

Abstract
The bacterial community in partially purified water, which is prepared by ion exchange from tap water and is used in pharmaceutical manufacturing processes, was analyzed by denaturing gradient gel electrophoresis (DGGE). 16S ribosomal DNA fragments, including V6, -7, and -8 regions, were amplified with universal primers and analyzed by DGGE. The bacterial diversity in purified water determined by PCR-DGGE banding patterns was significantly lower than that of other aquatic environments. The bacterial populations with esterase activity sorted by flow cytometry and isolated on soybean casein digest (SCD) and R2A media were also analyzed by DGGE. The dominant bacterium in purified water possessed esterase activity but could not be detected on SCD or R2A media. DNA sequence analysis of the main bands on the DGGE gel revealed that culturable bacteria on these media were Bradyrhizobium sp., Xanthomonas sp., and Stenotrophomonas sp., while the dominant bacterium was not closely related to previously characterized bacteria. These data suggest the importance of culture-independent methods of quality control for pharmaceutical water.
Several grades of water are used during the pharmaceutical manufacturing process both as a component of pharmaceutical products and for washing equipment. The grade of water should be selected according to its role in the process.
Control of the microbiological quality of these waters is important, since microorganisms may survive and proliferate in water systems and become a source of microbial or pyrogen contamination. If contaminated water is used in the final pharmaceutical product, these microorganisms or their metabolic products may eventually cause adverse consequences.
The validation of a manufacturing process is particularly important in order to assure the quality of pharmaceutical products. Validating a manufacturing support system, including water supplies, reduces dependence on intensive in-process testing and finished product testing. Water constitutes a key material for the production of medical supplies. Water must therefore meet strict quality standards, and that quality should be maintained until use.
It is necessary to provide sufficient information to control the microbiological quality of the water produced, rapidly and accurately. Any monitoring methods should be capable of elucidating the numbers and types of organisms that have been deemed significant relative to system control and product impact for each individual system. Identifying the bacteria in water is important in instances where specific waterborne microorganisms may be detrimental and potentially harmful to the products or processes in which the water is used. Microorganism information such as this may also be useful when identifying the source of microbial contamination in a product or process. Generally, some type of microbial isolate characterization should be a required element of water system monitoring. Identification based solely on culture yields valuable information. However, the formation of colonies by microorganisms is dependent on growth conditions, such as nutrient media and incubation temperature. Additionally, it is well recognized that most aquatic microorganisms cannot be cultivated under conventional conditions. These organisms enter an altered physiologic state termed the viable but nonculturable (VNC) state (2, 15). This is particularly important to remember when assessing pathogenic bacteria, since they may be undetectable by standard cultivation methods but may remain viable.
We have already reported the useful techniques for routine enumeration of physiologically active bacteria, including those in a VNC state, in purified water used in pharmaceutical manufacturing processes within one working day (8). These techniques make it possible to control pharmaceutical water within a time frame that permits adjustment. For better control of water supplies, identifying the bacteria in pharmaceutical water must be determined, as described above.
Recently, several molecular techniques have been developed in order to study natural samples. These molecular techniques identify microorganisms without isolation and reveal the enormous extent of microbial diversity. Specifically, denaturing gradient gel electrophoresis (DGGE) has emerged as a powerful tool (1, 3, 4, 7, 10, 11, 14). By DGGE of PCR-amplified fragments coding for 16S rRNA, DNA fragments of the same length but different base pair sequences can be separated. This method has recently been introduced into molecular microbial ecology to determine the genetic diversity of natural microbial communities and to identify the phylogenetic affiliation of community members.
DGGE was used in this study to analyze the bacteria, including those in a VNC state, in purified water (one type of pharmaceutical-grade water, used during many manufacturing processes) as the next stage of microbiological monitoring.
MATERIALS AND METHODS
Water samples.
Purified water samples prepared by ion exchange from tap water were collected in sterile bottles from the same point in the flow of the pharmaceutical water supply system. The sample water temperature was ca. 24°C. The purified water sample was stored on ice and was analyzed within an hour except for cell sorting of bacteria. Collection of bacteria by cell sorting was completed within a few hours.
Culture method.
