

Abstract
Internalization of a G protein-coupled receptor (GPCR) is essential to the desensitization, endocytosis and signal transduction of the receptor. It has been the general view that conventional homologous internalization of a GPCR requires activation of the G protein(s) coupled to the receptor. However, whether and how GPCR-mediated G protein-independent signals trigger receptor internalization remains unknown although G protein-independent internalization has been reported. Here we show that an angiotensin II (Ang II) AT1 receptor mutant incapable of activating any G protein still undergoes normal internalization. Substitution of Asp125 with Ala and Arg126 with Leu at the highly conserved DRY motif of the AT1 receptor disabled the ability of the receptor to activate G proteins as shown by various Ang II binding studies, GDP-GTP exchange and IP production assays. Surprisingly, the mutant internalized normally in the presence of Ang II and transactivated the EGF receptor (EGFR). Similar to the wild-type receptor, overexpression of a dominant negative K220R mutant GRK2 diminished the internalization of D125A-R126L, but not the transactivation of EGFR. These data indicate that G protein-independent specific signals may also trigger homologous internalizations of the AT1 receptor through β-arrestin-dependent and -independent pathways, suggesting a possible mechanism for G protein-independent activation of G protein-coupled receptor kinases (GRKs). This may represent a general mechanism for triggering GPCR internalization.
Keywords: AT1 receptor, internalization, G protein, angiotensin II, transactivation
Abbreviations: GPCR, G protein-coupled receptor; PKA, protein kinase A; PKC, protein kinase C; GRK, G protein-coupled receptor kinase; RGS, regulator of G protein signaling; Ang II, angiotensin II (Asp1-Arg2-Val3-Tyr4-Ile5-His6-Pro7-Phe8); IP, inositol (1,4,5)triphosphate; ERK, extracellular signal-regulated kinases; EGFR, EGF receptor; PDGFRβ, PDGF receptor β; GDT, Guanosine diphosphate; GTP, Guanosine triphosphate; GTPγS: Guanosine 5’-[γ-thiol]triphosphate
Introduction
Agonist binding to a GPCR induces conformational changes in the receptor, leading to activation of Gαβγ heterotrimers. One function of the activated G proteins is to activate GRKs that in turn phosphorylate the specific receptor for desensitization. Subsequently, β-arrestins bind to the GRK-phosphorylated motifs of the receptor and induce the receptor internalization. This homologous GPCR desensitization/internalization is agonist-specific and GRK-dependent. This type of feedback regulation is conventional, since it requires activation of classic G proteins 1–3. Homologous internalization of GPCRs can also take place through β-arrestin-independent pathway. However, the underlying mechanism and, especially, the role of G protein activation in this mode of internalization remain poorly understood 4–6. The second messenger-stimulated kinases PKA and PKC also phosphorylate the receptor and induce heterologous desensitization/internalization that is not agonist-specific 1–3. It is known that a GPCR activates not only multiple G protein pathways but also non-G protein pathways. However, it remains unknown whether activation of non-G protein signal pathways also exert homologous feedback regulations that are agonist-specific.
GPCRs share common structural features such as seven transmembrane apanning regions (TMs) and preserve highly conserved consensus motifs, such as DRY motif at the boundary of TM3 and the second cytoplasmic loop. As a result, the GPCRs are thought to utilize similar mechanisms for G protein coupling and activation 7, 8. For example, the DRY motif is thought to play pivotal roles in all G protein coupling and activation. A salt bridge formed between the Asp and Arg of the motif may serve as a switch for GDP-GTP exchange, an essential process required for any G protein activation. Mutation of this motif in rhodopsin, especially, the conserved Arg residue, has been shown to totally inhibit the receptor from G protein coupling and activation 9.
Ang II AT1 receptors exert complex and diverse physiological actions associated with many diseases or disorders such as hypertension, hypertrophy, fibrosis, thrombosis and atherosclerosis. In addition to activation of multiple G proteins including Gq/11, Gi, and G13 10–15, Ang II AT1 receptors activate the Jak2/STAT pathway and transactivate EGF and PDGF receptors 14–18. The AT1 receptors even activate signal pathways in a more complex fashion by forming homo- or heterodimerization with the AT1 19–22, Ang II AT2 19, bradykinin B2 23, β2-adrenergic 24, and dopamine D1 25, 26 receptors. In the case of these complex activations, whether and how the AT1-initiated discrete signals regulate the homologous internalization of the receptor, what signals trigger the homologous internalization and whether G protein-independent signals are also capable of initiating homologous internalization remain to be addressed. It has been reported that mutations of a highly conserved Asp74 residue in TM2 of the AT1 receptor impaired IP3 production, but not agonist-induced internalization. However, the Asp74 mutants still produced minimal amount of IP3 27, 28 and remained sensitive to GTPγS 29. In this report, we show that double mutations of Asp125 to Ala and Arg126 to Leu (D125A-R126L) at the highly conserved DRY motif of the AT1 receptor induced uncoupling of the receptor from all G proteins but had little effect on the receptor internalization and the EGFR transactivation.
Materials and Methods
Materials
Oligonucleotides were synthesized by Sigma-Genosys (Woodlands, TX) or MWG Biotech (High Point, NC). Ang II and [Sar1]Ang II were purchased from Bachem (King of Prussia, PA). Other peptide analogues of [Sar1]Ang II were synthesized by GeneMed Synthesis (South San Francisco, CA). Losartan, EXP3174, and Candesartan were gifts from DuPont Merck Co. (Wilmington, DE). 125I-[Sar1,Ile8]Ang II (2200 Ci/mmol) was purchased from The Peptide Radioiodination Center of Washington State University (Pulman, WA). [3H]myo-inositol (22 mCi/ml) was purchased from Amersham Biosciences (Piscataway, NJ). The monoclonal antibody 1D4 was purchased from the Cell Culture Center (Endotronics Inc.), the AG1478 from Calbiochem (EMD Biosciences, Inc., La Jolla, CA) and the Antibodies for Erk and phospho-Erk from Cell Signaling Technology (Beverly, MA), GTPγS from Sigma, and [35S]GTPγS from PerkinElmer Life Science Products (Boston, MA).
