments were done at 25 °C in solutions containing 42 mm NaCl, 9.6 mm imidazole-HCl, pH 7.0, 4.6 mm MgCl2, 0.25 mm EGTA, and 1 mm DTT. The rabbit skeletal actin concentration was 10 μm and the turkey gizzard tropomyosin, when present, was 2.5 μm.
Fluorescence Measurements
Fluorescence intensities of pyrene-labeled actin were measured in the Spex Fluorolog spectrophotometer (Spex, Industries, Inc., Edison, NJ) as previously described (Miller et al., 1988). The excitation and emission monochromators were set at 368 and 407 nm, respectively. Prior to measurements, pyrene-labeled actin (4.0 μm) was polymerized by addition of 40 mm NaCl and 2 mm MgCl2 in G buffer. The polymerization process was followed by fluorescence measurements. After the fluorescence intensity of F-actin was recorded, the sample was titrated with either caldesmon or tropomyosin. Subsequent changes in fluorescence were monitored as a function of titrant concentration.
Stern-Volmer constants were obtained from measurements of fluorescence quenching upon the addition of nitromethane to polymerized modified actin (4.0 μm) either alone or in the presence of tropomyosin and/or caldesmon.
Experiments with fluorescein-modified actin were performed as described above. The excitation and emission monochromators were set at 490 and 520 nm, respectively. Unless indicated otherwise, all fluorescence measurements were carried out in the same solvents as used in the binding experiments.
Proteolytic Removal of Labeled Cys-374 from Actin
In order to remove the last 3 COOH-terminal residues of actin, F-actin modified at Cys-374 with 1,5-IAEDANS was cleaved by trypsin (5:1 molar ratio of actin:trypsin) at 25 °C for 20 min (Mornet and Ue, 1984). The reaction was stopped with excess soybean trypsin inhibitor. Removal of the last 3 residues (including the labeled Cys-374) without any significant damage to actin was verified by comparisons of Coomassie-stained bands and fluorescence bands of the modified proteins on SDS gels. Samples of modified actin, before and after cleavage, were run on 10% SDS-polyacrylamide gels and stained with Coomassie Blue R. Identical gels (unstained) were examined under UV illumination. Co-sedimentations of caldesmon and actin were performed with unmodified actin, 1,5-IAEDANS-modified (uncleaved) actin, and 1,5-IAEDANS-modified (cleaved) actin as described under Airfuge Binding Experiments.
RESULTS
Binding Experiments
The possible involvement of the COOH-terminal region of actin in the binding of caldesmon was tested with fluorescently labeled actin. The rationale for these experiments was to observe changes in the fluorescence of probes placed at Cys-374 of actin upon the binding of caldesmon to this protein. We first determined if the addition of the bulky pyrene group on the penultimate residue of actin affected the binding of caldesmon to actin. The inset to Fig. 1A shows that modification of actin with pyrene-iodoacetamide causes a noticeable decrease in the amount of caldesmon pelleted with actin in a simple co-sedimentation assay. It should be noted that the modification of actin with pyrene had no effect on the amount of actin present in the supernatant in these assays.
Fig. 1. Effect of pyrene modification of actin on the binding of caldesmon to actin and actin-tropomyosin.