Colony-forming bacteria were detected by the filtration method. Bacteria in purified water were trapped on filters (pore size, 0.2 μm) and were incubated at approximately 30°C for 7 days on soybean casein digest (SCD) agar (SCDA; 15 g of peptone for casein, 5 g of peptone for soybean, 5 g of sodium chloride, and 15 g of agar per 1,000 ml of laboratory-quality water), R2A (0.5 g of yeast extract, 0.5 g of peptones [pancreatic digest of casein, 50%; and peptic digest of animal tissue, 50%], 0.5 g of acid hydrolysate of casein, 0.5 g of dextrose, 0.5 g of soluble starch, 0.3 g of dipotassium phosphate, 0.024 g of magnesium sulfate [anhydrous], 0.3 g of sodium pyruvate, and 15 g of agar per 1,000 ml of laboratory-quality water), and the filter paper was soaked with SCD broth (SCDB; 17 g of peptone for casein, 3 g of peptone for soybean, 5 g of sodium chloride, 2.5 g of potassium hydrogen phosphate, and 2.5 g of glucose per 1,000 ml of laboratory-quality water). SCD medium was purchased from Eiken Chemical Co., Ltd. (Tokyo, Japan), and R2A medium was purchased from Difco.
DNA extraction for 16S ribosomal DNA (rDNA) analysis.
DNA was extracted from bacterial cells in purified water adapted as follows. Bacterial cells in purified water were vacuum filtered onto polycarbonate white filters (pore size, 0.2 μm; ADVANTEC, Tokyo, Japan). These filters were placed into a sterilized tube with 500 μl of sterile DNA-free water. The suspensions were mixed thoroughly and were subsequently frozen in liquid nitrogen and were then thawed at room temperature three times (total) in succession. The suspensions were used for PCR amplification. The efficiency of lysis was determined by direct microscopic count with SYBR green I staining.
DNA was also extracted from bacterial colonies on each culture medium adapted as follows: colonies on each medium were picked up and suspended in 500 μl of DNA-free water and were mixed thoroughly. Subsequently, three cycles of freezing in liquid nitrogen and thawing at room temperature were conducted to release DNA from the microbial cells.
Primers and PCR amplification.
16S rDNA fragments were amplified with primers GC-clamp-EUB f933 and EUB r1387 (Table 1), which are specific for universally conserved bacterial 16S rDNA sequences. For DGGE analysis of the PCR product, a 40-bp GC-rich sequence (GC-clamp) was attached to the 5" end of primer GC-clamp-EUB f933. Amplification PCRs were performed with the reagents supplied with Ampli Taq Gold (Applied Biosystems). The PCR mixture, containing 0.25 μl of Ampli Taq Gold, 20 pmol of each primer, 6 μl of 25 mM MgCl2 solution, 5 μl of a 2 mM concentration of each deoxyribonucleoside triphosphate, 5 μl of 10× PCR buffer, and 0.5 μl of 2.5-mg/ml 8-methoxypsoralen (Sigma), was made up to 40 μl with DNA-free water. A DNA suspension was added last in a 10-μl volume after irradiation of the PCR mixture with UV light. 8-Methoxypsoralen was dissolved in dimethyl sulfoxide. The tubes containing the PCR mixture were irradiated from above at a distance of 1 cm with long-wave (365 nm) UV at room temperature (9). A hot-start PCR was performed at 95°C for 10 min, and touchdown PCR was performed as follows: the annealing temperature was initially set at 66°C and was then decreased by 0.5°C every cycle until it was 56°C. Twenty additional cycles were carried out at 56°C. Denaturing was carried out at 94°C for 1 min. Primer annealing was performed using the scheme described above for 1 min, and primer extension was performed at 72°C for 3 min. The final extension step was 7 min at 72°C.
TABLE 1.
Primer sequences and positions
| Primer | Position | Target | Sequence | Reference |
|---|---|---|---|---|
| EUB f933 | 933-954 | Bacteria, regions V6-V8 | 5"-GC-clamp-GCACAAGCGGTGGAGCATGTGG-3" | 7 |
| EUB r1387 | 1387-1368 | Bacteria, regions V6-V8 | 5"-GCCCGGGAACGTATTCACCG-3" | 7 |
| GC-clamp | 5"-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG |
DGGE analysis.
PCR products were loaded onto a 6.5% (wt/vol) polyacrylamide gel in 1× buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA at pH 8.0). The 6.5% (wt/vol) polyacrylamide gels (acrylamide/bisacrylamide ratio, 37.5:1) were made with denaturing gradients ranging from 40 to 60% for 16S rDNA fragments. Denaturant (100%) contained 7 M urea and 40% formamide. The electrophoresis was run at 60°C, for 10 min at 20 V, and subsequently for 12 h at 100 V. After electrophoresis, the gels were stained for 20 min with SYBR Gold nucleic acid gel stain (Molecular Probes) as specified by the manufacturer. DGGE gels were scanned with FluorImager (Molecular Dynamics) using a 488-nm argon laser, and digital images were detected by Image QuaNT (version 4-2-J) to generate a densitometric profile.