Mutagenesis, expression, sample preparation, and ligand binding of the AT1 receptors
The human GRK2 mutant K220R was constructed using site directed mutagenesis and was subcloned into a phCMV1 vector (GTS Inc) with Hind III and Not I sites. The synthetic rat AT1 receptor gene was described earlier 30. AT1 mutants were prepared by the restriction fragment-replacement and the polymerase chain reaction methods. The DNA sequence analysis was performed by Cleveland Genomics Inc. to confirm the mutations. To express the AT1 receptor protein, 3 μg of column (Qiagen) purified plasmid DNA per 107 cells was used in the transfection. COS-1 cells (ATCC), cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, were transfected with the GenePORTER™ transfection reagents (GTS Inc). Transfected cells cultured for 48 h were harvested and cell membranes were prepared using the nitrogen PARR bomb disruption method 30. For preparation of a membrane fraction without Mg2+, the HME buffer (50 mM HEPES, pH 7.4, 12.5 mM MgCl2, and 1.5 mM EGTA, containing protease inhibitors) was prepared without MgCl2. The receptor expression was assessed in each case by immunoblot analysis (not shown) and by 125I-[Sar1,Ile8]Ang II saturation binding analysis. 125I-[Sar1,Ile8]Ang II binding experiments were described earlier 30.
GTPγS and GDP exchange assay.
[35S]GTPγS binding and immunoprecipitation were performed as described previously 31, 32. Briefly, each tube contained 0.5 μM GDP, 0.1 nM [35S]GTPγS, 50 μg membrane protein, and various concentrations of Ang II in a final volume of 250 μl. The binding assay was carried out in an assay buffer consisting of 50 mM HEPES, 100 mM NaCl, and 10 mM MgCl2, pH 7.4. Non-specific binding of [35S]GTPγS was determined in the presence of 10 μM of unlabeled GTPγS. The tubes were assembled on ice and the reaction started by incubation of tubes at 30°C. After 20 min of incubation, the reaction was terminated by the addition of 750 μl of ice-cold assay buffer. The reaction mixture was pelleted by centrifugation at 20,000 g for 5 min at 4°C. The pellets were re-suspended in 100 μl of solubilization buffer (50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1.25% (v/v) NP-40 and 0.2% (v/v) SDS), containing protease inhibitors. The solubilized pellets were diluted further, with 100 μl of solubilization buffer minus SDS, and pre-cleared by the addition of 1.3% (v/v) rabbit serum and 30 μl of protein A Sepharose, for 1 h at 4°C. The samples were centrifuged at 20,000 g, 4°C, for 5 min. The supernatant (100 μl) was removed and added to a mixture of anti-Gαq/11, anti-Gαi, and anti-Gαs antibodies (Santa Cruz). Samples were incubated at 4°C overnight before addition of 50 μl of protein A Sepharose. After a further incubation at 4°C for 2 h the Sepharose beads were washed three times with 0.5 ml of solubilization buffer minus SDS. Finally the solubilization buffer was removed and scintillation fluid added before quantification by liquid scintillation counting.
Production of total inositol phosphates (IP).
The COS-1 cells, cultured in 60 mm Petri dishes, were labeled for 24 h with [3H]myo-inositol (1μCi/ml) at 37°C in inositol-free DMEM containing 10% bovine calf serum 24 h after transfection. For the IP assay (i.e. 48 h after transfection), the labeled cells were washed three times with serum-free medium and incubated with DMEM containing 10 mM LiCl for 20 min. Then medium alone or ligands were added to the cells. After incubation for 45 min at 37°C, the medium was removed, and total soluble IP was extracted from the cells by a perchloric acid extraction method as described previously 30. The amount of [3H]-IP eluted from the column was counted and a concentration-response curve generated using iterative nonlinear regression analysis 30.
Measurement of AT1 receptor internalization.
Internalization of the AT1 receptors was measured by a method as described elsewhere 33. Briefly, COS-1 cells, transiently transfected with AT1 receptors in 12-well plates, were stimulated with and without Ang II (0.03 and 100 nM) for 10 min at 37°C. Surface-bound ligands were removed by a gentle acid wash (50 mM sodium citrate, 0.2 mM sodium phosphate, 90 mM NaCl, 0.1% bovine serum albumin, pH 5.0) for 10 min at 4°C, which did not affect subsequent receptor binding. Then a radio-ligand binding assay was performed (5 h at 4°C) to quantify receptors remaining at the cell surface. Internalized receptors are expressed as a percentage loss of cell surface binding compared with cells not exposed to Ang II.
Immunoblots.
Cells that were serum-deprived overnight were treated with Ang II in the presence and absence of 100 nM of the EGFR-specific inhibitor, AG1478 for 20 min. Following this, the cells were washed twice with ice-cold Dulbecco’s Phosphate-buffered saline. The cells were then lysed on ice with ice-cold lysis buffer (50 mM Tris, pH 7.2, 1% (vol/vol) Triton X-100, 1 mM Na3VO4, 1 mM EGTA, 0.2 mM phenylmethanesulfonyl fluoride, 25 μg/ml leupeptin and 10 μg/ml aprotinin). The samples were then centrifuged at 14,000 g for 10 min. Protein content in the supernatants was determined by the BCA assay according to the manufacture’s instructions (Pierce, Rockford, IL). Fifteen μg of total cell lysate protein were subjected to SDS-PAGE and then transferred to a polyvinylidene difluoride membrane by electroblotting at 200 mA for 1.5 h. The membrane was immunoblotted according to a standard Western Blot protocol as furnished by the manufacturers, and the immunoreactive proteins were detected by enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech Inc., Piscataway, NJ). An autoradiograph of the blot was analyzed by an OS-Scan Image Analysis System to obtain the densitometry data.
