

Abstract
Background
Two putative methionine aminopeptidase genes, map (essential) and yflG (non-essential), were identified in the genome sequence of Bacillus subtilis. We investigated whether they can function as methionine aminopeptidases and further explored possible reasons for their essentiality or dispensability in B. subtilis.
Results
In silico analysis of MAP evolution uncovered a coordinated pattern of MAP and deformylase that did not correlate with the pattern of 16S RNA evolution. Biochemical assays showed that both MAP (MAP_Bs) and YflG (YflG_Bs) from B. subtilis overproduced in Escherichia coli and obtained as pure proteins exhibited a methionine aminopeptidase activity in vitro. Compared with MAP_Bs, YflG_Bs was approximately two orders of magnitude more efficient when assayed on synthetic peptide substrates. Both map and yflG genes expressed in multi-copy plasmids could complement the function of a defective map gene in the chromosomes of both E. coli and B. subtilis. In contrast, lacZ gene transcriptional fusions showed that the promoter activity of map was 50 to 100-fold higher than that of yflG. Primer extension analysis detected the transcription start site of the yflG promoter. Further work identified that YvoA acted as a possible weak repressor of yflG expression in B. subtilis in vivo.
Conclusion
Both MAP_Bs and YflG_Bs are functional methionine aminopeptidases in vitro and in vivo. The high expression level of map and low expression level of yflG may account for their essentiality and dispensality in B. subtilis, respectively, when cells are grown under laboratory conditions. Their difference in activity on synthetic substrates suggests that they have different protein targets in vivo.
Background
Ribosome-mediated protein synthesis is always initiated with either methionine (in eukaryotes) or N-formylmethionine (in prokaryotes and eukaryotic organelles) [1]. However, after removal of the N-formyl group from the polypeptide by peptide deformylase (DEF, EC 3.5.1.88), the N-terminal methionine of a large number of proteins is cleaved by methionine aminopeptidase (MAP, EC 3.4.11.18) [2,3]. The efficiency of removal of the initiator methionine is defined by a highly conserved local substrate specificity, which is determined by both methionine and its adjacent residue. MAP hydrolytically removes the N-terminal methionine only when the penultimate residue bears a small and uncharged side chain (Gly, Ala, Ser, Thr, Pro, Val, or Cys) [4-9], and residues downstream of the penultimate residue have little impact on the reaction. All MAPs studied to date were reported to be cobalt-dependent metalloproteases [3]. However, some reports showed that MAPs exhibited activity in the presence of other divalent ions, such as Zn(II), Fe(II) or Mn(II) [10-12]. According to their sequence homology, MAPs are grouped into two subtypes, type I and type II [13,14]. An insertion of approximately 60 residues in the C-terminal domain of type II methionine aminopeptidases is the only difference between the two types [14]. Eukaryotes possess at least two map genes, of both type I and type II [3], while there is only one map gene in most prokaryotic genomes, either type I (Archaea) or type II (Bacteria) [15].
MAP is distributed throughout living organisms, where it plays an important physiological role. The deletion of the single map gene in prokaryotes such as Escherichia coli [16] or Salmonella typhimurium [17] is lethal. In yeast, the deletion of any one of the two map genes (type I or type II) causes a slow growth phenotype, while deletion of both genes is lethal [18]. Why might MAP activity have an essential role in living cells? Previous studies showed that many proteins needed to have their N-terminal methionine removed to have normal biological activity, proper subcellular localization and eventual degradation (reviewed in [19]). As a result, it is possible that MAP is essential because of the essentiality of its downstream targets. Besides essentiality, MAP changes the dynamics of the sulfur containing metabolites, which may have an important role in the homeostasis of the cell.
In general, there is only one map gene in the genome of prokaryotes, with the exception of a cyanobacteria strain, which has three functional methionine aminopeptidases [20]. Surprisingly, in the genomes of Bacilli, two or even more putative MAP genes can be detected by sequence alignment. In B. subtilis, two putative genes responsible for MAP activity, map and yflG, can be identified [21]. Kobayashi et al. [22] reported that map is an essential gene in B. subtilis while yflG is not, an apparent paradox if yflG codes for a methionine aminopeptidase. In an attempt to better understand the functional role of MAP, we studied the evolutionary trend of MAPs in silico and demonstrated that both the map and yflG genes from B. subtilis code for methionine aminopeptidases in vitro and in vivo; furthermore, the finding of a high expression level for map in parallel with a low expression of yflG in B. subtilis, may account for their essentiality and dispensability, respectively.
Results
Sequence alignment and evolutionary pattern analyses
In the genome sequence of B. subtilis, two genes could code for methionine aminopeptidases, map and yflG, respectively [21] (Figure 1). The sequence similarity and identity between E. coli MAP (MAP_Ec) and B. subtilis MAP (MAP_Bs) is 65% and 46%, respectively. Similarity between MAP_Ec and YflG from B. subtilis (YflG_Bs) is 55% and their identity is 34% and similarity between MAP_Bs and YflG_Bs is 58% and their identity is 36%. However, the sequence of YflG_Bs differs slightly from the PROSITE consensus for type I MAPs (PS00680), which is [MFY]-x-G-H-G- [LIVMC]-[GSH]-x(3)-H-x(4)-[LIVM]-x-[HN]-[YWVH]. In particular it displays one extra residue next to the conserved GHG metal-binding motif. Interestingly, MAP_Bs is more similar to MAP_Ec and YflG_Bs is more similar to the only putative methionine aminopeptidase of Staphylococcus aureus (labelled as "YMAP" in Figure 1). Overall the proteins are very similar in their physico-chemical properties (they have closely similar length and are slightly acidic). Moreover, as shown in Figure 1, the metal (presumably cobalt) binding site (highlighted residues) is conserved. It was therefore important to investigate whether both proteins had methionine aminopeptidase activity.
Figure 1.

Sequences alignment between MAP from E. coli, B. subtilis, and S. aureus. The first line shows the protein sequence of MAP from B. subtilis (MAP_Bs), the second shows MAP from E. coli (MAP_Ec), the third shows YflG from B. subtilis (YflG_Bs) and the last shows YflG/MAP from Staphylococcus aureus (YMAP_Sa). Identical amino acids are shadowed in dark and conservative amino acids are shadowed in grey. The highlighted residues stand for those conserved amino acids important for MAP activity (bold and italic residues are conservative basic ones, while bold only are acidic ones). [MFY]-x-G-H-G-[LIVMC]-[GSH]-x(3)-H-x(4)-[LIVM]-x-[HN]-[YWVH] (where H is a cobalt ligand) is the conserved motif within the protein sequences (placed under the alignment).
This difference in evolution of proteins considered as essential in related organisms (Firmicutes) is intriguing, and we explored the evolution pattern of map in a study meant to identify persistent genes in bacteria [23]. When matched with the evolution of 16S RNA, the map gene does not follow a linear course of evolution, as do most ribosomal proteins [23], but shows an erratic pattern (Figure 2). This suggests that MAP might be involved in different biological processes besides its role as methionine aminopeptidase according to the organisms. This prompted us to explore the evolution of MAP with proteins that might be functionally related, using the criteria we used to identify persistent genes. We defined persistent genes as the genes present as orthologs in more than 85% of the genomes of a clade (here 26 Firmicutes), after removing obligatory endosymbionts from the list (see Methods). The protein divergence (sequence similarity divided by sequence length difference) of MAP in B. subtilis and its orthologs from other Firmicutes were compared with bacterial evolutionary distance (measured by 16S rRNA distance). Similar analyses were performed with a general protease, ClpP, DEF, ribosomal protein S4, RpsD and YkrB, a second deformylase present in B. subtilis (Figure 2). Interestingly, in contrast to the expected evolutionary pattern of RpsD, MAP exhibited the same erratic evolutionary way as Def and YkrB, the two functional deformylases in B. subtilis, suggesting co-evolution of methionine aminopeptidase and deformylase.
Figure 2.
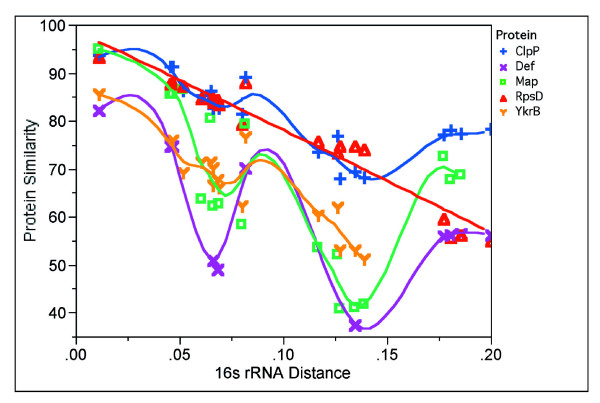
Evolutionary trend analyses of MAP, ClpP, Def, RpsD and YkrB. Map, ClpP, Def, RpsD and YkrB were picked up from B. subtilis. Their BBHs from other Firmicutes were detected. Their protein divergence (sequence similarity divided by sequence length difference) in B. subtilis and their orthologs from other Firmicutes were compared with bacterial evolutionary distance individually (measured by 16S rRNA evolution).