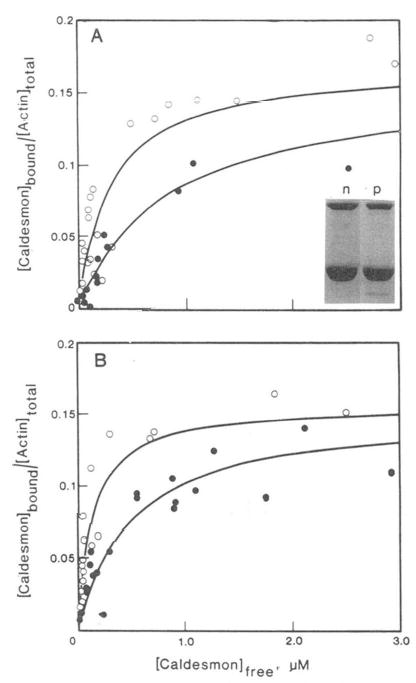
Varied concentrations of 14C-labeled caldesmon were incubated with 10 μm actin (○) or pyrene-labeled actin (•) at 25 °C in solutions containing 42 mm NaCl, 9.6 mm imidazole-HCl, pH 7.0, 4.6 mm MgCl2,0.25 mm EGTA, and 1 mm DTT. A, absence of tropomyosin: apparent association constants are 3.6 × 106 and 1.3 × 106 M−1 for normal and pyrenyl actin, respectively. Inset: SDS gels of co-sedimentation of unlabeled caldesmon with normal (n) and pyrenyl (p) actin. B, presence of 2.5 μm smooth muscle tropomyosin. Apparent association constants are 7.5 × 106 and 2.0 × 106 M−1 for normal and pyrenyl actin.
To determine the changes in binding resulting from labeling the penultimate residue in actin, the binding of [14C] iodoacetamide-labeled caldesmon to modified and unmodified actin was measured. As shown earlier, the labeling of caldesmon with iodoacetamide did not significantly alter its binding to actin (Velaz et al, 1989). Fig. 1A shows the fraction of actin with bound caldesmon as a function of the free caldesmon concentration. Both unmodified and pyrene-actin are saturated with caldesmon at a stoichiometry of 1 caldesmon to 6 actin monomers. This differs slightly from our earlier estimate of 1:7–10 (Velaz et al., 1989) because of the use of a lower value for the molecular weight of caldesmon in the present case. The affinity of caldesmon for actin is clearly depressed by the pyrene label; the concentration of free caldesmon required for half-saturation is 2.8-fold higher for pyrene-actin than for unmodified actin. No attempt was made to determine if this change in apparent binding constant is due to a change in the cooperativity parameter or in the binding constant to an isolated binding site (McGhee and von Hippie, 1974). Actin which was subjected to a mock modification, in which the pyrenyl-iodoacetamide was omitted, was identical to unmodified untreated actin.
Fig. 1B shows that in the presence of smooth muscle tropomyosin, the binding of caldesmon to unmodified actin is 3.7-fold stronger than the binding to pyrene-actin. Comparison of Fig. 1A and B, shows that tropomyosin has a similar stimulatory effect on the binding of caldesmon to both actin and to pyrene-actin. Thus, modification of the penultimate residue of actin with a bulky group weakens but does not abolish the binding of caldesmon to actin.
Caldesmon-induced Changes in the COOH-terminal Site of Actin
Since modification of the COOH-terminus of actin weakens the affinity of caldesmon for actin it is not surprising that the environment of the fluorescent probe is altered by caldesmon. Fig. 2 shows the titration of 4.0 μm actin with caldesmon in both the absence (2A) and presence (2B) of smooth muscle tropomyosin under conditions similar to those used in the co-sedimentation studies. The fluorescence intensity decreases by 15% in the absence of tropomyosin and by about 25% in the presence of tropomyosin. It is interesting to note that tropomyosin, which potentiates the inhibitory action of caldesmon, also increases the caldesmon-induced conformational change in the COOH-terminal region of actin.
Fig. 2. Effect of caldesmon on the fluorescence of pyrene-labeled actin.
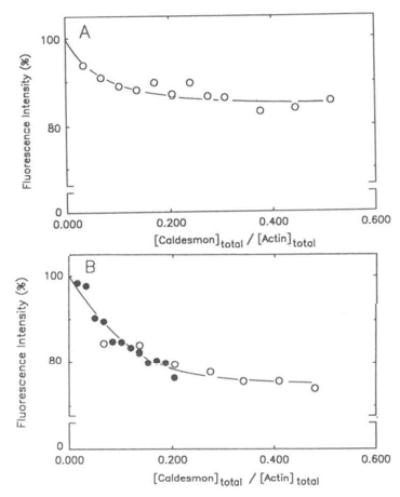
Pyrene-labeled skeletal F-actin (4.0 μm) was titrated with up to 1.7 μm caldesmon in the absence (A) and presence (B) of smooth muscle tropomyosin (2.0 μm). Pyrene-actin was polymerized with 2 mm MgCl2 and 40 mm NaCl in G buffer before titrations with caldesmon. Fluorescence measurements were monitored as described under “Materials and Methods.” Relative fluorescence intensities are plotted as a function of molar ratios of caldesmon added to actin. The open and closed symbols refer to two separate sets of experiments.