Sequencing of DGGE fragments.
Bands in the DGGE gel of 16S rRNA fragments were excised with a razor blade. The DNA was retrieved from the acrylamide block with Centriluter (Amicon) at 150 V for 3 h. The DNA was reamplified with EUB f933 and EUB r1387. PCR products were purified with a Wizard DNA Clean-up system (Promega). The clones were PCR amplified again with primers GC-clamp-EUB f933 and EUB r1387 and were analyzed by DGGE. The clones that produced a DGGE band at the same position as the excised band were selected for subsequent sequence analysis. The sequence analysis of selected clones was performed on an ABI 310 automated DNA sequencer (Perkin-Elmer) with EUB 1387r, EUB 1114f, and EUB 1099r primers. The dye terminator cycle-sequencing reactions were performed according to the manufacturer's guidelines.
6CFDA staining and cell sorting of bacteria.
The physiologic activity of dominant bacteria in purified water was determined by cell sorting. 6-Carboxyfluorescein diacetate (6CFDA) staining was carried out as described by Tanaka et al. (17). Approximately 50 liters of purified water was concentrated 1,000-fold with an ultrafiltration system (Filtron; Pall Co., Ltd). The concentrated sample was mixed with a half-volume of 6CFDA buffer (0.3 M phosphate buffer, [pH 8.5], 15% [wt/vol] NaCl, and 1.5 mM EDTA). 6CFDA stock (1% [wt/vol] in acetone; Sigma) was applied (final concentration: 10 μg/ml), and bacterial cells were stained for 5 min at room temperature under dark conditions. The samples were analyzed immediately by flow cytometry on an Epics Elite flow cytometer (Beckman Coulter, Fullerton, Calif.). Side scatter was measured in order to discriminate bacterial cells from other particles. The green fluorescence of 6-carboxyfluorescein was determined using the PMT2 channel (515 to 525 nm). Sorting was carried out under the following conditions: frequency of droplet formation, 19 kHz; deflection amplitude, 80%; number of drops sorted per event, 2; and drop delay, 26 drops. Areas for sorting of cells with and without esterase activity were determined in each sample.
Nucleotide sequence accession number.
The sequence obtained in this study has been deposited in the GenBank database under accession no. AF412384.
RESULTS
DGGE analysis of bacterial community structure in purified water.
The efficiencies of both freeze-thaw and phenol-chloroform methods were first compared in the extraction of bacterial DNA from purified water used in pharmaceutical manufacturing processes. A previous report suggested that the freeze-thaw method resulted in more bands than did chemical disruption during DGGE analysis of a 16S rDNA fragment (5, 7). Our results for 16S rDNA analysis indicated that a difference between freeze-thaw and phenol-chloroform methods in DGGE profiles could not be detected (data not shown). Therefore, in this study, we relied on the freeze-thaw method for DGGE analysis of PCR-amplified 16S rDNA fragments on account of its convenience.
Contamination of exogenous bacterial DNA in PCR reagents represents a serious problem for amplification of eubacterial 16S rDNA sequences with broad-range eubacterial 16S rDNA primers. Contamination of eubacterial DNA in commercial preparations of Taq DNA polymerase has been reported earlier (6, 19). We used 8-methoxypsoralen in combination with long-wave UV treatment for 6 min to eliminate contaminating DNA in PCR reagents without significantly affecting the activity of Taq DNA polymerase (9).
Purified water samples were collected from February to April 2000. There were no significant differences in the total bacterial number among six purified water samples (1.5 × 103 to 5.5 × 103 cells/ml). Figure 1 shows the DGGE profiles of 16S rDNA fragments targeting the domain bacteria obtained from purified water in pharmaceutical manufacturing processes. They revealed some distinguishable bands per lane, reflecting the structure of the microbial community. The overall 6-day pattern was similarly indicative of minimal diversity in the bacterial community in purified water. A single band indicated by the signpost in Fig. 1 appeared to be the most intense band in all cases.
FIG. 1.
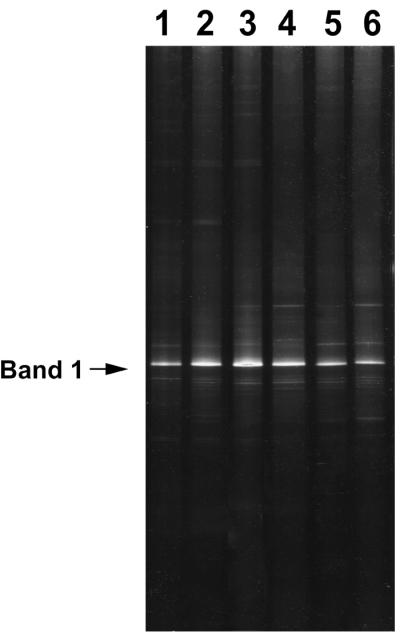
DGGE analysis of 16S rDNA fragments obtained from purified water samples. Lanes: 1, 17 February 2000; 2, 24 February 2000; 3, 25 February 2000; 4, 2 March 2000; 5, 9 March 2000; and 6, 13 April 2000.