Enzyme activity in vitro
It is often recognized that gene function identification solely based on sequence comparisons could be misleading [24,25]. Therefore, we first determined whether map and yflG were authentic MAP genes by overproducing the proteins in E. coli and characterizing their enzymatic activity. Both products of map and yflG genes were purified to more than 90% homogeneity. Mass spectrometry showed that the molecular mass of MAP_Bs and YflG_Bs was 27409.17 ± 0.92 Da and 27209.35 ± 2.14 Da, respectively. Interestingly, both proteins purified from E. coli retained their initial methionine, consistent with the nature of the second residue, isoleucine in both cases.
As seen in Table 1, both enzymes exhibited a methionine aminopeptidase activity in vitro with the synthetic peptide substrates tested. They differed widely, however, in the extent of their activity: the specific activity of YflG_Bs was about 90- and 20-fold higher than that of MAP_Bs with the tetrapeptide MGMM and tripeptide MAS as the substrates, respectively. Both enzymes preferred the tetrapeptide MGMM as the substrate. With MAS and MG as the substrates, MAP_Bs activity was 76.9% and 46.2% of that with MGMM as the substrate, respectively, while YflG_Bs retained only 17.7% and 0.6% relative activity, respectively, with these substrates.
Table 1.
Reaction rate of MAP_Bs and YflG_Bs with different peptides as the substrates
| Specific Activity (U/mg) | ||
| Peptide (4 mM) | MAP_Bs | YflG_Bs |
| MGMM | 2.6 ± 0.3 | 232.0 ± 8.2 |
| MAS | 2.0 ± 0.1 | 41.0 ± 5.5 |
| MG | 1.2 ± 0.1 | 1.3 ± 0.1 |
Three measurements for each of the proteins with different substrates were made.
The effect of divalent ions (0.2 mM) on enzyme activity was investigated (Figure 3). MAP_Bs had significant activity with both Mn2+ and Co2+. MAP_Bs retained appreciable activity (26–50%) after replacement of Co2+ with Cd2+, Zn2+, Ca2+, Mg2+, Cu2+, Sr2+ and Ba2+, but not Ni2+. In contrast, YflG_Bs showed a strong preference for Co2+ with a decrease in activity to below 20% with any other divalent ions tested.
Figure 3.
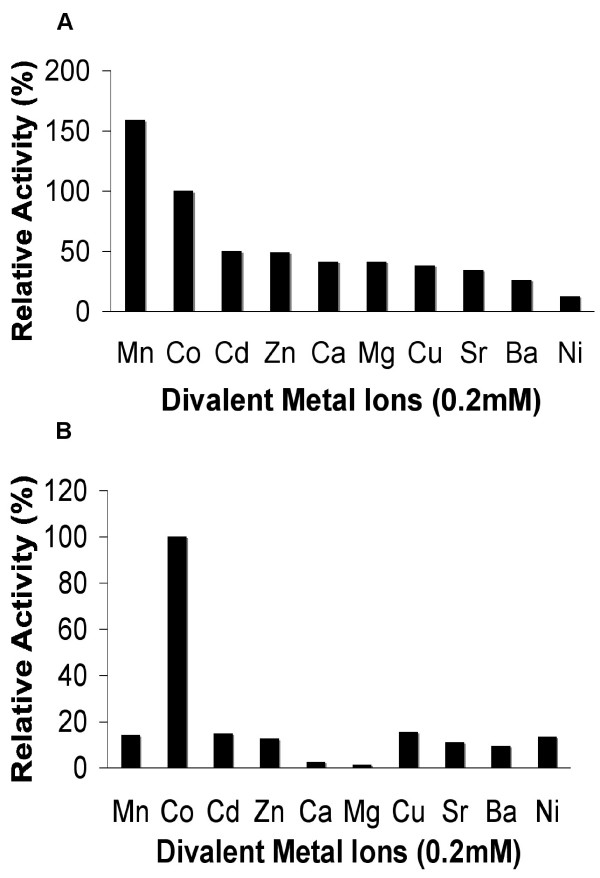
Relative activity of (A) MAP_Bs and (B) YflG_Bs in the present of different ions. MGMM (4 mM) was used as the substrate. All ions were tested at 0.2 mM concentration. Each of their relative activity was defined as 100% in the presence of Co2+ (MAP_Bs: 2.6 U/mg; YflG_Bs: 232.0 U/mg).
Subcellular localization
To investigate whether MAP_Bs and YflG_Bs had any difference in their subcellular localization, we constructed N-terminal green fluorescent protein (GFP) fusions of both proteins. As previously reported, the catalytic domain of the methionine aminopeptidase is located in the C-terminal part of the polypeptide chain [20,26-29], therefore N-terminal GFP fusions of both proteins were not expected to interfere with their functions. In addition, we also constructed C-terminal GFP fusions of both MAP_Bs and YflG_Bs, but because of the possible degradation of the fusion proteins in vivo or some unknown reasons, we failed to detect any fluorescence in either case (data not shown). Cells from strains BSIP8001 (MAP::GFP fusion) and BSIP8002 (YflG::GFP fusion) in the mid-exponential growth phase were collected and visualized by fluorescence microscopy, respectively. Both GFP-MAP_Bs (Figure 4A) and GFP-YflG_Bs (Figure 4B) proteins distributed evenly all over the cells. As was expected, the intensity of the fluorescence of both proteins increased with the increasing concentration of xylose used to induce expression of the GFP fusions (0.1%- 1% (w/v)) (data not shown).
Figure 4.

Localization of GFP-MAP_Bs and GFP-YflG_Bs fusions. A. Localization of GFP-MAP_Bs in strain BSIP8001 grown in minimal medium with 0.5% xylose (w/v); B. Localization of GFP-YflG_Bs in strain BSIP8002 grown in minimal medium with 1% xylose (w/v).
MAP-defective mutants are rescued by MAP_Bs or YflG_Bs supplied in trans
The E. coli mutant EM9 contains an engineered map gene, whose expression is under the control of isopropyl β-D-thiogalactoside (IPTG), so that the strain cannot grow unless IPTG is added to the growth medium [16]. Using the replicative vector pBAD, which holds an arabinose-inducible promoter, the map and yflG genes from B. subtilis were individually introduced into strain EM9. As shown in Figure 5, the transformants could complement the map defect when IPTG was omitted (leading to a MAP deficiency) if a high concentration of arabinose was supplied, in the case of plasmids carrying either B. subtilis map or yflG genes. Lower concentrations of arabinose only partially supported growth. This demonstrated that both map and yflG from B. subtilis can supply the MAP function to E. coli in vivo.
Figure 5.
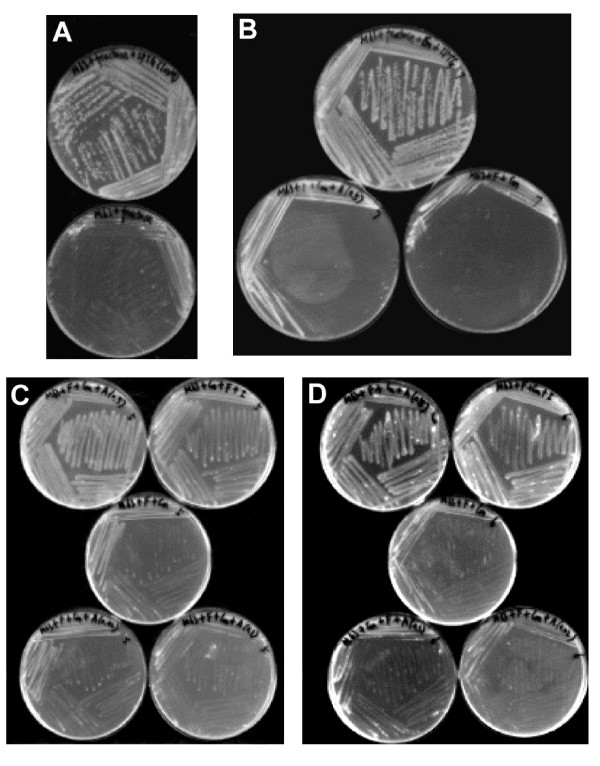
Genetic rescue experiments. The bacteria were streaked on M63 minimal solid medium and the plates were incubated at 37°C overnight. The concentration of IPTG was 1 mM. The order of the plates in each picture described below is from left to right and from top to bottom. A. EM9 host strain on M63 with IPTG; EM9 on M63 without IPTG. B. EM9 strain with pBAD vector alone (ECHP1007) on M63 with IPTG; ECHP1007 on M63 with arabinose (0.5% (W/V)); ECHP1007 on M63 without IPTG and arabinose. C. EM9 strain caring pBAD map vector (ECHP1005) on M63 with arabinose (0.5% (W/V)); ECHP1005 on M63 with IPTG; ECHP1005 on M63 without IPTG; ECHP1005 on M63 with arabinose (0.02% (W/V)); ECHP1005 on M63 with arabinose (0.1% (W/V)). D. EM9 strain caring pBAD yflG vector (ECHP1006) on M63 with arabinose (0.5% (W/V)); ECHP1006 on M63 with IPTG; ECHP1006 on M63 without IPTG; ECHP1006 on M63 with arabinose (0.1% (W/V)); ECHP1006 on M63 with arabinose (0.02% (W/V)).
