

The Case: A previously healthy 24-year-old man presented to the emergency department with sudden onset of severe periumbilical and right costovertebral pain accompanied by nausea and vomiting. His vital signs, including oxygen saturation, were normal. His physical exam was remarkable for decreased abdominal sounds with epigastric and right costovertebral tenderness. The patient had no history of tobacco or illicit drug abuse. His mother had died at age 41 from a pulmonary embolism believed to have been caused by an unspecified hypercoagulable disorder.
Except for leukocytosis (leukocyte count 15.0 [normal 4.0– 11.0] ∞ 109/L), his complete blood count, serum blood chemistry, and liver and renal function test results were normal. An abdominal CT scan with contrast medium showed a wedge-shaped hypodense lesion in the right kidney, indicating infarction (Fig. 1A, arrow), as well as a filling defect in the lumen of the superior mesenteric artery (SMA), consistent with a thrombus or thromboembolus (Fig. 1B, arrowhead), and edema of the wall of the small intestine (Fig. 1B, arrow).
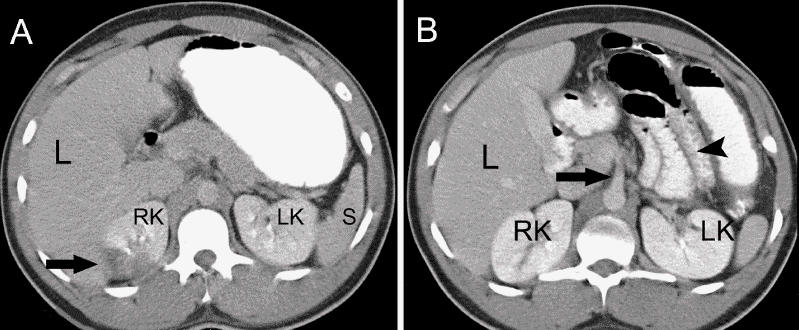
Fig. 1: Abdominal CT scans with contrast medium. Left: Hypodense lesion in right kidney (arrow), indicating infarction. Right: Filling defect in lumen of superior mesenteric artery (arrowhead), consistent with thrombus or thromboembolus, and edema of wall of small intestine (arrow). L = liver, RK/LK = right/left kidney, S = spleen.
Embolectomy with patch angioplasty (dissection of the native artery with removal of the thrombus followed by arterial repair using a patch of bovine arterial material) of the SMA was performed. Intraoperative transesophageal echocardiography (TEE), performed to search for the source of the SMA embolus, showed a mobile thrombus 1.5 ∞ 2 cm in size in the descending thoracic aorta near the origin of the left subclavian artery (Fig. 2, arrow). Intravenous heparin therapy was begun; higher than usual doses were required to achieve adequate anticoagulation (an activated partial thromboplastin time > 2.5 ∞ control). Therefore, a presumptive diagnosis of antithrombin deficiency was made, which was confirmed by an antithrombin–heparin cofactor level of 65% (normal pooled plasma activity 80%– 120%). Results of all other tests for prothrombotic states, including measurement of protein C and S, homocysteine, antinuclear antibodies and antiphospholipid antibodies, were negative. Warfarin was added to the anticoagulation therapy on postoperative day 2, and after 3 days of concomitant administration it replaced the heparin therapy.