Fluorescence titration studies were also done in the presence of 100 mm NaCl (data not shown). The magnitudes of changes in fluorescence intensity were similar to those shown in Fig. 2 for 40 mm NaCl, although approximately 10-fold higher total caldesmon concentrations were required to reach the same titration end points. Thus, increasing the ionic strength considerably weakens the binding of caldesmon to actin but does not affect the involvement of the COOH-terminal region in this interaction.
Titrations of pyrene-labeled actin from smooth muscle verified that the effects of caldesmon on the environment of Cys-374 are not specific to the α-skeletal protein. Caldesmon quenched the fluorescence of smooth pyrene-actin by 10 and 20% in the absence and presence of tropomyosin, respectively (not shown). Although these effects were somewhat smaller than those observed for α-skeletal actin (Fig. 2), they saturated at similar molar ratios of caldesmon to smooth and skeletal actins.
The decrease in fluorescence resulting from the binding of caldesmon to actin could be due to several factors. One common reason for decreased fluorescence is increased exposure of the probe to solvent quenchers. To determine if caldesmon functions in this manner, the quenching of both pyrene and fluorescein probes on Cys-374 by nitromethane was measured. Fig. 3 shows Stern-Volmer plots for nitro-methane quenching of the fluorescence of pyrene-actin and fluorescein-actin in the presence (solid symbols) and absence (open symbols) of caldesmon. The Stern-Volmer quenching constants (slopes of the curves) for both pyrene-actin and fluorescein-actin are greater in the presence of caldesmon than in its absence (Table I). Thus, caldesmon does increase the exposure of the COOH-terminal segment of actin to the solvent.
Fig. 3. Stern-Volmer plots of pyrene-actin (A) and fluorescein-actin (B) quenching by nitromethane.
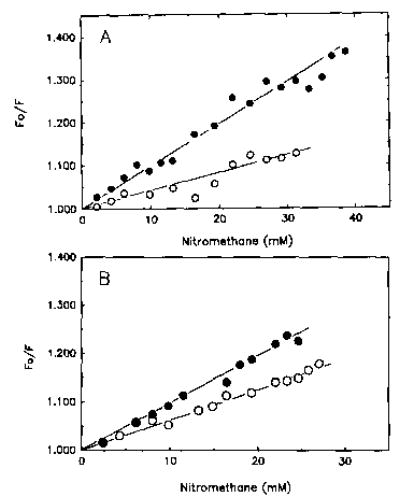
A, pyrene-modified F-actin (4.0 μm) was titrated with nitromethane in the presence of (•) and absence (○) of caldesmon (1.5 μm). Fluorescence changes are expressed as a ratio of initial fluorescence (F0) to the fluorescence after addition of nitromethane (F). Table I lists Stern-Volmer constants for all quenching experiments. Solvent conditions were identical to those described in the legend to Fig. 2. B, actin modified with 5-(iodoacetamido)-fluorescein at Cys-374 was polymerized with 2 mm MgCl2 and 40 mm NaCl in G buffer. The polymerized fluorescein-actin (4.0 μm) was titrated with nitromethane in the presence (•) and absence (○) of caldesmon (1.5 μm). Solvent conditions were the same as described in the legend to Fig. 2.
Table I. Stern-Volmer quenching constants (Ksv) for titrations of pyrene- and fluorescein-actin with nitromethane.