In B. subtilis, map is an essential gene, while yflG is not [22]. However, with a back-up map or yflG copy under the control of IPTG provided in multi-copy plasmid pDG148 (30–50 copies using the pUB110 replicon from B. subtilis [30]), map::lacZ disruptants in the chromosome of B. subtilis were obtained (BSHP7046 and BSHP7037). Nevertheless, as witnessed in both cases by the systematic formation of variegated blue white streaked colonies on X-gal plates, the clones were extremely unstable, leading to the loss of transformants after a few generations (data not shown).
map expression is considerably higher than yflG expression
The fact that map is essential while yflG is not [22], together with the ability of yflG gene to rescue MAP_Bs function when supplied at a high level, points to substantial differences in the expression level of the two genes. To test this possibility, the expression of map and yflG genes in B. subtilis in vivo was studied using lacZ as the reporter gene.
Strains BSHP7042 and BFS4611 were grown in minimal medium and were used to monitor the expression of map and yflG, respectively. The map gene showed the higher promoter activity (Figure 6). Its activity increased gradually during the log phase of growth and a nearly four-fold increase was detected (109–379 U/mg) between early exponential and stationary growth phases. In contrast, the yflG gene showed very low activity under all the conditions tested (4–8 U/mg) (Figure 6). In summary, the map gene promoter showed 50 to 100-fold higher activity than that of yflG using lacZ as the reporter gene.
Figure 6.
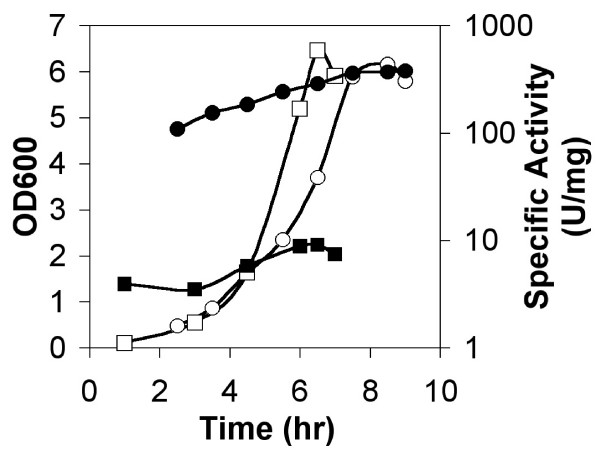
Expression of map and yflG lacZ transcriptional fusions. β-galactosidase expression of map and yflG lacZ transcriptional fusion during the growth curve of strains BSHP7042 and BFS4611 in minimal medium. OD600 (○) and β-galactosidase activity (●) were measured for strain BSHP7042 with map lacZ transcription fusion. OD600 (□) and β-galactosidase activity (■) were measured for strain BFS4611 with yflG lacZ transcription fusion.
Promoter localization of the yflG gene
In the B. subtilis genome, the yflG gene is located upstream of the yflH gene, followed by a putative Rho-independent transcription terminator. RT-PCR experiments demonstrated that yflG and yflH belonged to a common transcription unit, making an operon (data not shown). Furthermore, lacZ transcription fusion with yflH gene (strain BSHP7043) showed the same activity changes as that of yflG gene during all the conditions tested (data not shown), substantiating the RT-PCR results. In order to identify the promoter of the yflG-yflH operon, we carried out primer extension analysis. Two primers complementary to two different regions inside the yflG gene were used yielding identification of the same transcription start point. As shown in Figure 7, the start is located 31 nt upstream of the ATG translation start codon and regions weakly similar to consensus -35 (TTCCTA) and -10 (TAAGCT) regions are found upstream of this start point, separated by 18 nt, in an AT-rich region. It is difficult at this point to correlate the structure of this promoter with the poor expression of yflG in all conditions tested.
Figure 7.
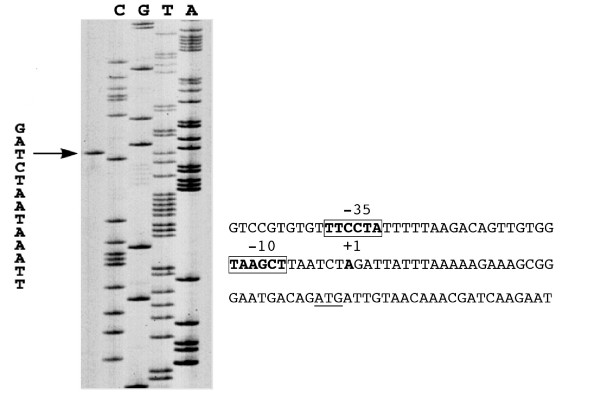
Identification of the transcription start point of the yflG promoter. Primer extension and sequencing reaction were performed with the same primer. The size of the extended product is compared to a DNA-sequencing ladder of the corresponding region. An arrow indicates the +1 site. Predicted -10 and -35 regions are in bold and boxed; the experimental transcription start site is in bold; the translational start site is underlined.
YvoA showed a slight repression effect on yflG expression
As the expression of yflG in B. subtilis in vivo was very low, we endeavoured to uncover conditions to enhance its expression. No significant difference was found when the carbon, nitrogen and sulfur sources were changed, and no difference was found in conditions of sporulation or germination (data not shown). As genes proximal in the chromosome often code for proteins with related functions, the neighbourhood of yflG was analysed. Gene nagP, which encodes a putative phosphoenolpyruvate-dependent transport system N-acetylglucosamine-specific enzyme IICB component, is located upstream of yflG and transcribed divergently (Figure 8A). It could share common control elements. Exploring the corresponding intergenic region with a sliding window 19 nt-long we uncovered sequence AATTGGTATAGATCACTAG which has a very significant counterpart (AGCTGGTCTAGATCACTAG) upstream of the nagAB operon, encoding two N-acetylglucosamine metabolic genes and a putative transcriptional regulator (GntR family) gene, yvoA (Figure 8A). This prompted us to investigate whether YvoA was a possible regulator for yflG. Using a lacZ fusion, we monitored the expression of yflG in a yvoA-disrupted strain, BSIP8004.
Figure 8.
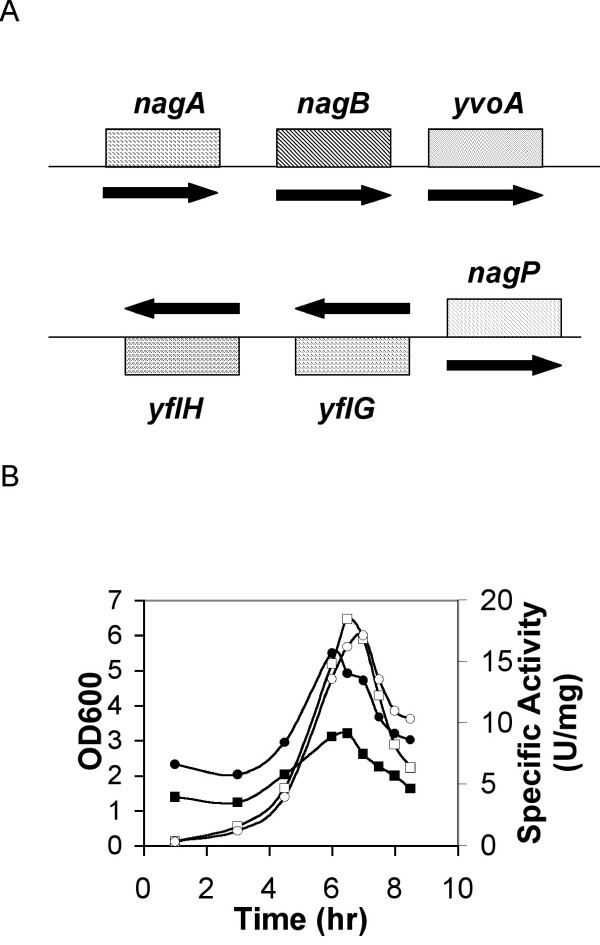
Expression of yflG lacZ transcriptional fusions in yvoA disrupted strain. A. gene organization around yflG and yvoA. The arrows indicate the direction of gene transcription; B. β-galactosidase expression yflG lacZ transcriptional fusion in yvoA disrupted strain BSIP8004 and yvoA wild type strain BFS4611 grown in minimal medium. OD600 (○) and β-galactosidase activity (●) were measured for strain BSIP8004. OD600 (□) and β-galactosidase activity (■) were measured for strain BSIP8004 and strain BFS4611.