Statistical analysis.
The results are expressed as the mean ± S.E.M. of three to five independent determinations. The significance in measured values was evaluated using an unpaired Student’s t-test.
Results
AT1 receptor mutant: characterization and ligand binding.
To abolish all G protein activation activity of the AT1 receptor, double mutations were introduced into an AT1 receptor in which the highly conserved Asp125 and Arg126 residues were substituted with Ala125 and Leu126 as illustrated in Figure 1. The expression of the wild type and mutant receptors in COS-1 cells was confirmed by ligand binding and Western blots with 1D4 monoclonal antibody (data not shown). Recombinant expression in transiently transfected COS-1 cells was employed for characterization of the AT1 receptors as described previously 30, 34. Expression in each case was measured by immunoblotting with a C-terminal epitope-directed monoclonal antibody 1D4, followed by 125I-[Sar1,Ile8]Ang II saturation binding and competition binding to the AT1 receptor ligands, along with IP production to measure function. In the total membrane fraction, the high affinity binding of the expressed wild-type and mutant AT1 receptors was 0.50±0.07 nM and 0.53±0.09 nM, respectively, for the cold peptide antagonist 127I-[Sar1,Ile8]Ang II, and 11.8±1.7 nM and 11.2±1.4 nM, respectively, for AT1 receptor-selective non-peptide antagonist losartan. Similar to the wild-type AT1 receptor, the Kd values of the mutant receptor estimated from competition binding for the agonist [Sar1]Ang II and the native hormone Ang II were 0.46 ± 0.06 nM and 1.51 ± 0.1 nM, respectively. These values were generated with EDTA-washed membranes, and therefore represent the intrinsic affinity of the receptor in the absence of G protein coupling. The Bmax values, estimated for the two receptors, were close in value (~4 pmol/mg membrane protein), consistent with the levels of receptor expression estimated by immunoblot analysis. Scatchard plot analysis of the 125I-[Sar1,Ile8]Ang II saturation binding indicated a single affinity class for both receptors (Kd, 0.49±0.07 and 0.51±0.08 for wild type and the mutant, respectively). Competition binding studies, employing Ang II, [Sar1]Ang II and Losartan demonstrated that the receptors expressed in COS-1 cells preserve the selectivity and affinity profile previously described for both native tissue receptors and recombinant- expressed receptors.
Figure 1.
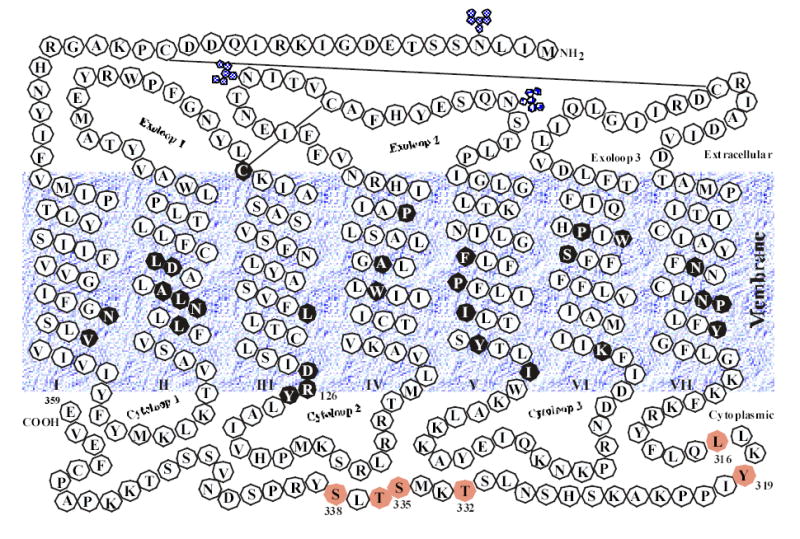
The secondary structure model of the rat AT1 angiotensin II receptor. Putative -helical transmembrane domains I–VII, three potential glycosylation sites, and two disulfide bonds are shown. Residues and motifs in closed black circles are highly conserved in more than 90% of GPCRs. The amino acids in the cytoplasmic tail of the receptor that have been suggested to be important for receptor internalization are shaded. The membrane interface boundaries for all seven TM helices are tentative.
Mutant D125A-R126L fails to induce GTP-GDP exchange.
The ability of the AT1 receptor to activate IP production in COS-1 cells has been demonstrated before in this laboratory 34. In sharp contrast to the wild-type AT1 receptors, mutant D125A-R126L failed to produce IP in the presence of Ang II at various concentrations (Figure 2). Moreover, the basal IP production was negligible, even when the mutant was over-expressed in the COS-1 cells, whereas the wild-type receptor-expressing COS-1 cells produced 2.6% basal IP activity, in the absence of any exogenous agonist (Figure 2). These IP results were the net IP production, after deduction of IP values (about 2.5% or 1000 cpm/106 cells) from mock-transfected COS-1 cells.
Figure 2.
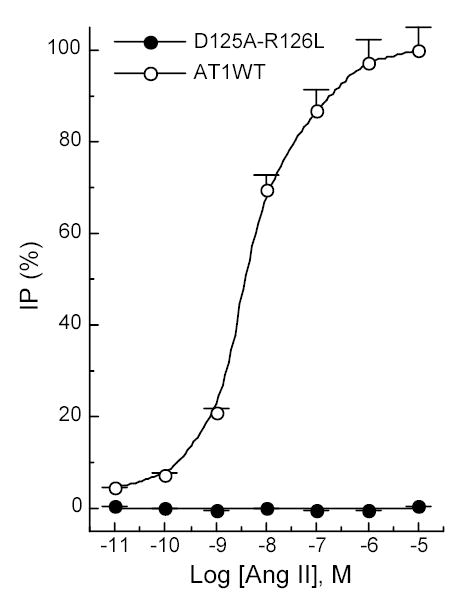
Total IP productions of COS-1 cells transfected with the AT1 receptors. Expression levels of the two receptors were about 4.0 pmol/mg protein. The net IP production was obtained after subtraction of the IP values from that for mock-transfected COS-1 cells. The maximal IP activity (100%) was elicited by stimulating the wild-type AT1 receptor with Ang II at 10−5 M concentration. The absolute maximal IP production was 39,000 cpm/106 cells. Values are means ± S.E.M. of at least three independent experiments in duplicate.