Genetic diversity of different cell fractions in purified water.
The genetic diversity of bacteria from the PCR-DGGE analysis was investigated, including total, esterase-active, and culturable bacteria on selective media to determine the diversity of bacteria possessing physiologic activity (Fig. 2). Esterase-active cells were sorted by flow cytometry on the basis of the intensity of 6CFDA fluorescence. All colonies formed on R2A, SCDA, and SCDB were retrieved in unison and were suspended into one tube each. Each suspension was analyzed by PCR-DGGE. Several bands corresponding to esterase-active cells, including the band originating from the dominant bacterium in the DGGE profile of the whole community, were present (Fig. 2, lanes 1 and 2; band 1). These results show that the dominant bacteria in purified water possess esterase activity. Several bands were observed, but the band originating from the dominant bacterium (Fig. 2, band 1), found universally in the purified water used in this study, could not be detected on culture media.
FIG. 2.

Bacterial diversity in both total and physiologically active populations in purified water. Lanes: 1, total bacteria; 2, esterase-active bacteria; 3, mixture of DNA originating from randomly collected colonies on R2A media; 4, mixture of DNA originating from randomly collected colonies on SCDA media; and 5, mixture of DNA originating from randomly collected colonies on SCDB media.
Community analysis of culturable bacteria in purified water.
We analyzed approximately 50 colonies isolated on each of three different kinds of media (R2A, SCDA, and SCDB). Isolated colonies were suspended, and the DNA was extracted by the freeze-thaw method. PCR-amplified 16S rDNA fragments were analyzed by DGGE. Figure 3a shows the DGGE profile of 16S rDNA fragments targeting the domain bacteria obtained from R2A, SCDA, and SCDB, respectively. The DGGE pattern of isolated bacteria from R2A media (lane 2) shows eight separate bands. Of the total isolate DNA, 33% appeared as band 2; 19% of the total isolate DNA appeared as band 3; and 15% of the total isolate DNA appeared as band 4. The DGGE pattern of isolated bacteria from SCDA media (lane 3) showed four separate bands. Of the total isolate DNA, 78% appeared as band 5. The DGGE pattern of isolated bacteria from SCDB media (lane 4) showed five separate bands, and 85% of the total isolate DNA appeared as band 6.
FIG. 3.

Identification of bands originating from bacteria cultured on various media from purified water. Lanes: 1, total bacteria; 2, mixture of DNA originating from randomly collected colonies on R2A media; 3, mixture of DNA originating from randomly collected colonies on SCDA media; and 4, mixture of DNA originating from randomly collected colonies on SCDB media.
Sequence analysis of culturable bacteria.
The partial 16S rDNA fragments of five isolate bacteria (numbered DGGE bands in Fig. 3a) were sequenced. Comparison of 16S rDNA sequences with sequences available in GenBank databases revealed high similarity values for these bacterial isolates. Figure 3b shows related organisms, their detection rate on each medium for the bacteria corresponding to the separated DGGE bands, and their similarities. Analysis revealed that DGGE bands 2 and 4 are closely related to the genus Bradyrhizobium, whereas band 3 is related to the genus Xanthomonas. Sequences obtained from DGGE bands 5 and 6 are related to those of species from the genus Xanthomonas and the genus Stenotrophomonas.
Phylogenetic affiliation of dominant bacteria.
Several fragments detected in all purified water samples (bands 1 in Fig. 1 to 3) were excised and reamplified by PCR. Partial 16S rDNA sequences of approximately 450 bases were obtained from the DNA originating from each dominant band. All of the partial 16S rDNA sequences were the same. These sequence data confirm that the bacteria appearing in bands 1 in Fig. 1 to 3 were the same. It was verified that the main band shown as band 1 originated from the dominant bacterium in purified water. This sequence was compared with sequences available in GenBank, and the phylogenetic affiliation of the sequences was further analyzed with the sequences of bands 2 to 6 in Fig. 3 by Clustal W 1.74 (Fig. 4). The phylogenetic tree shown in Fig. 4 indicates that the organism represented by the main band (Fig. 4, band 1) belonged to the Alphaproteobacteria and was not closely related to previously characterized bacteria. The bacterial species most closely related was an environmental clone.