As shown in Figure 8B, the promoter of yflG showed a low but consistently higher activity in the yvoA gene disrupted strain BSIP8004 when compared with the wild type parent BFS4611 (two-fold higher activity). This suggested that YvoA could contribute to the regulation of yflG expression.
Discussion
In prokaryotes, it is usually accepted that there is only one gene responsible for methionine aminopeptidase. However, the B. subtilis genome program predicted the existence of two methionine aminopeptidase genes, map and yflG [21]. We have shown that both MAP_Bs and YflG_Bs are functional methionine aminopeptidases in vitro (Table 1 and Figure 3). Furthermore, gene rescue experiments showed that both map and yflG genes expressed on replicative plasmids could supply the MAP function in E. coli and B. subtilis, demonstrating that both MAP_Bs and YflG_Bs are functional methionine aminopeptidases in vivo (Figure 5). Interestingly, Kobayashi et al. [22] reported that disruption of the map gene in B. subtilis is lethal, while the deletion of yflG is not. In the present study, we obtained B. subtilis map mutants when map or yflG genes were expressed in multi-copy replicative plasmids under the control of an IPTG induced promoter (strains BSHP7046 and BSHP7037). This observation suggests that the single copy of the yflG gene expressed in the chromosome could not supply the MAP function in B. subtilis, substantiating that under our conditions its expression is extremely low, making MAP the only significant methionine aminopeptidase available. This is corroborated by the lacZ fusion studies showing that the map gene is expressed some 50–100-fold higher than yflG under all conditions tested. Despite the weak expression of yflG, however, we detected a sigma A promoter using primer extension (Figure 7). We further showed that this expression is modulated by YvoA (Figure 8), suggesting a possible connection between N-acetylglucosamine metabolism and methionine aminopeptidase. In this respect it is worth noting that in E. coli glucosamine-6-phosphate deaminase activity is modulated by the amino-terminal methionine of the enzyme [31]. We tried several growth conditions involving N-acetylglucosamine as a carbon or nitrogen source, or both, but did not find any modulation of YflG activity (data not shown). We also explored expression of yflG::lacZ fusions during sporulation and germination. No enhancement was observed (data not shown). As a consequence, we surmise that the high expression level of map accounts for its essentiality in B. subtilis.
The published MAP_Ec activity [6,11] is about 5–10 times higher than that of MAP_Bs in our assay conditions. This may be related to the observation that the map gene in E. coli does not belong to a highly expressed ribosome protein operon, in contrast to the situation in B. subtilis [32]. As a result, the relative expression level of map in E. coli might be lower than that of the map gene in B. subtilis. For still unknown reasons it might be impossible for the yflG gene to function as the main methionine aminopeptidase in B. subtilis in vivo in spite of the high activity of YflG_Bs in vitro. As shown in Table 2, the enzyme activity of MAPs in crude cell extracts of E. coli (BL5), B. subtilis (168) and yflG disrupted B. subtilis strain (BFS4611) showed not much difference in vitro under all the condition tested. Different expression levels of map genes in a genome carrying more than one methionine aminopeptidase genes have been previously suggested to be the main reason for their different physiological roles in vivo, but no direct evidence was provided [20,33]. Methionine aminopeptidases in vivo are active on proteins, i.e. on complex polypeptides [34,35] and not on the short peptides in our in vitro assays. It could well be that specific targets require a specific aminopeptidase. A similar conclusion holds for other apparently paralogous genes. For example, there are also two functional peptide deformylases in B. subtilis, but only one is probably the predominant deformylase with concomitant high gene expression [36].
Table 2.
MAP activity in crude cell extracts of E. coli and B. subtilis
| Specific Activity (U/mg) | |||
| Peptide (4 mM) | E. coli (BL5) | B. subtilis wild type (168) | B. subtilis yflG mutant (BFS4611) |
| MGMM | 1.5 ± 0.2 | 1.5 ± 0.1 | 1.5 ± 0.1 |
| MAS | 1.1 ± 0.2 | 1.1 ± 0.1 | 1.1 ± 0.2 |
| MG | 0.6 ± 0.1 | 0.8 ± 0.2 | 0.4 ± 0.1 |
Cells in stationary growth phase of each of the strains were collected and sonically disrupted. Supernatants of each of the cell debris were used to measure MAP activity. Totally, three times of measurements were carried out.
The finding of the co-evolutionary parallel trend of methionine aminopeptidase and deformylase in Firmicutes (Figure 2) is consistent with functional relationship in vivo, which correlates well with the fact that deformylase removes all N-formyl groups as a prerequisite for the subsequent function of MAP [33]. This co-evolution may be the signature of protein-protein interactions [37,38], an intriguing conjecture, which seems to be supported by the consistent duplication of deformylases in genomes with more than one methionine aminopeptidase (Table 3).
Table 3.
Distribution of def and map genes in selected genomes
| Numbers in Each Genome | |||
| Genome Name | def | map | EMBL ID |
| Streptomyces coelicolor | 4 | 3 | AL645882 |
| Streptomyces avermitilis | 3 | 4 | BA000030 |
| Nocardia farcinica IFM 10152 | 3 | 3 | AP006618 |
| Shewanella oneidensis MR-1 | 3 | 1 | AE014299 |
| Silicibacter pomeroyi DSS-3 | 3 | 1 | CP000031 |
| Bacillus cereus ZK | 2 | 3 | CP000001 |
| Bacillus thuringiensis serovar konkukian | 2 | 3 | AE017355 |
| Bacillus anthracis 'Ames Ancestor' | 2 | 3 | AE017334 |
| Bacillus anthracis Sterne | 2 | 3 | AE017225 |
| Bacillus cereus ATCC 10987 | 2 | 3 | AE017194 |
| Bacillus anthracis Ames | 2 | 3 | AE016879 |
| Bacillus cereus ATCC 14579 | 2 | 3 | AE016877 |
| Leifsonia xyli subxyli CTCB07 | 2 | 3 | AE016822 |
| Synechocystis PCC 6803 | 1 | 3 | BA000022 |
| Bacillus clausii KSM-K16 | 1 | 3 | AP006627 |
Conclusion
We proved that both MAP_Bs and YflG_Bs are functional methionine aminopeptidases in B. subtilis and we suggested that map gene is essential because of its high expression level, while yflG is nonessential possibly because of its low expression level making that it can not take over or compensate the function of map when map is not expressed, or because of specific targets dedicated to only one of the two MAPs. Conservation of several map-like genes in Bacilli suggests involvement in a process specific to this group of organisms, which may involve also peptide deformylase.