The complete inability of D125A-R126L in activating the Gq/IP pathway may not preclude the possibility that the defective mutant receptor is capable of activating other G proteins, directly or indirectly, through other mechanisms. To examine this possibility, a series of binding experiments were performed. Figure 3 shows that the mutant receptor was completely insensitive to Mg2+ (Figure 3B) and GTPγS (Figure 3C), whereas the wild-type receptor displayed an increased binding affinity for the agonist Ang II (Figure 3A) in the presence of Mg2+ and a decreased binding affinity for the agonist 125I-Ang II in the presence of GTPγS (Figure 3C). This result indicates that the mutant receptor did not directly couple to any G proteins.
Figure 3.
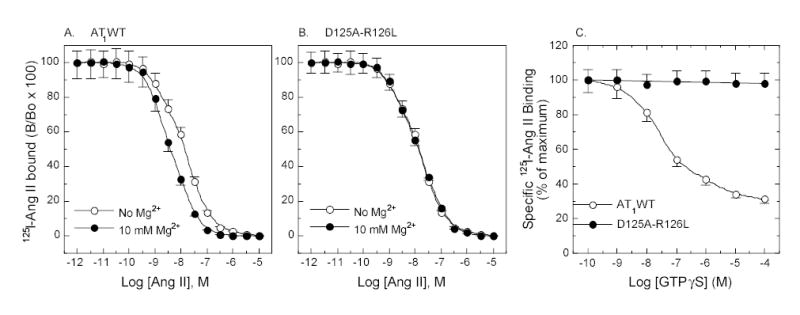
Ligand binding property of the AT1 receptors. Competition binding of 125I-[Sar1,Ile8]Ang II with agonist Ang II in the presence and absence of 10 mM Mg2+ (A and B). Binding of agonist 125I-Ang II in the presence of GTPγS (C). The membrane preparations used for binding assays with 10 mM Mg2+ and GTPγS were prepared in the presence of Mg2+ without EDTA wash. Values are means ± S.E.M. of at least three independent experiments in duplicate.
To examine whether the mutant receptors directly or indirectly induced GTP-GDP exchange, GTPγS and GDP exchange assays were performed. Consistent with the above binding assays, stimulation of the mutant receptor with Ang II failed to induce any GTP-GDP exchange mediated by G proteins, suggesting no activation of any Gα proteins. By contrast, the wild-type receptor elicited an apparent GTP-GDP exchange, indicating activation of G proteins (Figure 4).
Figure 4.
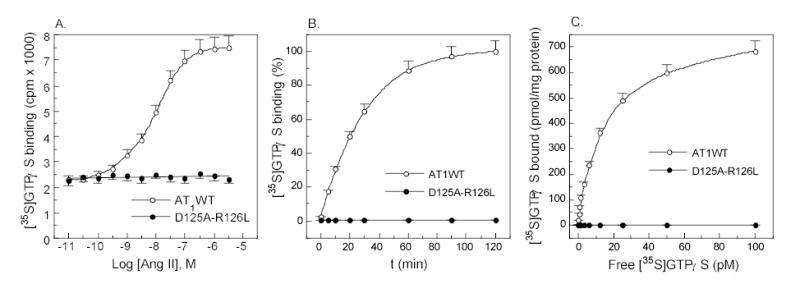
AT1 receptor-mediated activation of Gα proteins detected by [35S]GTPγS and GDP exchange assay. (a) [35S]GTPγS (0.1 nM) binding in the presence of increasing amounts of Ang II for 20 min. (b) Time dependent [35S]GTPγS binding in the presence of 100 nM Ang II. (c) Saturation binding of [35S]GTPγS in the presence of 100 nM Ang II for 20 min. The membrane proteins used for GTPγS and GDP exchange assay were prepared in the presence of Mg2+ without EDTA wash. Values are means ± S.E.M. of at least three independent experiments.
Mutant D125A-R126L still internalizes.
In view of the apparent difference in G protein coupling and activation between mutant D125A-R126L and the wild-type AT1 receptors, we examined the capacity of the mutant to undergo homologous internalization. Treatment of the wild type and D125A-R126L mutant with 100 nM Ang II for 10 min induced 51.6% and 45.8% receptor internalization of, respectively (Figure 5A). The kinetics of the internalization of the mutant was similar to the wild type although the level of the internalized mutant receptors was 5.8% lower (p<0.05). Similar internalization profiles were observed for both wild type and the mutant receptors when Ang II at low level (0.03 nM) was utilized (data not shown). The inactive Ang II analog, [Sar1,Ile4,Ile8]Ang II, failed to induce internalization of either receptor (Figure 5A), consistent with a previous report 33. Since GRK2 is known to play an important role in initiation of homologous internalization of AT1 receptors, over-expression of a dominant negative R220K mutant GRK2 was employed in the study and reduced the internalization to 24.2% and 18.3% for the wild type and mutant receptors, respectively (Figure 5). These reductions reflected 53.1% and 60% decreases in the capacity of receptor internalization. Interestingly, the magnitude of the reduction in the mutant receptor was 6.9% greater with comparison to the wild type (p<0.05). These changes in magnitude are also apparent when the kinetics of receptor internalization is plotted as shown in figure 5B. However, no changes in the time-course of internalization were observed between the wild type and mutant receptors in the presence and absence of R220K expression. Also AG1478, an EFGR kinase-specific inhibitor, failed to affect internalization of either receptor.
Figure 5.