Stern-Volmer constants correspond to the slopes of the curves shown in Figs. 3 and 4 or in similar plots.
|
Ksv |
|||
|---|---|---|---|
| Probe attached to Cys-374 on actin | Protein added | Skeletal actin | Smooth actin |
| m−1 | |||
| Pyrene | 4.1 | 4.4 | |
| Caldesmon (1.5 μm) | 10.3 | 7.3 | |
| Tropomyosin (2 μm) | 3.1 | ||
| Caldesmon (1.5 μm) + tropomyosin (2.0 μm) | 8.6 | ||
| Fluorescein | 6.5 | ||
| Caldesmon (1.5 μm) | 9.6 | ||
Qualitatively similar effects of caldesmon on the accessibility of the pyrene probe to collisional quenching were observed also for smooth muscle actin (Fig. 4). The smaller changes in Stern-Volmer coefficients noted with smooth actin than those measured for skeletal actin (Table I) were consistent with the corresponding fluorescence changes induced in these proteins by caldesmon. The involvement of tropomyosin in potentiating caldesmon-induced changes in actin (Fig. 2) was readily detected also in quenching titration experiments (Fig. 4). While tropomyosin alone decreased the accessibility of the pyrene probe on actin to nitromethane, this effect was reversed in the presence of caldesmon and tropomyosin (Fig. 4).
Fig. 4. Stern-Volmer plot of fluorescence quenching of pyrene-labeled actin from smooth muscle.
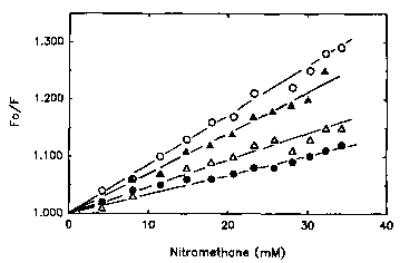
Polymerized pyrene-actin (4.0 μm) from smooth muscle was titrated with nitromethane (▵). The quenching of pyrene fluorescence was also monitored in the presence of 2.0 μm tropomyosin (•), 1.5 μm caldesmon (▴), and 2.0 μm tropomyosin and 1.5 μm caldesmon simultaneously (○).
Removal of the Probe Reverses Modification-induced Changes in the Binding of Caldesmon to Actin
The experiments described so far do not indicate whether modification of Cys-374 with a fluorescent probe eliminates a favorable interaction between actin and caldesmon (either by direct or indirect effects) or whether the presence of the label introduces an unfavorable interaction (either direct or indirect). To examine this point, we compared the binding of caldesmon to pyrene-actin and to cleaved pyrene-actin. It might be argued that if the COOH-terminal residues of actin interact favorably with caldesmon then their proteolytic removal (together with the probe) should not restore the normal binding between these proteins and could even impair it further as in the case of actin and profilin (Malm et al., 1980). The cleaved pyrene-actin was prepared by a limited tryptic digestion of F-actin which removed the last 3 COOH-terminal residues of this protein (Mornet and Ue, 1984). Fig. 5A (lanes 1′ and 2′) shows that the digestion with trypsin totally removes the fluorescent probe from actin without any appreciable proteolytic damage to actin (Fig. 5A, lanes 1 and 2). The minor cleavage of actin to yield a small fraction of lower molecular weight fragments is inconsequential to the binding experiments since these fragments do not pellet with F-actin. Fig. 5B shows that the amounts of caldesmon which co-sediment with actin are restored to control levels after cleavage of the pyrene group. Thus, in contrast to actin-profilin interactions (Malm et al., 1980), the modification of the penultimate cysteine residue does not have the same effect on the binding of caldesmon as removing the 3 COOH-terminal residues from actin. This suggests that the COOH-terminus of actin has an indirect role in the interactions with caldesmon.
Fig. 5. Binding of caldesmon to unmodified, modified, and cleaved actin.