Methods
Bacterial strains, plasmids, and culture conditions
E. coli and B. subtilis strains as well as plasmids used in this work are listed in Table 4. E. coli TG1 and XL1-Blue were used for cloning experiments (TG1 for single cross-over recombination or replicative plasmids propagation and XL1-Blue for double cross-over recombination) and E.coli BL5 for IPTG-induced protein overproducing. B. subtilis 168 was used as the wild type strain in this study. E. coli cells were grown in Luria-Bertani (LB) medium [39] or in M63 minimal medium (KH2PO4, 4.4 mM; K2HPO4, 8 mM; (NH4)2SO4, 15 mM; MgSO4, 2 mM; ferric citrate, 34 μM; VitB1, 0.0001% (w/v); fructose, 0.4% (w/v); sodium citrate, 0.3 mM; CaCl2, 50 μM; MnCl2, 5 μM; ZnCl2, 12 μM; CuCl2, 2.5 μM; CoCl2, 2.5 μM; Na2MoO4, 2.5 μM) supplemented with a final concentration of arabinose from 0.02% to 0.5% (w/v) when needed. B. subtilis cells were grown in SP medium or in minimal medium (K2HPO4, 8 mM; KH2PO4, 4,4 mM; glucose, 27 mM; Na3-citrate, 0.3 mM; L-glutamine, 15 mM; L-tryptophan, 0.244 mM; ferric citrate, 33.5 μM; MgSO4 or L-methionine, 1 mM; MgCl2, 0.61 mM; CaCl2, 49.5 μM; FeCl3, 49.9 μM; MnCl2, 5.05 μM; ZnCl2, 12.4 μM; CuCl2, 2.52 μM; CoCl2, 2.5 μM; Na2MoO4, 2.48 μM). Antibiotics were added to the following concentrations when required: ampicillin, 100 μg/ml; spectinomycin, 80 μg/ml; chloramphenicol, 5 μg/ml; phleomycin, 5 μg/ml; kanamycin, 100 μg/ml for E. coli and 5 μg/ml for B. subtilis. IPTG was added at 1 mM concentration or as stated when needed. Solid media were prepared by adding 1.5% agar (w/v) to the respective liquid media. Bacteria were grown at 37°C. The optical density (OD) of bacterial cultures was measured at 600 nm. All experiments were performed in accordance with the European regulation requirements concerning the contained use of Genetically Modified Organisms of Group-I (French agreement N° 2735).
Table 4.
Bacterial strains and plasmids used or created in this study
| Strains | Genotype | Source or Reference |
| Escherichia coli | ||
| XL1-Blue | K12 supE44 hsdR17 recA1 endA1 gyrA46 thi relA1 lac- F'[proAB+ lacIq lacZΔM15 Tn10(tetR)] | Laboratory collection |
| TG1 | K12 supE hsdΔ5 thi Δ (lac-proAB) F'[traD36 proA+ proB+ lacIq lacZΔM15] | Laboratory collection |
| BL5(DE3) | F- | Invitrogen |
| EM9 | PT7-map (on chromosome) | Chiron Culture Collection Centre |
| ECIP8003 | F- pIPP8003 | This work |
| ECIP8004 | F- pIPP8004 | This work |
| ECHP1005 | EM9 pHPP1017 | This work |
| ECHP1006 | EM9 pHPP1018 | This work |
| ECHP1007 | EM9 pBAD | This work |
| Bacillus subtilis | ||
| 168 | trpC2 | Laboratory collection |
| BFS4611 | trpC2 yflG::lacZ | Functional analysis projecta |
| BFS817 | trpC2 yvoA::lacZ | Functional analysis projecta |
| BSHP7042 | trpC2 map-lacZ | This work |
| BSHP7045 | trpC2 pDG148-map | This work |
| BSHP7046 | trpC2 map::lacZ pDG148-map | This work |
| BSHP7037 | trpC2 map::lacZ pDG148-yflG | This work |
| BSHP7038 | trpC2 pDG148-yflG | This work |
| BSHP7043 | trpC2 yflH::lacZ | This work |
| BSIP8001 | trpC2 amyE::gfp-map | This work |
| BSIP8002 | trpC2 amyE::gfp-yflG | This work |
| BSIP8003 | trpC2 yvoA::[(lacZ-ery)::pEC23(Kan)] | This work |
| BSIP8004 | trpC2 yflG::lacZ yvoA::[(lacZ-ery)::pEC23(Kan)] | This work |
| Plasmids | Description | Source or Reference |
| pET24b(+) | expression vector, KanR | Novagen |
| pBAD-abg | genetic rescue vector, AmpR | [43] |
| pDG148 | expression vector, AmpR KanR | [44] |
| pJM783 | cloning vector, CmR | [45] |
| pSG1729 | N-terminal GFP fusion vector, AmpR SpecR | [42] |
| pEC23 | integrative plasmid, KanR | M. Simon and P. Stragier, unpublished |
| pIPP8003 | pET24b map | This work |
| pIPP8004 | pET24b yflG | This work |
| pHPP1017 | pBAD map | This work |
| pHPP1018 | pBAD yflG | This work |
| pHPP7017 | pJM783 map-lacZ | This work |
| pHPP7018 | pJM783 yflH::lacZ | This work |
| pHPP7021 | pDG148 map | This work |
| pHPP7022 | pDG148 yflG | This work |
| pHPP7025 | pJM783 map::lacZ | This work |
| pIPP8001 | pSG1729 map | This work |
| pIPP8002 | pSG1729 yflG | This work |
a This strain has been constructed during the frame of the project for the functional characterization of the genome of B. subtilis in Japan [46].
DNA techniques
DNA purification, restriction enzyme digestion, ligation and transformation of E. coli were performed according to standard protocols [40]. For cloning purpose, YieldAce or Pfu DNA polymerase (Stratagene) was used. All the DNA sequences in the plasmids constructed in this work were determined using the dideoxy-chain termination method and Thermo Sequenase Kit (Amersham Pharmacia Biotech). B. subtilis cells were transformed with plasmid DNA following the two-steps protocol described previously [41]. Transformants were selected on LB plates containing corresponding antibiotics and IPTG when needed.
Plasmid and strain constructions
In order to clone the genes from B. subtilis for protein producing in E. coli, both map and yflG genes were amplified by PCR from genomic DNA of B. subtilis 168 and cloned into expression vector pET24b(+) (Novagen) using NdeI and XhoI restriction sites. The amplified fragments included nucleotides +1 to +744 and nucleotides +1 to +747 relative to the translational start point of map and yflG, respectively. The resulting constructs, plasmids pIPP8003 and pIPP8004, were transformed into E. coli BL5 giving strains ECIP8003 and ECIP8004 for overproducing MAP and YflG, respectively.
To construct map and yflG GFP fusion plasmids and strains, both map and yflG genes were amplified by PCR from genomic DNA of B. subtilis 168 and cloned into N-terminal GFP fusion vector pSG1729 [42] using XhoI and EcoRI restriction sites. The amplified fragments included nucleotides +1 to +744 and nucleotides +1 to +747 relative to the translational start point of map and yflG, respectively. The resulting plasmids, pIPP8001 and pIPP8002, were linearized by the ScaI restriction enzyme and were transformed into B. subtilis 168. Clones were selected for the amylase deficient phenotype after double cross-over recombination at amyE site [42], giving strains BSIP8001 and BSIP8002, respectively.
To obtain map and yflG expression plasmids for further map gene rescue analysis in a conditional map gene deletion E. coli strain, EM9 [16], both map (nucleotides -30 to +744 relative to the map translation start point) and yflG (nucleotides -21 to +754 relative to the yflG translation start point) genes were amplified by PCR from genomic DNA of B. subtilis 168 with the creation of BamHI and BglII sites. Both purified PCR fragments were cleaved by BamHI and BglII restriction enzymes and inserted between the same two sites in plasmid pBAD containing arabinose inducible promoter (a modified pBAD-αβγ plasmid containing an additional BamHI cloning site was used after removing a BamHI and BglII insert [43]), producing plasmids pHPP1017 and pHPP1018, respectively. The proper orientation was checked by the ability of cloned fragment to be recovered by double digestion using BamHI and BglII restriction enzymes. Plasmids pHPP1017, pHPP1018 and the pBAD vector were transformed into E. coli strain EM9 individually and selected on LB plates containing ampicillin, chloramphenicol and IPTG, giving strains ECHP1005, ECHP1006 and ECHP1007, respectively.
To obtain a back-up map and yflG copies for further conditional map disruptant construction in B. subtilis, the map gene (nucleotides -43 to +744 relative to the map translation start point) and yflG gene (nucleotides -54 to 733 relative to the yflG translation start point) was amplified by PCR from genomic DNA of B. subtilis 168 using primers introducing a SalI cloning site at the 5' end and a SphI cloning site at the 3' end, respectively. Both fragments were then inserted into the SalI and SphI sites of the IPTG-inducible replicative vector pDG148 [44], producing plasmid pHPP7021 and pHPP7022, respectively. The wild type strain B. subtilis 168 was transformed with these plasmids, giving strains BSHP7045 and BSHP7038, respectively.
To disrupt the map gene, a 246 bp-long fragment within the map gene (nucleotides +142 to +388 relative to the map translation start point) was amplified by PCR from genomic DNA of B. subtilis 168 using primers introducing an EcoRI cloning site at the 5' end and a BamHI cloning site at the 3' end of the fragment. This fragment was then inserted into the EcoRI and BamHI sites of plasmid pJM783 [45], which contains a promoterless lacZ reporter gene, producing plasmid pHPP7025. The plasmid was introduced into the chromosome of BSHP7045 and BSHP7038 strains by a single cross-over event, giving strains BSHP7046 and BSHP7037, respectively. To ascertain that they were correctly inserted, clones were checked by PCR.