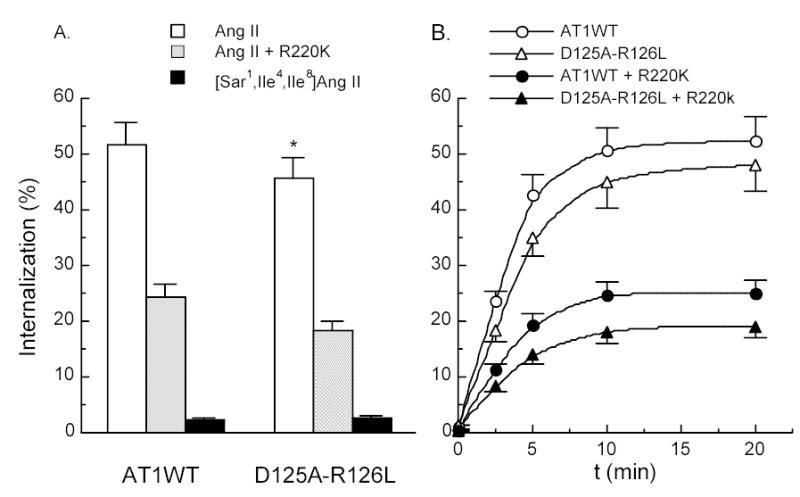
Internalization of the AT1 receptors. (A) In the presence of active Ang II and inactive Ang II analog [Sar1,Ile4,Ile8]Ang II. (B) Internalization kinetics of the AT1 receptors in the presence of 100 nM Ang II. 100 nM Ang II-induced internalization of the AT1 receptors in COS-1 cells co-transfected with 5 μg dominant negative K220R mutant GRK2 DNA per 60 mm dish for both internalization studies. * and †, compared with the wild-type for Ang II treatment and Ang II plus R220K treatment, respectively.
In the absence of Ang II, the heterologous internalization of both the wild type and mutant D125A-R126L was negligible under the experimental conditions as detected by saturation binding assays with 125I-[Sar1,Ile8]Ang II.
Mutant D125A-R126L receptor transactivates EGF receptors.
Since D125A-R126L still underwent almost normal homologous internalization it must have initiated certain specific signals that preceded the internalization. To identify the specific D125A-R126L-initiated signals, transactivation of EGFR was examined. In the presence or absence of EGFR kinase-specific inhibitor AG1478, the wild type and mutant receptors displayed no difference in transactivation of EGFR upon [Sar1]Ang II or Ang II stimulation (Figure 6). This suggests that the AT1 receptor-mediated transactivation of EGFR is independent of G protein activation. Over-expression of the dominant-negative R220K mutant GRK2 did not diminish the transactivation of EGFR by either receptor (data not shown). The inactive Ang II analog [Sar1,Ile4,Ile8]Ang II also failed to induce the transactivation of EGFR in COS-1 cells expressing the wild-type and mutant AT1 receptors (data not shown).
Figure 6.
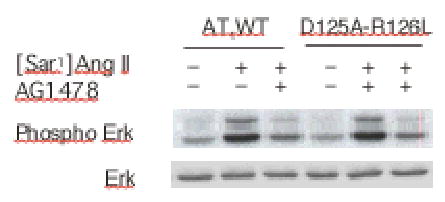
Transactivation of EGF receptors mediated by the AT1 receptors. COS-7 cells were transfected with AT1 receptors and treated with 10 nM of Ang II for 3 min in the presence or absence of EGF receptor tyrosine kinase inhibitor AG 1478 (100 nM). Phosphorylation of ERK was detected with anti-phospho-ERK antibodies after 10% SDS-PAGE separation of the COS-7 cell lysates.
Discussion
Internalization plays an important role in receptor desensitization, endocytosis, and signal transduction. Heterologous internalization of a GPCR is a passive process that does not require a specific agonist binding to the receptor. However, homologous internalization of a GPCR is an active process that requires the following: (1) specific ligand binding; (2) a conformational change of the receptor; (3) GRK-mediated phosphorylation of the receptor; and (4) signal transductions initiated by the activated receptor. It is generally believed that conventional homologous internalization of a GPCR depends on the activation of G proteins, since activation of GRKs requires pre-activation of G proteins (Figure 7). In order to determine whether non-G protein signals, also activated by a GPCR, might also trigger homologous internalization of the receptor, a mutant receptor that would still interact with the agonist but fail to activate G proteins was required. The AT1 mutant receptor D125A-R126L was constructed to serve this purpose (Figure 1). This mutant receptor showed unimpaired agonist binding capacity but failed to activate any G protein. Therefore it would anticipated that the mutant receptor would no longer be able to undergo conventional homologous internalization since it no longer activated G proteins or GRKs. To our surprise, the mutant receptor showed almost normal homologous internalization in the presence of agonist Ang II at both high (100 nM) and low (0.03 nM) levels. The finding at low level of Ang II is consistent to previous observations made by Gaborik et al with a similar double mutant DRY/AAY 35, 36. This result indicates that homologous internalization of the AT1 receptor is also inducible through mechanisms distinct from G protein activation. The type of homologous internalization induced through G protein activation-independent mechanisms is therefore “unconventional” (Figure 7).
Figure 7.