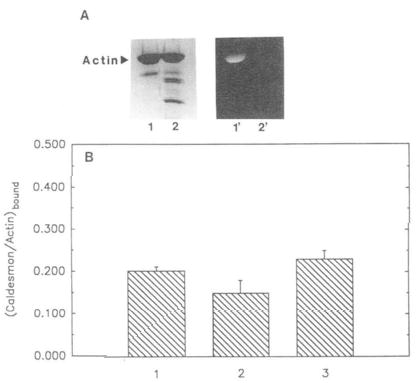
1,5-IAEDANS-labeled actin (at Cys-374) and tryptically cleaved F-actin (to remove the COOH-terminal residues) were prepared as described under “Materials and Methods.” A, lanes 1 and 2, Coomassie stained, 1,5-IAEDANS modified, uncleaved and cleaved actin, respectively. Lanes 1′ and 2′ correspond to the same bands viewed under UV illumination. B, comparison of caldesmon binding to unmodified, modified, and cleaved actin. Unmodified, modified, and cleaved actin (4.0 μm each) were allowed to independently incubate with caldesmon (1.5 μm) Pelleted samples from co-sedimentation experiments (see “Materials and Methods’) were run on SDS gels, Coomassie stained, and analyzed by densitometer. Molar ratios of caldesmon bound to actin are represented in the form of a bar graph. 1, caldesmon bound to unmodified actin (0.20 ± 0.01); 2, caldesmon bound to 1,5-IAEDANS modified (uncleaved) actin (0.15 ± 0.03); 3, caldesmon bound to 1,5-IAEDANS modified (cleaved) actin (0.23 ± 0.02). Standard deviation (n = 5) is indicated by error bars above each bar column.
DISCUSSION
The interaction between caldesmon and actin is interesting for several reasons. First, the binding of caldesmon to actin results in the inhibition of ATP hydrolysis by actomyosin which could have regulatory significance. Second, like the tropomyosin-troponin complex, caldesmon appears to bind along the actin filament (Lehman et al., 1989) with each caldesmon monomer covering several actin monomers (Velaz et al., 1989, and references therein). However, in contrast to tropomyosin, caldesmon does not have repeating actin-binding structural units and it is unclear how binding of the COOH-terminal region of caldesmon stabilizes its binding along 7 or more actin monomers. Third, while caldesmon and tropomyosin-troponin both inhibit the actin-activated ATP-ase of myosin subfragments they operate by distinct mechanisms. Tropomyosin-troponin has little effect on the binding of myosin subfragments (S1 or heavy meromyosin) to actin during steady-state ATP hydrolysis but does inhibit the Vmax of hydrolysis (Chalovich et al, 1981; Chalovich and Eisenberg, 1982). Caldesmon, however, inhibits the ATPase activity and the binding of S1-ATP to actin in parallel (Chalovich et al, 1987; Hemric and Chalovich, 1988; Velaz et al, 1990). Finally, it has been shown that caldesmon binds to the NH2-terminal region of actin, residues 1–7 (Bartegi et al, 1990; Levine et al, 1990; Adams et al, 1990), which is also important for the activation of myosin ATPase by actin (DasGupta and Reisler, 1989; Bertrand et al, 1989). Caldesmon may also interact weakly with the region in the actin sequence from residues 18 to 28 (Adams et al, 1990) which are involved in rigor binding between myosin and actin (Mejean et al, 1987).
To further define the sites of actin involved in the binding of caldesmon and map their overlap with the myosin-binding site, we have examined the possibility of an interaction between caldesmon and the COOH-terminal region of actin. The present results show that the COOH-terminal region of actin influences its interaction with caldesmon. Modification of Cys-374 with a pyrene group weakens the binding constant by 3- or 4-fold. Reduced binding of caldesmon to actin has been observed also in co-sedimentation experiments with 1,5-IAEDANS (Fig. 5) and fluorescein-labeled actins. The complementary fluorescence measurements reveal that the conformation of the COOH-terminal region of actin changes upon binding to caldesmon. This interaction results in greater exposure of the probe to solvent. Thus, the fluorescence of the pyrenyl probe decreases upon binding to caldesmon and this is accompanied by an increase in quenching as seen by the Stern-Volmer analysis. Again, as noted for the binding affinities, the perturbation of the probe environment by caldesmon is not specific to the pyrene group or the α-skeletal actin. Similar effects have been observed for fluorescein actin and smooth muscle actin.