To construct a map transcriptional fusion with the lacZ gene, a DNA fragment downstream from the map gene (nucleotides from + 423 relative to the translation start point to the gene's stop codon) was amplified by PCR from genomic DNA of B. subtilis 168 using primers introducing an EcoRI cloning site at the 5' end and a BamHI cloning site at the 3' end of the fragment, then inserted into the EcoRI and BamHI sites of plasmid pJM783 [45], producing plasmid pHPP7017. The plasmid was introduced into the chromosome of B. subtilis 168 by a single crossover event, giving strain BSHP7042.
The yflG transcriptional fusion with the lacZ gene (yflG::lacZ disruptant, strain BFS4611) was constructed within the framework of the European Union and Japanese project for the functional analysis of the genome of B. subtilis, where more than 2000 genes have been disrupted by fusion with lacZ reporter gene [22,46,47].
To construct a yflH transcriptional fusion with the lacZ gene, a DNA fragment downstream from the yflH gene (nucleotides from + 32 to +310 relative to the translation start point) was amplified by PCR from genomic DNA of B. subtilis 168 using primers introducing an EcoRI cloning site at the 5' end and a BamHI cloning site at the 3' end of the fragment, then inserted into the EcoRI and BamHI sites of plasmid pJM783 [45], producing plasmid pHPP7018. The plasmid was introduced into the chromosome of B. subtilis 168 by a single cross-over event, giving strain BSHP7043.
To obtain the disruptant yvoA strain compatible with BFS4611, the BFS817 strain (yvoA::lacZ disruptant, from the European Union and Japanese consortium, see above) was transformed with the ScaI linearized pEC23 plasmid (which carries a kanamycin resistance gene, M. Simon and P. Stragier, unpublished) for replacement with the kanamycin resistance gene of the lacZ and Erm genes belonging to the pMutin plasmid (erythromycin resistant) integrated into the genome, by homologous recombination. The resulting clones were checked for their inability to grow on erythromycin and chloramphenicol. The resulting strain was named BSIP8003. The chromosomal DNA of BSIP8003 was prepared and used to transform the BFS4611 strain, producing the double yvoA and yflG disrupted strain BSIP8004.
Expression and purification of MAP_BS and YflG_Bs proteins
The strains ECIP8003 and ECIP8004 were grown at 37°C in LB medium containing kanamycin (25 μg/ml) and induced with 0.5 mM IPTG for four hours. Cell pellets were harvested by centrifugation and stored at -20°C. Cells were suspended in 20 mM Tris-HCl (pH 8.0) and sonically disrupted. The cellular debris was removed by centrifugation at 12,000 g for 40 min. The supernatants were applied to a DEAE-sephacel column chromatography (Amersham). The protein (MAP_Bs or YflG_Bs) was eluted with 0.1 M NaCl in the same buffer. Protein concentration was determined by the Bradford's method [48] using protein assay kit (Bio-Rad Laboratories). The purity of isolated proteins was determined by SDS-PAGE [40]. Ion mass spectrometry was used to measure the molecular mass of each purified protein as well as checking the existence of the first methionine in each of the proteins.
Enzyme assays
The methionine aminopeptidase assay was performed as described by Arie Ben-Bassat et al. [6] with small modifications. 10 μl protein solution was added to 90 μl of the substrate solution (4 mM peptide in 0.1 M K2HPO4 (pH7.5), and 0.2 mM CoCl2), then incubated at 37°C for 10 min. The reaction was stopped by incubation at 100°C for 2 min. After addition of 900 μl of the colour development mixture (containing 0.2 mg of L-amino acid oxidase, 0.03 mg of horseradish peroxidase, and 0.2 mg of o-dianisidine dihydrochloride in 0.1 M Tris-HCl (pH 7.4)), the tube was incubated at 37°C for 10 min, then the absorbance at 440 nm was recorded. Standard curve with known concentration of L-methionine in the colour development buffer was plotted for quantitative analysis. The absorbance of 1 μmol of methionine per ml at 440 nm is equivalent to 8.6 [6]. One Unit of activity was defined as 1 μmol of methionine produced per min under the assay conditions used. Preference of metal ions was tested by changing the cobalt in the substrate solution into other divalent metal ions at a final concentration of 0.2 mM.
Amylase activity was detected after growth of B. subtilis strains on Tryptose Blood Agar Base (TBAB, Difco) supplemented with 10 g/l hydrolyzed starch (Sigma). Starch degradation was detected by sublimating iodine onto the plates [49].
β-galactosidase specific activity was measured as described by Miller [50]. One Unit of β-galactosidase activity was defined as the amount of enzyme that produced 1 nmol of o-nitrophenol per min at 28°C. Specific activity was expressed in Units per mg protein. The experiments were performed in triplicates.
Subcellular localization analysis
To visualize GFP-MAP_Bs and GFP-YflG_Bs fusions locations in living cells, BSIP8001 and BSIP8002 strains were grown in minimal medium with 0.5% fructose as the sole carbon source supplemented with xylose to different final concentrations (0.1% to 1% (w/v)). Cells from the mid-exponential growth phase were collected and visualized by fluorescence microscopy. Microscope Axiovert 135 TV (ZEISS, Germany) was used in the experiment and fluorescence filter sets used to visualize GFP were obtained from Chroma Technology Corp (USA). Cells were visualized on 1% agarose (w/v) slides [51] using Axiovision 4 system (Allied High Tech Products, Inc.) with exposure time of 3–10 s.
Genetic rescue experiment
The growth of EM9, ECHP1005 (containing B. subtilis map gene), ECHP1006 (containing B. subtilis yflG gene) and ECHP1007 (containing pBAD vector alone), was compared on M63 minimal medium plates (37°C, overnight) in the presence or absence of IPTG (1 mM) with or without arabinose (0.02 %, 0.1 % and 0.5 %).
RNA isolation and manipulation
Total cellular RNA was extracted from B. subtilis 168 cells growing in minimal medium to an OD600 of 0.5 using High Pure RNA Isolation Kit from Roche. The RNA concentration was determined by light absorption at 260 nm and 280 nm. 2 μg of RNA were loaded onto 1.2% agarose gel to check the RNA purity and integrity. RT-PCR experiments were performed using RT-PCR System (Promega) as specified by the manufacturer.
Primer extension analysis was carried out using reverse transcriptase AMV (Roche) as described [52]. Two oligonucleotides (+33 to +62 and +97 to +126 relative to translation start point of yflG) were used for the identification of yflG promoter. The same primers were used for the generation of sequence ladders. Reaction products were separated on 7% denaturing polyacrylamide gel containing 8 M urea. DNA sequences were determined using Sanger's dideoxy chain-termination method with "Thermo Sequenase radiolabeled terminator cycle sequencing kit" from Amersham Pharmacia Biotech.
DNA sequence and in silico analysis
DNA sequences were analyzed using the DNA Strider software [53]. The program BLAST [54] was used to search for homologous sequences in the database. The program CLUSTALW was used for multiple alignment [55]. B. subtilis sequences were analyzed using the SubtiList database [56,57]. Accepted consensus sequences were extracted from PROSITE [58,59].
Evolutionary pattern analyses
First, MAP orthologs (orthologs were defined using the BBH (Bi-directional Best Hit) strategy, i.e. if gene a of genome A is the most similar hit of gene b in genome B and vice versa, gene a and b are regarded as a pair of orthologs [60]) were retrieved between each pair of bacterial genomes. Secondly, the BBHs, with protein sequence similarity >50% and protein length difference <1.3, of the gene map in B. subtilis were collected. Thirdly, the orthologs of each of the BBHs were collected in all the genomes (a total of 206 bacterial genomes in EMBL as of July 1st, 2005), after recursive identification until the members in this cluster is no longer extended. Last, the protein divergence (sequence similarity divided by sequence length difference) of MAP in B. subtilis and its orthologs from other Firmicutes were compared with bacterial evolutionary distance (measured by 16S rRNA evolution). Same analyses were carried out on ClpP, Def, RpsD and YkrB.
Authors' contributions
CHY carried out MAPs enzyme activity analysis, subcellular observation, expression level comparisons, and drafted the manuscript. HYL did genetic rescue experiment in E. coli and primer extension analysis. AS designed the initial experiments, completed the genetic rescue experiment in B. subtilis, constructed lacZ fusion mutants of B. subtilis, and wrote part of the manuscript. GF did the evolutionary pattern analyses. YPW helped to analyse the experimental data and draft manuscript. AMG supervised the MAPs enzyme activity analysis, and helped to draft the manuscript. AD designed and supervised the whole study.