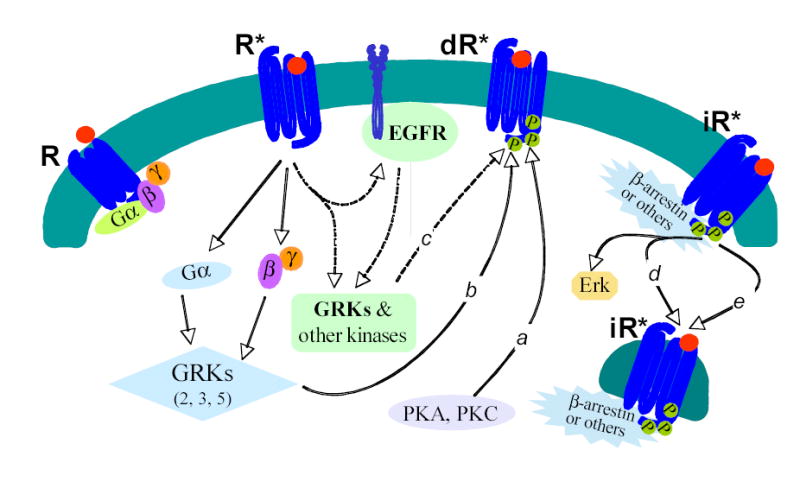
Internalization-triggering signals and internalization of the Ang II AT1 receptors. Label a, mechanism of heterologous internalization initiated by PKA and PKC; b, mechanism of conventional homologous internalization initiated by an activated G proteins; c, mechanism of unconventional homologous internalization initiated by G protein-independent signals; d, β-arrestin-dependent internalization (binding of β-arrestins to a GPCR is essential); e, β-arrestin-independent internalization (binding of other non-β-arrestin molecules such as caveolin or GRK2 is essential). Here R*, dR*, and iR* means activated, desensitized, and internalized AT1 receptors, respectively. In this illustration GRKs play an important role in both G protein-dependent conventional homologous internalization and G protein-independent unconventional homologous internalization. Other kinases may also be involved in the initiation of homologous internalizations of the AT1 receptor. Transactivation of EGFR is not only independent of AT1 receptor internalization but also independent of G protein activation.
Phosphorylation of an activated GPCR by GRKs is a tightly regulated process. Activation of these GRKs often requires activated G proteins. For example, activation of GRK2 requires double binding of the activated Gαq and Gβγ dimer to the N-terminal RGS-like domain and the C-terminal PH domain of GRK2 37–39. The K220R mutant GRK2 is deficient only in its protein kinase activity. Its binding capacity to proteins such as Gαq and Gβγ remains intact. It is known that GRK2, GRK3, and GRK5 phosphorylate activated AT1 receptors, and expression of the dominant-negative K220R mutant GRK2 causes about 50% inhibition of the wild-type AT1 internalization 40. Our results suggest, however, that GRKs still played an important role in inducing internalization of the mutant D125A-R126L. This suggests that the GRKs could be activated through G protein-independent mechanisms since the dominant-negative K220R mutant GRK2 also diminished “unconventional” homologous internalization of the mutant D125A-R126L. This calls for further study of the mechanisms of GRK activation. Indeed, a recent report has shown that the N-terminal RGS-like domain of the GRK2 also binds to the metabotropic glutamate receptor 1a 39. Other examples suggesting G protein-independent activation of GRKs are the findings that GRK2 desensitizes the PDGFβ receptor and GRK3 mediates P2X7 receptor internalization since activation of neither PDGF receptor, nor P2X7 receptor, activates G proteins 41, 42.
It has been documented that homologouse internalization of AT1 receptors can take place through both β-arrestin-dependent and -independent pathways depending on the concentration of Ang II. The β-arrestin-dependent pathway plays a major role at low concentration (0.03 nM) whereas β-arrestin-independent pathway becomes predominant at saturation concentration of Ang II (100 nM) 6, 35, 43. Our results show that the mutant D125A-R126L receptor, similar to the wild type, can utilize both pathways for homologous internalization. Most importantly, the β-arrestin-independent internalization of the mutant receptor occurs independently of G protein activation. The inability of [Sar1,Ile4,Ile8]Ang II to induce internalization of the AT1 receptors may further support the presence of β-arrestin-independent pathway since this inactive Ang II analog can induce association of the receptors with β-arrestin as reported by Wei et al 44.
It is at least theoretically possible that binding of agonist Ang II to mutant D125A-R126L may result in an indirect activation of G proteins. This assumption is based on the fact that AT1 receptors may act in the form of a homo- and heterodimer with Ang II AT1, Ang II AT2, bradykinin B2, β2-adrenergic, and dopamine D1 receptors 19–26. Defective mutant AT1 receptors may restore its wild-type-like full function through forming homodimers to rescue its activity, as previously reported 22. However, our results do not support this possibility since there was no GTP-GDP exchange detected under our experimental conditions.
Similar to many GPCRs, AT1 receptors also initiate multiple signals transduced through activation of classical trimeric G proteins and transactivation of receptor tyrosine kinase EGFR and PDGFR 14–18. Subsequent internalization of the activated AT1 receptors leads to activation of Erk1/2 44, 45. However, the signals that trigger homologous internalization of the AT1 receptor remain unknown although the G-protein-independent homologous internalization has been reported 36, 44. It is also unknown whether altered or activated conformation alone of the AT1 receptor can trigger homologous internalization by recruiting β-arrestins or other molecules such as caveolin and GRKs. Our results suggest that the G protein-independent homologous internalization of the mutant D125A-R126L could be initiated by G protein-independent signals that transactivate EGFR. However, in the present study we were unable to identify the exact signal that might have triggered the receptor internalization since the detailed mechanism leading to EGFR transactivation remains largely unknown. The fact that the EGFR-specific inhibitor, AG1478, failed to block the homologous internalization of the mutant AT1 receptor suggests that the internalization-triggering signal is upstream of the transactivation of EGFR.
Perspectives
This study showed that both β-arrestin-dependent and -independent homologous internalizations could take place independently of G protein activation. The G protein-independent signals initiated by AT1 receptors may also trigger homologous internalization. This may represent a general mechanism for triggering GPCR internalization. Our results also suggest that GRKs might be activated through G protein-independent mechanisms and the AT1-mediated transactivation of EGFR is not only G protein-independent but also internalization-independent.
Acknowledgments
This work was supported by a SDG grant 0030019N of the American Heart Association and a RO1 grant HL65492 of the National Heart, Lung, and Blood Institute to YHF.