The above evidence for the involvements of the COOH-terminal region of actin in the binding of caldesmon was subjected to further scrutiny by examining the co-sedimentation of caldesmon with a truncated actin. The observation that the proteolytic removal of the 1,5-IAEDANS probe (together with residues 373–375) from actin (Mornet and Ue, 1984) abolished the probe-induced inhibition of caldesmon binding, sheds light on the nature of caldesmon interaction with the COOH-terminal residues of actin. It is unlikely that these residues interact directly and favorably with caldesmon as they do with profilin (Malm et al, 1980). In the case of a favorable interaction the loss of the 3 COOH-terminal residues of actin would not yield a protein which binds caldesmon at least as well as the intact actin. Also, direct contact between caldesmon and the COOH-terminal residues of actin appears unlikely in view of the increased exposure of the COOH-terminal region to solvent by caldesmon.
The most likely explanation of the results of this study is that of interdependence between the caldesmon-binding site on actin and the conformation of its COOH-terminal region. Probe-induced changes in that segment of actin affect the binding of caldesmon and reciprocally, the binding of caldesmon to actin changes the conformation of the COOH-terminal region. This article appears significantly tighter in the presence of tropomyosin since caldesmon-induced changes in actin are greater in the presence than in the absence of tropomyosin. This tropomyosin effect cannot be explained by simple enhancement of caldesmon binding to actin since binding saturation of actin is readily achieved with caldesmon alone. Thus, for example, stronger binding (in 40 mm NaCl) and weaker binding caldesmon (in 100 mm NaCl) elicited the same final changes in pyrene-actin fluorescence, albeit at different caldesmon concentrations. Evidently, tropomyosin potentiates a conformational change in actin caused by the binding of caldesmon. Since tropomyosin potentiates also the inhibition of actomyosin-ATPase by caldesmon, the two effects may be correlated.
The functional implication of the conformational coupling between the COOH-terminal region and the caldesmon site on actin is at present speculative but interesting. Both caldesmon and S1 interact with the NH2-terminal region of actin. The binding of S1 to pyrene-actin results in a 70% decrease in its fluorescence (Criddle et al, 1985) and light chains A1 appear to bind to the COOH-terminal region on actin (Sutoh, 1982). These direct or indirect effects of myosin on the COOH-terminal region of actin could modulate the interaction of caldesmon with its binding sites on actin.
Acknowledgments
We wish to thank Dr. Laly Velaz for assistance in preliminary studies related to this work and to Caryl E. Benson and Michael Vy Freedman for expert technical assistance.
Footnotes
This work was supported by United State Public Health Service Grants AR 22031 (to E. R.) and AR 35216 (to J. M. C.).
The abbreviations used are: DTT, dithiothreitol; S1, myosin subfragment-1; pyrene-actin, actin labeled at Cys-374 with N-(1-pyrenyl)iodoacetamide; fluorescein-actin, actin labeled at Cys-374 with 5-(iodoacetamido) fluorescein; 1,5-IAEDANS, 5-(iodoacetamidoethyl) aminonaphthalene-l-sulfonic acid; SDS, sodium dodecyl sulfate; EGTA, [ethylenebis(oxyethylenenitrilo)] tetraacetic acid.