Fig. 2: Intraoperative transesophageal echocardiogram, showing mobile thrombus (arrow) 1.5 ∞ 2 cm in size in descending thoracic aorta near origin of left subclavian artery.
The patient had an uneventful recovery, with resolution of the aortic thrombus seen on TEE at 8 weeks' follow-up. The antithrombin–heparin cofactor level at that time was 65%, which confirmed the diagnosis of antithrombin deficiency. The patient was informed that he would have to continue the warfarin therapy indefinitely.
Clinically significant thrombotic processes may result from a primary hypercoagulable state caused by deficiencies in one of the components in the coagulation–anticoagulation system or from an acquired hypercoagulable state caused by precipitating factors such as trauma, immobilization or advanced age. Genetically susceptible people may have thrombosis after exposure to exogenous stimuli such as pregnancy, surgery or estrogen use. When testing for primary (inherited) hypercoagulable disorders in patients presenting with thrombosis, physicians should take into consideration whether the thrombosis is arterial or venous (Box 1) and whether there are precipitating factors or a family history of thrombosis (Box 2). Initial laboratory evaluation should include a complete blood count and peripheral blood smear to rule out polycythemia vera, essential thrombocythemia and disseminated intravascular coagulopathy; renal function tests and urinalysis to rule out nephrotic syndrome; liver function tests to rule out impaired protein synthesis; and prothrombin and partial thromboplastin times.1 Antinuclear antibody testing and measurement of the erythrocyte sedimentation rate should be performed if autoimmune diseases such as systemic lupus erythematosus or vasculitidies (giant cell arteritis, Takayasu's arteritis) are suspected as the cause of the thrombosis. Tests for specific hypercoagulable disorders should include a screening clotting assay for factor V Leiden mutation (using plasma deficient in factor V) and, if needed, confirmatory genetic testing, genetic testing for prothrombin gene G20210A mutation, functional assays for protein C and S deficiencies, tests for antiphospholipid antibody syndrome (lupus anticoagulant, anticardiolipin and, if available, anti-β2 glycoprotein I antibodies) and antithrombin–heparin cofactor assay for antithrombin deficiency. Patients who present with spontaneous, unexplained, arterial thrombosis should be tested for the presence of antiphospholipid antibody syndrome (primary, or secondary to other autoimmune disorders such as systemic lupus erythematosus) and hyperhomocysteinemia.1
Box 1.

Box 2.

Antithrombin deficiency was found to be the cause of the thrombosis in our patient. Antithrombin is the main inhibitor of thrombin and the other clotting factors involved in the intrinsic and common coagulation pathways. Antithrombin deficiency may be inherited or acquired. The inherited form, first described in 1965 in a Norwegian family,2 is an autosomal dominant trait with a prevalence of about 1 per 2000 and presents early in life with venous and, rarely, arterial thrombosis.1,3 A family history of thrombotic disorders is present in most cases.1 Secondary pathophysiologic conditions associated with reduced antithrombin concentration should be excluded before a diagnosis of inherited deficiency can be established (Box 3). Inherited antithrombin deficiency can be one of 2 types: type 1 (quantitative abnormality) is the result of reduced synthesis of biologically normal protein, and type 2 (qualitative abnormality) is the result of normal synthesis of a deficient protein.
Box 3.

The best screening test for inherited antithrombin deficiency is the antithrombin–heparin cofactor assay. This assay measures factor Xa inhibition, which is reduced in both quantitative and qualitative antithrombin defects.1
Ideally, blood for investigation of coagulation disorders should be obtained before the initiation of heparin or warfarin therapy. In addition, warfarin may decrease protein C and S levels and transiently increase antithrombin levels. Heparin increases the anticoagulant activity of antithrombin and thus may decrease its levels by 30%, which may lead to erroneous diagnosis of antithrombin deficiency. Because of the reduced levels or biological activity of antithrombin, large quantities of heparin are often required to achieve adequate anticoagulation. Such “heparin resistance” may alert the clinician to consider antithrombin deficiency as the underlying cause of thrombosis before the results of confirmatory tests become available. Patients with thrombosis due to inherited antithrombin deficiency should receive life-long anticoagulation with warfarin, which impairs the synthesis of vitamin K–dependent clotting factors (factors VII, IX and X), thus allowing appropriate anticoagulation irrespective of the antithrombin level.1,3 The infusion of antithrombin concentrates may be useful in patients with antithrombin deficiency who have recurrent thrombosis despite adequate anticoagulation.3 However, prophylactic anticoagulation is not generally indicated in asymptomatic cases unless the patient has been exposed to prothrombotic situations such as surgery or prolonged immobilization.1,3
Constantin B. Marcu Vrije University Medical Center Amsterdam, The Netherlands Thomas J. Donohue Andre E. Ghantous Hospital of Saint Raphael Yale University School of Medicine New Haven, Conn.
REFERENCES
- 1.Bauer KA. Hypercoagulable states. In: Hoffman R, Benz EJ, Shattil SJ, editors. Hematology: basic principles and practice. New York: Churchill Livingstone; 2005. p. 2197-224.
- 2.Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh 1965;13:516. [PubMed]
- 3.Bick RL. Prothrombin G20210A mutation, antithrombin, heparin cofactor II, protein C, and protein S defects. Hematol Oncol Clin North Am 2003;17(1):9-36. [DOI] [PubMed]