Acknowledgments
Acknowledgements
The initial stage of the work was supported by the HKU-Pasteur Research Centre (SAR Hong Kong, China) and we would like to thank HF Kung and KY Yuen for their interest at that early moment. CH You was supported by EGIDE (France). We would like to thank JD Huang for his kind gift of pBAD-αβγ vector, Chiron Culture Collection Center for conditional map gene deletion E. coli mutant, EM9, U Mechold for kind suggestions to both the experiment and manuscript drafting, and YL Yap for contribution in silico analyses and discussion.
Contributor Information
CongHui You, Email: cyou@pasteur.fr.
HongYan Lu, Email: lv_hongyan@hotmail.com.
Agnieszka Sekowska, Email: AS208663@dsvidf.cea.fr.
Gang Fang, Email: fangg@pasteur.fr.
YiPing Wang, Email: wangyp@pku.edu.cn.
Anne-Marie Gilles, Email: amgilles@pasteur.fr.
Antoine Danchin, Email: adanchin@pasteur.fr.
References
- Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol Rev. 1983;47:1–45. doi: 10.1128/mr.47.1.1-45.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, Robison K, Church GM. Comparing the predicted and observed properties of proteins encoded in the genome of Escherichia coli K-12. Electrophoresis. 1997;18:1259–1313. doi: 10.1002/elps.1150180807. [DOI] [PubMed] [Google Scholar]
- Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem Sci. 1998;23:263–267. doi: 10.1016/S0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- Huang S, Elliott RC, Liu PS, Koduri RK, Weickmann JL, Lee JH, Blair LC, Ghosh-Dastidar P, Bradshaw RA, Bryan KM. Specificity of cotranslational amino-terminal processing of proteins in yeast. Biochemistry. 1987;26:8242–8246. doi: 10.1021/bi00399a033. [DOI] [PubMed] [Google Scholar]
- Moerschell RP, Hosokawa Y, Tsunasawa S, Sherman F. The specificities of yeast methionine aminopeptidase and acetylation of amino-terminal methionine in vivo. Processing of altered iso-1-cytochromes c created by oligonucleotide transformation. J Biol Chem. 1990;265:19638–19643. [PubMed] [Google Scholar]
- Ben-Bassat A, Bauer K, Chang SY, Myambo K, Boosman A, Chang S. Processing of the initiation methionine from proteins: properties of the Escherichia coli methionine aminopeptidase and its gene structure. J Bacteriol. 1987;169:751–757. doi: 10.1128/jb.169.2.751-757.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunasawa S, Stewart JW, Sherman F. Amino-terminal processing of mutant forms of yeast iso-1-cytochrome c. The specificities of methionine aminopeptidase and acetyltransferase. J Biol Chem. 1985;260:5382–5391. [PubMed] [Google Scholar]
- Miller CG, Strauch KL, Kukral AM, Miller JL, Wingfield PT, Mazzei GJ, Werlen RC, Graber P, Movva NR. N-terminal methionine-specific peptidase in Salmonella typhimurium. Proc Natl Acad Sci U S A. 1987;84:2718–2722. doi: 10.1073/pnas.84.9.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirel PH, Schmitter MJ, Dessen P, Fayat G, Blanquet S. Extent of N-terminal methionine excision from Escherichia coli proteins is governed by the side-chain length of the penultimate amino acid. Proc Natl Acad Sci U S A. 1989;86:8247–8251. doi: 10.1073/pnas.86.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker KW, Bradshaw RA. Yeast methionine aminopeptidase I can utilize either Zn2+ or Co2+ as a cofactor: a case of mistaken identity? Protein Sci. 1998;7:2684–2687. doi: 10.1002/pro.5560071224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza V M, Holz RC. The methionyl aminopeptidase from Escherichia coli can function as an iron(II) enzyme. Biochemistry. 1999;38:11079–11085. doi: 10.1021/bi990872h. [DOI] [PubMed] [Google Scholar]
- Wang J, Sheppard GS, Lou P, Kawai M, Park C, Egan DA, Schneider A, Bouska J, Lesniewski R, Henkin J. Physiologically relevant metal cofactor for methionine aminopeptidase-2 is manganese. Biochemistry. 2003;42:5035–5042. doi: 10.1021/bi020670c. [DOI] [PubMed] [Google Scholar]
- Bazan JF, Weaver LH, Roderick SL, Huber R, Matthews BW. Sequence and structure comparison suggest that methionine aminopeptidase, prolidase, aminopeptidase P, and creatinase share a common fold. Proc Natl Acad Sci U S A. 1994;91:2473–2477. doi: 10.1073/pnas.91.7.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arfin SM, Kendall RL, Hall L, Weaver LH, Stewart AE, Matthews BW, Bradshaw RA. Eukaryotic methionyl aminopeptidases: two classes of cobalt-dependent enzymes. Proc Natl Acad Sci U S A. 1995;92:7714–7718. doi: 10.1073/pnas.92.17.7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Cui YM, Chen LL, Gu M, Li J, Nan FJ, Ye QZ. Mutations at the S1 sites of methionine aminopeptidases from Escherichia coli and Homo sapiens reveal the residues critical for substrate specificity. J Biol Chem. 2004;279:21128–21134. doi: 10.1074/jbc.M401679200. [DOI] [PubMed] [Google Scholar]
- Chang SY, McGary EC, Chang S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J Bacteriol. 1989;171:4071–4072. doi: 10.1128/jb.171.7.4071-4072.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CG, Kukral AM, Miller JL, Movva NR. pepM is an essential gene in Salmonella typhimurium. J Bacteriol. 1989;171:5215–5217. doi: 10.1128/jb.171.9.5215-5217.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chang YH. Amino-terminal protein processing in Saccharomyces cerevisiae is an essential function that requires two distinct methionine aminopeptidases. Proc Natl Acad Sci U S A. 1995;92:12357–12361. doi: 10.1073/pnas.92.26.12357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther WT, Matthews BW. Structure and function of the methionine aminopeptidases. Biochim Biophys Acta. 2000;1477:157–167. doi: 10.1016/s0167-4838(99)00271-x. [DOI] [PubMed] [Google Scholar]
- Atanassova A, Sugita M, Sugiura M, Pajpanova T, Ivanov I. Molecular cloning, expression and characterization of three distinctive genes encoding methionine aminopeptidases in cyanobacterium Synechocystis sp. strain PCC6803. Arch Microbiol. 2003;180:185–193. doi: 10.1007/s00203-003-0576-x. [DOI] [PubMed] [Google Scholar]
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Dusterhoft A, Ehrlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim SY, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guiseppi G, Guy BJ, Haga K, Haiech J, Harwood CR, Henaut A, Hilbert H, Holsappel S, Hosono S, Hullo MF, Itaya M, Jones L, Joris B, Karamata D, Kasahara Y, KlaerrBlanchard M, Klein C, Kobayashi Y, Koetter P, Koningstein G, Krogh S, Kumano M, Kurita K, Lapidus A, Lardinois S, Lauber J, Lazarevic V, Lee SM, Levine A, Liu H, Masuda S, Mauel C, Medigue C, Medina N, Mellado RP, Mizuno M, Moestl D, Nakai S, Noback M, Noone D, OReilly M, Ogawa K, Ogiwara A, Oudega B, Park SH, Parro V, Pohl TM, Portetelle D, Porwollik S, Prescott AM, Presecan E, Pujic P, Purnelle B, Rapoport G, Rey M, Reynolds S, Rieger M, Rivolta C, Rocha E, Roche B, Rose M, Sadaie Y, Sato T, Scanlan E, Schleich S, Schroeter R, Scoffone F, Sekiguchi J, Sekowska A, Seror SJ, Serror P, Shin BS, Soldo B, Sorokin A, Tacconi E, Takagi T, Takahashi H, Takemaru K, Takeuchi M, Tamakoshi A, Tanaka T, Terpstra P, Tognoni A, Tosato V, Uchiyama S, Vandenbol M, Vannier F, Vassarotti A, Viari A, Wambutt R, Wedler E, Wedler H, Weitzenegger T, Winters P, Wipat A, Yamamoto H, Yamane K, Yasumoto K, Yata K, Yoshida K, Yoshikawa HF, Zumstein E, Yoshikawa H, Danchin A. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Debarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C, Fukushima T, Haga K, Harwood CR, Hecker M, Hosoya D, Hullo MF, Kakeshita H, Karamata D, Kasahara Y, Kawamura F, Koga K, Koski P, Kuwana R, Imamura D, Ishimaru M, Ishikawa S, Ishio I, Le Coq D, Masson A, Mauel C, Meima R, Mellado RP, Moir A, Moriya S, Nagakawa E, Nanamiya H, Nakai S, Nygaard P, Ogura M, Ohanan T, O'Reilly M, O'Rourke M, Pragai Z, Pooley HM, Rapoport G, Rawlins JP, Rivas LA, Rivolta C, Sadaie A, Sadaie Y, Sarvas M, Sato T, Saxild HH, Scanlan E, Schumann W, Seegers JF, Sekiguchi J, Sekowska A, Seror SJ, Simon M, Stragier P, Studer R, Takamatsu H, Tanaka T, Takeuchi M, Thomaides HB, Vagner V, van Dijl JM, Watabe K, Wipat A, Yamamoto H, Yamamoto M, Yamamoto Y, Yamane K, Yata K, Yoshida K, Yoshikawa H, Zuber U, Ogasawara N. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Rocha E, Danchin A. How essential are non-essential genes? Mol Biol Evol, in press. [DOI] [PubMed]