References
- Adams S, DasGupta G, Chalovich JM, Reisler E. J Biol Chem. 1990;265:19652–19657. [PubMed] [Google Scholar]
- Ball EH, Kovala T. Biochemistry. 1988;27:6093–6098. doi: 10.1021/bi00416a039. [DOI] [PubMed] [Google Scholar]
- Bartegi A, Fattoum A, Derancourt J, Kassab R. J Biol Chem. 1990;265:15231–15238. [PubMed] [Google Scholar]
- Bertrand R, Chaussepied P, Audemard E, Kassab R. Eur J Biochem. 1989;181:747–754. doi: 10.1111/j.1432-1033.1989.tb14787.x. [DOI] [PubMed] [Google Scholar]
- Boyer M, Feinberg J, Hue HK, Capony JP, Benyamin Y, Roustan C. Biochem J. 1987;248:359–364. doi: 10.1042/bj2480359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Bretcher A. J Biol Chem. 1984;259:12873–12880. [PubMed] [Google Scholar]
- Bryan J, Imai M, Lee R, Moore P, Cook RG, Lin WG. J Biol Chem. 1989;264:13873–13879. [PubMed] [Google Scholar]
- Bryan J, Lee R. J Muscle Res Cell Motil. 1991;12:372–375. doi: 10.1007/BF01738592. [DOI] [PubMed] [Google Scholar]
- Chalovich JM, Eisenberg E. J Biol Chem. 1982;257:2432–2437. [PMC free article] [PubMed] [Google Scholar]
- Chalovich JM, Chock PB, Eisenberg E. J Biol Chem. 1981;256:575–578. [PMC free article] [PubMed] [Google Scholar]
- Chalovich JM, Cornelius P, Benson CE. J Biol Chem. 1987;262:5711–5716. [PubMed] [Google Scholar]
- Cooper JA, Walker SB, Pollard TD. J Muscle Res Cell Motil. 1983;4:253–262. doi: 10.1007/BF00712034. [DOI] [PubMed] [Google Scholar]
- Criddle AH, Geeves MA, Jeffries T. Biochem J. 1985;232:343–349. doi: 10.1042/bj2320343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska R, Goch A, Galazkiewicz B, Osinska H. Biochim Biophys Acta. 1985;842:70–75. doi: 10.1016/0304-4165(85)90295-8. [DOI] [PubMed] [Google Scholar]
- DasGupta G, Reisler E. J Mol Biol. 1989;207:833–836. doi: 10.1016/0022-2836(89)90249-0. [DOI] [PubMed] [Google Scholar]
- Doi Y, Banba M, Vertut-Doi A. Biochemistry. 1991;30:5769–5777. doi: 10.1021/bi00237a020. [DOI] [PubMed] [Google Scholar]
- Fujii T, Imai M, Rosenfeld GC, Bryan J. J Biol Chem. 1987;262:2757–2763. [PubMed] [Google Scholar]
- Grabarek Z, Gergely J. Biophys J. 1987;51:331a. [Google Scholar]
- Hayashi K, Fujio Y, Kato I, Sobue K. J Biol Chem. 1991;266:355–361. [PubMed] [Google Scholar]
- Hemric ME, Chalovich JM. J Biol Chem. 1988;263:1878–1885. [PubMed] [Google Scholar]
- Hemric ME, Chalovich JM. J Biol Chem. 1990;265:19672–19678. [PMC free article] [PubMed] [Google Scholar]
- Ikebe M, Reardon S. J Biol Chem. 1988;263:3055–3058. [PubMed] [Google Scholar]
- Kabsch W, Mannherz HG, Suck D, Pai EF, Holmes KC. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- Kouyama T, Mihashi T. Eur J Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lash JA, Sellers JR, Hathaway DR. J Biol Chem. 1986;261:16155–16160. [PubMed] [Google Scholar]
- Lehman W, Craig R, Lui J, Moody C. J Muscle Res Cell Motil. 1989;10:101–112. doi: 10.1007/BF01739966. [DOI] [PubMed] [Google Scholar]
- Levine BA, Moir AJG, Perry SV. Eur J Biochem. 1988;172:389–397. doi: 10.1111/j.1432-1033.1988.tb13899.x. [DOI] [PubMed] [Google Scholar]
- Levine BA, Moir AJG, Audemard E, Mornet D, Patchell VB, Perry SV. Eur J Biochem. 1990;193:687–696. doi: 10.1111/j.1432-1033.1990.tb19388.x. [DOI] [PubMed] [Google Scholar]