References
- 1.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. . Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 2.Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. . Mol Pharmacol. 2003;63:9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- 3.Lodowski DT, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ. Keeping G proteins at bay: a complex between G protein-coupled receptor kinase 2 and Gbetagamma. . Science 23. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- 4.van Koppen CJ, Jakobs KH. Arrestin-independent internalization of G protein-coupled receptors. . Mol Pharmacol. 2004;66:365–367. doi: 10.1124/mol.104.003822. [DOI] [PubMed] [Google Scholar]
- 5.Pao CS, Benovic JL. Phosphorylation-independent desensitization of G protein-coupled receptors? Sci STKE. 2002;2002:PE42. doi: 10.1126/stke.2002.153.pe42. [DOI] [PubMed] [Google Scholar]
- 6.Gaborik Z, Hunyady L. Intracellular trafficking of hormone receptors. . Trends Endocrinol Metab. 2004;15:286–293. doi: 10.1016/j.tem.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin JM. The probable arrangement of the helices in G protein-coupled receptors. . Embo J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. . Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 9.Acharya S, Karnik SS. Modulation of GDP release from transducin by the conserved Glu134-Arg135 sequence in rhodopsin. . J Biol Chem. 1996;271:25406–25411. doi: 10.1074/jbc.271.41.25406. [DOI] [PubMed] [Google Scholar]
- 10.Shirai H, Takahashi K, Katada T, Inagami T. Mapping of G protein coupling sites of the angiotensin II type 1 receptor. . Hypertension. 1995;25:726–730. doi: 10.1161/01.hyp.25.4.726. [DOI] [PubMed] [Google Scholar]
- 11.Macrez-Lepretre N, Kalkbrenner F, Morel JL, Schultz G, Mironneau J. G protein heterotrimer Galpha13beta1gamma3 couples the angiotensin AT1A receptor to increases in cytoplasmic Ca2+ in rat portal vein myocytes. . J Biol Chem. 1997;272:10095–10102. doi: 10.1074/jbc.272.15.10095. [DOI] [PubMed] [Google Scholar]
- 12.Maturana AD, Casal AJ, Demaurex N, Vallotton MB, Capponi AM, Rossier MF. Angiotensin II negatively modulates L-type calcium channels through a pertussis toxin-sensitive G protein in adrenal glomerulosa cells. . J Biol Chem. 1999;274:19943–19948. doi: 10.1074/jbc.274.28.19943. [DOI] [PubMed] [Google Scholar]
- 13.Smith RD, Baukal AJ, Dent P, Catt KJ. Raf-1 kinase activation by angiotensin II in adrenal glomerulosa cells: roles of Gi, phosphatidylinositol 3-kinase, and Ca2+ influx. . Endocrinology. 1999;140:1385–1391. doi: 10.1210/endo.140.3.6600. [DOI] [PubMed] [Google Scholar]
- 14.Harris RC. Signaling and cellular effects of angiotensin II. In: Epstein M, and Brunner, H., ed. Angiotensin II receptor antagonists Philadelphia: Hanley & Belfus; 2000:49–68.
- 15.Feng YH, and Douglas, J. G. Angiotensin receptors: an overview. In: Epstein M, and Brunner, H., ed. Angiotensin II receptor antagonists Philadelphia: Hanley & Belfus; 2000:29–48.
- 16.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. . Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 17.Pierce KL, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. . Oncogene. 2001;20:1532–1539. doi: 10.1038/sj.onc.1204184. [DOI] [PubMed] [Google Scholar]
- 18.Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. . Oncogene. 2001;20:1594–1600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- 19.AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. . J Biol Chem. 2001;276:39721–39726. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 20.AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. . Nat Med. 2001;7:1003–1009. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- 21.AbdAlla S, Lother H, Langer A, el Faramawy Y, Quitterer U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. . Cell. 2004;119:343–354. doi: 10.1016/j.cell.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Monnot C, Bihoreau C, Conchon S, Curnow KM, Corvol P, Clauser E. Polar residues in the transmembrane domains of the type 1 angiotensin II receptor are required for binding and coupling. Reconstitution of the binding site by co-expression of two deficient mutants. . J Biol Chem. 1996;271:1507–1513. doi: 10.1074/jbc.271.3.1507. [DOI] [PubMed] [Google Scholar]
- 23.AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. . Nature. 2000;407:94–98. doi: 10.1038/35024095. [DOI] [PubMed] [Google Scholar]
- 24.Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. . Circulation. 2003;108:1611–1618. doi: 10.1161/01.CIR.0000092166.30360.78. [DOI] [PubMed] [Google Scholar]
- 25.Zeng C, Asico LD, Wang X, Hopfer U, Eisner GM, Felder RA, Jose PA. Angiotensin II regulation of AT1 and D3 dopamine receptors in renal proximal tubule cells of SHR. . Hypertension. 2003;41:724–729. doi: 10.1161/01.HYP.0000047880.78462.0E. [DOI] [PubMed] [Google Scholar]
- 26.Zeng C, Luo Y, Asico LD, Hopfer U, Eisner GM, Felder RA, Jose PA. Perturbation of D1 dopamine and AT1 receptor interaction in spontaneously hypertensive rats. . Hypertension. 2003;42:787–792. doi: 10.1161/01.HYP.0000085334.34963.4E. [DOI] [PubMed] [Google Scholar]
- 27.Hunyady L, Baukal AJ, Balla T, Catt KJ. Independence of type I angiotensin II receptor endocytosis from G protein coupling and signal transduction. . J Biol Chem. 1994;269:24798–24804. [PubMed] [Google Scholar]