- Brenner SE. Errors in genome annotation. Trends Genet. 1999;15:132–133. doi: 10.1016/S0168-9525(99)01706-0. [DOI] [PubMed] [Google Scholar]
- Gilks WR, Audit B, De Angelis D, Tsoka S, Ouzounis CA. Modeling the percolation of annotation errors in a database of protein sequences. Bioinformatics. 2002;18:1641–1649. doi: 10.1093/bioinformatics/18.12.1641. [DOI] [PubMed] [Google Scholar]
- Yang G, Kirkpatrick RB, Ho T, Zhang GF, Liang PH, Johanson KO, Casper DJ, Doyle ML, Marino JPJ, Thompson SK, Chen W, Tew DG, Meek TD. Steady-state kinetic characterization of substrates and metal-ion specificities of the full-length and N-terminally truncated recombinant human methionine aminopeptidases (type 2) Biochemistry. 2001;40:10645–10654. doi: 10.1021/bi010806r. [DOI] [PubMed] [Google Scholar]
- Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science. 1998;282:1324–1327. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- Griffith EC, Su Z, Turk BE, Chen S, Chang YH, Wu Z, Biemann K, Liu JO. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem Biol. 1997;4:461–471. doi: 10.1016/S1074-5521(97)90198-8. [DOI] [PubMed] [Google Scholar]
- Zuo S, Guo Q, Ling C, Chang YH. Evidence that two zinc fingers in the methionine aminopeptidase from Saccharomyces cerevisiae are important for normal growth. Mol Gen Genet. 1995;246:247–253. doi: 10.1007/BF00294688. [DOI] [PubMed] [Google Scholar]
- McKenzie T, Hoshino T, Tanaka T, Sueoka N. The nucleotide sequence of pUB110: some salient features in relation to replication and its regulation. Plasmid. 1986;15:93–103. doi: 10.1016/0147-619X(86)90046-6. [DOI] [PubMed] [Google Scholar]
- Lara-Gonzalez S, Dixon HB, Mendoza-Hernandez G, Altamirano MM, Calcagno ML. On the role of the N-terminal group in the allosteric function of glucosamine-6-phosphate deaminase from Escherichia coli. J Mol Biol. 2000;301:219–227. doi: 10.1006/jmbi.2000.3937. [DOI] [PubMed] [Google Scholar]
- Suh JW, Boylan SA, Oh SH, Price CW. Genetic and transcriptional organization of the Bacillus subtilis spc-alpha region. Gene. 1996;169:17–23. doi: 10.1016/0378-1119(95)00757-1. [DOI] [PubMed] [Google Scholar]
- Giglione C, Serero A, Pierre M, Boisson B, Meinnel T. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. Embo J. 2000;19:5916–5929. doi: 10.1093/emboj/19.21.5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD, Gagnon J, Walsh KA. Ovalbumin: a secreted protein without a transient hydrophobic leader sequence. Proc Natl Acad Sci U S A. 1978;75:94–98. doi: 10.1073/pnas.75.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R, Hunter T. Role of methionine in the initiation of haemoglobin synthesis. Nature. 1970;227:672–676. doi: 10.1038/227672a0. [DOI] [PubMed] [Google Scholar]
- Haas M, Beyer D, Gahlmann R, Freiberg C. YkrB is the main peptide deformylase in Bacillus subtilis, a eubacterium containing two functional peptide deformylases. Microbiology. 2001;147:1783–1791. doi: 10.1099/00221287-147-7-1783. [DOI] [PubMed] [Google Scholar]
- Goh CS, Cohen FE. Co-evolutionary analysis reveals insights into protein-protein interactions. J Mol Biol. 2002;324:177–192. doi: 10.1016/S0022-2836(02)01038-0. [DOI] [PubMed] [Google Scholar]
- Ramani AK, Marcotte EM. Exploiting the co-evolution of interacting proteins to discover interaction specificity. J Mol Biol. 2003;327:273–284. doi: 10.1016/S0022-2836(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 1951;62:293–300. doi: 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J FEGMT. Molecular Cloning: a Laboratory Manual. NY, Cold Spring Harbor; 1989. [Google Scholar]
- Kunst F, Rapoport G. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J Bacteriol. 1995;177:2403–2407. doi: 10.1128/jb.177.9.2403-2407.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PJ, Marston AL. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene. 1999;227:101–110. doi: 10.1016/S0378-1119(98)00580-0. [DOI] [PubMed] [Google Scholar]
- Muyrers JP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999;27:1555–1557. doi: 10.1093/nar/27.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stragier P, Bonamy C, Karmazyn-Campelli C. Processing of a sporulation sigma factor in Bacillus subtilis: how morphological structure could control gene expression. Cell. 1988;52:697–704. doi: 10.1016/0092-8674(88)90407-2. [DOI] [PubMed] [Google Scholar]
- Perego M. Integrational vectors for genetic manipulation in Bacillus subtilis. In: Sonenshein AL HJALR, editor. Bacillus subtilis and other Gram-positive bacteria. Washington, D.C, Amercian Society for Microbiology; 1993. pp. 615–624. [Google Scholar]
- Bacillus Subtilis Genome Database http://bacillus.genome.ad.jp
- Micado Release 6.1 http://locus.jouy.inra.fr/cgi-bin/genmic/madbase/progs/madbase.operl
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Stulke J, Martin-Verstraete I, Zagorec M, Rose M, Klier A, Rapoport G. Induction of the Bacillus subtilis ptsGHI operon by glucose is controlled by a novel antiterminator, GlcT. Mol Microbiol. 1997;25:65–78. doi: 10.1046/j.1365-2958.1997.4351797.x. [DOI] [PubMed] [Google Scholar]
- Miller JH, Shinaberger JH, Gardner PW. Experience with a new plate type dialyzer. Proc Clin Dial Transplant Forum. 1972;2:50–51. [PubMed] [Google Scholar]
- Glaser P, Sharpe ME, Raether B, Perego M, Ohlsen K, Errington J. Dynamic, mitotic-like behavior of a bacterial protein required for accurate chromosome partitioning. Genes Dev. 1997;11:1160–1168. doi: 10.1101/gad.11.9.1160. [DOI] [PubMed] [Google Scholar]
- Pikielny CW, Rosbash M. mRNA splicing efficiency in yeast and the contribution of nonconserved sequences. Cell. 1985;41:119–126. doi: 10.1016/0092-8674(85)90066-2. [DOI] [PubMed] [Google Scholar]
- Marck C. 'DNA Strider': a 'C' program for the fast analysis of DNA and protein sequences on the Apple Macintosh family of computers. Nucleic Acids Res. 1988;16:1829–1836. doi: 10.1093/nar/16.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- Higgins DG, Bleasby AJ, Fuchs R. CLUSTAL V: improved software for multiple sequence alignment. Comput Appl Biosci. 1992;8:189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]
- Subtilist http://genolist.pasteur.fr/SubtiList
- Moszer I, Jones LM, Moreira S, Fabry C, Danchin A. SubtiList: the reference database for the Bacillus subtilis genome. Nucleic Acids Res. 2002;30:62–65. doi: 10.1093/nar/30.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJ, Hofmann K, Bairoch A. The PROSITE database, its status in 2002. Nucleic Acids Res. 2002;30:235–238. doi: 10.1093/nar/30.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROSITE http://www.expasy.ch/prosite/
- Tatusov RL, Koonin EV, Lipman DJ. A Genomic Perspective on Protein Families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]