- Lin TI, Dowben RM. J Biol Chem. 1983;258:5142–5150. [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Malm B, Nystrom LE, Lindberg U. FEBS Lett. 1980;113:241–244. doi: 10.1016/0014-5793(80)80601-6. [DOI] [PubMed] [Google Scholar]
- Marston SB, Lehman W. Biochem J. 1985;231:517–522. doi: 10.1042/bj2310517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD, von Hippel PH. J Mol Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- Mejean C, Boyer M, Labbe JP, Marlier L, Benyamin Y, Roustan C. Biochem J. 1987;244:571–577. doi: 10.1042/bj2440571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L, Kalnoski M, Yunossi Z, Bulinski JC, Reisler E. Biochemistry. 1987;26:6064–6070. doi: 10.1021/bi00393a018. [DOI] [PubMed] [Google Scholar]
- Miller L, Phillips M, Reisler E. J Biol Chem. 1988;263:1996–2002. [PubMed] [Google Scholar]
- Moody CJ, Marston SB, Smith CWJ. FEBS Lett. 1985;191:107–112. doi: 10.1016/0014-5793(85)81003-6. [DOI] [PubMed] [Google Scholar]
- Mornet D, Ue K. Proc Natl Acad Sci U S A. 1984;81:3680–3684. doi: 10.1073/pnas.81.12.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngai PK, Walsh MP. Biochem J. 1985;230:695–707. doi: 10.1042/bj2300695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owada MK, Hakura A, Iida K, Yahara L, Sobue K, Kakiuchi S. Proc Natl Acad Sci U S A. 1984;81:3133–3137. doi: 10.1073/pnas.81.10.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patchell VB, Perry SV, Moir AJG, Audemard E, Mornet D, Levine BA. Biochem Soc Trans. 1989;17:901. [Google Scholar]
- Penefsky H. J Biol Chem. 1977;252:2891–2899. [PubMed] [Google Scholar]
- Smith CWJ, Marston SB. FEBS Lett. 1985;184:115–119. doi: 10.1016/0014-5793(85)80665-7. [DOI] [PubMed] [Google Scholar]
- Sobue K, Muramoto Y, Fujita M, Kakiuchi S. Proc Natl Acad Sci U S A. 1981;78:5652–5655. doi: 10.1073/pnas.78.9.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobue K, Takahashi K, Wakabayashi I. Biochem Biophys Res Commun. 1985;132:645–651. doi: 10.1016/0006-291x(85)91181-7. [DOI] [PubMed] [Google Scholar]
- Spudich JA, Watt S. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- Strzelecka-Golaszewska H, Prochniewicz E, Nowak E, Zmorzynski S, Drabikowski W. Eur J Biochem. 1980;104:41–52. doi: 10.1111/j.1432-1033.1980.tb04397.x. [DOI] [PubMed] [Google Scholar]
- Sutoh K. Biochemistry. 1982;21:3654–3661. doi: 10.1021/bi00258a020. [DOI] [PubMed] [Google Scholar]
- Sutoh K, Mabuchi I. Biochemistry. 1984;23:6757–6761. [Google Scholar]
- Sutoh K, Yin HL. Biochemistry. 1989;28:5269–5275. doi: 10.1021/bi00438a052. [DOI] [PubMed] [Google Scholar]
- Szpacenko A, Dabrowska R. FEBS Lett. 1986;202:182–186. doi: 10.1016/0014-5793(86)80683-4. [DOI] [PubMed] [Google Scholar]
- Velaz L, Hemric ME, Benson CE, Chalovich JM. J Biol Chem. 1989;264:9602–9610. [PubMed] [Google Scholar]
- Velaz L, Ingraham RH, Chalovich JM. J Biol Chem. 1990;265:2929–2934. [PubMed] [Google Scholar]
- Wang CLA, Wang LWC, Lu RC. Biochem Biophys Res Commun. 1989;162:746–752. doi: 10.1016/0006-291x(89)92373-5. [DOI] [PubMed] [Google Scholar]
- Wang CLA, Wang LWC, Xu S, Lu RC, Saavedra-Alanis V, Bryan J. J Biol Chem. 1991;266:9166–9172. [PubMed] [Google Scholar]
- Yazawa M, Yagi K, Sobue K. J Biochem. 1987;102:1065–1073. doi: 10.1093/oxfordjournals.jbchem.a122144. [DOI] [PubMed] [Google Scholar]