- 28.Miserey-Lenkei S, Lenkei Z, Parnot C, Corvol P, Clauser E. A functional enhanced green fluorescent protein (EGFP)-tagged angiotensin II at(1a) receptor recruits the endogenous Galphaq/11 protein to the membrane and induces its specific internalization independently of receptor-g protein coupling in HEK-293 cells. . Mol Endocrinol. 2001;15:294–307. doi: 10.1210/mend.15.2.0600. [DOI] [PubMed] [Google Scholar]
- 29.Seta K, Nanamori M, Modrall JG, Neubig RR, Sadoshima J. AT1 receptor mutant lacking heterotrimeric G protein coupling activates the Src-Ras-ERK pathway without nuclear translocation of ERKs. . J Biol Chem. 2002;277:9268–9277. doi: 10.1074/jbc.M109221200. [DOI] [PubMed] [Google Scholar]
- 30.Noda K, Feng YH, Liu XP, Saad Y, Husain A, Karnik SS. The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. . Biochemistry. 1996;35:16435–16442. doi: 10.1021/bi961593m. [DOI] [PubMed] [Google Scholar]
- 31.Akam EC, Challiss RA, Nahorski SR. G(q/11) and G(i/o) activation profiles in CHO cells expressing human muscarinic acetylcholine receptors: dependence on agonist as well as receptor-subtype. . Br J Pharmacol. 2001;132:950–958. doi: 10.1038/sj.bjp.0703892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Young KW, Bootman MD, Channing DR, Lipp P, Maycox PR, Meakin J, Challiss RA, Nahorski SR. Lysophosphatidic acid-induced Ca2+ mobilization requires intracellular sphingosine 1-phosphate production. Potential involvement of endogenous EDG-4 receptors. . J Biol Chem. 2000;275:38532–38539. doi: 10.1074/jbc.M006631200. [DOI] [PubMed] [Google Scholar]
- 33.Thomas WG, Qian H, Chang CS, Karnik SS. Agonist-induced phosphorylation of the angiotensin II (AT(1A)) receptor requires generation of a conformation that is distinct from the inositol phosphate-signaling state. . J Biol Chem. 2000;275:2893–2900. doi: 10.1074/jbc.275.4.2893. [DOI] [PubMed] [Google Scholar]
- 34.Feng YH, Sun Y, Douglas JG. Gbeta gamma -independent constitutive association of Galpha s with SHP-1 and angiotensin II receptor AT2 is essential in AT2-mediated ITIM-independent activation of SHP-1. . Proc Natl Acad Sci U S A. 2002;99:12049–12054. doi: 10.1073/pnas.192404199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaborik Z, Szaszak M, Szidonya L, Balla B, Paku S, Catt KJ, Clark AJ, Hunyady L. Beta-arrestin- and dynamin-dependent endocytosis of the AT1 angiotensin receptor. . Mol Pharmacol. 2001;59:239–247. doi: 10.1124/mol.59.2.239. [DOI] [PubMed] [Google Scholar]
- 36.Gaborik Z, Jagadeesh G, Zhang M, Spat A, Catt KJ, Hunyady L. The role of a conserved region of the second intracellular loop in AT1 angiotensin receptor activation and signaling. . Endocrinology. 2003;144:2220–2228. doi: 10.1210/en.2002-0135. [DOI] [PubMed] [Google Scholar]
- 37.Eichmann T, Lorenz K, Hoffmann M, Brockmann J, Krasel C, Lohse MJ, Quitterer U. The amino-terminal domain of G-protein-coupled receptor kinase 2 is a regulatory Gbeta gamma binding site. . J Biol Chem. 2003;278:8052–8057. doi: 10.1074/jbc.M204795200. [DOI] [PubMed] [Google Scholar]
- 38.Lodowski DT, Barnhill JF, Pitcher JA, Capel WD, Lefkowitz RJ, Tesmer JJ. Purification, crystallization and preliminary X-ray diffraction studies of a complex between G protein-coupled receptor kinase 2 and Gbeta1gamma2. . Acta Crystallogr D Biol Crystallogr. 2003;59:936–939. doi: 10.1107/s0907444903002622. [DOI] [PubMed] [Google Scholar]
- 39.Dhami GK, Dale LB, Anborgh PH, O’Connor-Halligan KE, Sterne-Marr R, Ferguson SS. G Protein-coupled receptor kinase 2 regulator of G protein signaling homology domain binds to both metabotropic glutamate receptor 1a and Galphaq to attenuate signaling. . J Biol Chem. 2004;279:16614–16620. doi: 10.1074/jbc.M314090200. [DOI] [PubMed] [Google Scholar]
- 40.Oppermann M, Freedman NJ, Alexander RW, Lefkowitz RJ. Phosphorylation of the type 1A angiotensin II receptor by G protein-coupled receptor kinases and protein kinase C. . J Biol Chem. 1996;271:13266–13272. doi: 10.1074/jbc.271.22.13266. [DOI] [PubMed] [Google Scholar]
- 41.Hildreth KL, Wu JH, Barak LS, Exum ST, Kim LK, Peppel K, Freedman NJ. Phosphorylation of the platelet-derived growth factor receptor-beta by G protein-coupled receptor kinase-2 reduces receptor signaling and interaction with the Na(+)/H(+) exchanger regulatory factor. . J Biol Chem. 2004;279:41775–41782. doi: 10.1074/jbc.M403274200. [DOI] [PubMed] [Google Scholar]
- 42.Feng YH, Wang L, Wang Q, Li X, Zeng R, Gorodeski GI. ATP ligation stimulates GRK-3 – mediated phosphorylation and beta-arrestin-2- and dynamin-dependent internalization of the P2X7-receptor. Am J Physiol Cell Physiol. 2005;288:(in press). [DOI] [PMC free article] [PubMed]
- 43.Zhang J, Ferguson SS, Barak LS, Menard L, Caron MG. Dynamin and beta-arrestin reveal distinct mechanisms for G protein-coupled receptor internalization. . J Biol Chem. 1996;271:18302–18305. doi: 10.1074/jbc.271.31.18302. [DOI] [PubMed] [Google Scholar]
- 44.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. . Proc Natl Acad Sci U S A. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei H, Ahn S, Barnes WG, Lefkowitz RJ. Stable interaction between beta-arrestin 2 and angiotensin type 1A receptor is required for beta-arrestin 2-mediated activation of extracellular signal-regulated kinases 1 and 2. . J Biol Chem. 2004;279:48255–48261. doi: 10.1074/jbc.M406205200. [DOI] [PubMed] [Google Scholar]