FIG. 4.

Phylogenetic affiliations within the domain bacteria in purified water as revealed by comparative analysis of 16S rRNA sequences from DGGE bands and those stored in public nucleotide databases. Sequences determined in this study are shown in boldface (for location of bands, compare with Fig. 3). The scale bar corresponds to 0.1 substitution per nucleotide position.
DISCUSSION
The primary goal of successful microbiological quality control of pharmaceutical water is to gather sufficient information about bacterial number, species, and physiologic activity. Conventional culture methods have been used for routine monitoring of pharmaceutical water in manufacturing processes for over 50 years. However, it has been found that the conventional culture method has several disadvantages for monitoring. The number and species of bacteria detectable by culture are affected by choice of media, incubation temperature, and culture period. In the purified water used in this study, differences in the number and species of culturable bacteria on each media were observed. R2A media (13), which is composed of low levels of organic substances and various minerals, produced the highest number of colonies among the three media. DNA sequences of colony-forming bacterial populations obtained from DGGE analysis were compared with sequences available in GenBank and identified. It has been shown previously that relatively short sequences, such as those obtained from DGGE analysis, are sufficient for an approximate phylogenetic identification (11, 16, 18). As shown in Fig. 3, bacterial isolates from R2A media were mainly Bradyrhizobium sp. belonging to Alphaproteobacteria; bacterial isolates from SCDA media were mainly Xanthomonas sp.; and bacterial isolates from SCDB media were mainly Stenotrophomonas sp. belonging to Gammaproteobacteria. This may demonstrate that Alphaproteobacteria grow more easily than Gammaproteobacteria on nutrient-poor media such as R2A, with the reverse true on nutrient-rich media such as SCD. This selectivity of culturing methods means that none of the three culture media has advantages in terms of isolation of different species; thus, cell counts are underestimated when a single culture technique is used. The culture conditions used for routine monitoring of pharmaceutical water for manufacturing processes should be chosen carefully.
Recent studies describing the persistence of bacteria in aquatic environments have demonstrated that many of these organisms enter VNC states. We have already reported the presence of VNC bacteria in purified water (8). In the present study, several molecular techniques helped to identify microorganisms without cultivation and revealed the enormous extent of microbiological diversity.
The diversity of bacteria in purified water was determined by PCR-DGGE, and identification of them was performed by sequence analysis. DGGE patterns of direct DNA extracts showed that the diversity of the bacterial community in purified water in this study was quite lower than in the aquatic environment (3, 7). Each partial sequence of the dominant bacterium in several samples of purified water was confirmed to be the same strain. This is probably a direct consequence of the confinement and handling of this particular water supply system.
Moreover, several kinds of bands corresponding to esterase-active cells were present in the DGGE profile of the whole community. These results showed that some bacteria, including the dominant bacterium in purified water, possess physiologic activity. At the same time, the DGGE profile originating from colonies cultured on three kinds of media showed that the dominant bacterium in purified water could not form colonies on these media, while these media are commonly used to monitor pharmaceutical water supplies. Therefore, the dominant bacterium is VNC bacteria or is unsuitable for culture conditions.
The phylogenetic tree shown in Fig. 4 indicated that the dominant bacterium belonged to Alphaproteobacteria and formed a highly related group with no close relationship to any characterized bacteria described above.
It has been reported that most aquatic bacteria exist in a starved state but retain metabolic activity. Bacteria with physiologic activity can have major effects on pharmaceutical products from bacterial endotoxins, pathogenicity, and metabolic product (12). In addition, bacterial enzymes can alter the active ingredients of pharmaceuticals. Therefore, water supply systems used in pharmaceutical manufacturing processes should be diligently and accurately monitored for all bacteria, including those in VNC states. The PCR-DGGE approach will provide early warning of an impending problem and allow corrective action.
Acknowledgments
We thank Noriaki Shimokawa for generous support and fruitful discussion.
REFERENCES
- 1.Bernard, L., C. Courties, C. Duperray, H. Schäfer, G. Muyzer, and P. Lebaron. 2001. A new approach to determine the genetic diversity of viable and active bacteria in aquatic ecosystems. Cytometry 43:314-321. [PubMed] [Google Scholar]
- 2.Byrd, J. J., H.-S. Xu, and R. R. Colwell. 1991. Viable but nonculturable bacteria in drinking water. Appl. Environ. Microbiol. 57:875-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Casamayor, E. O., H. Schäfer, L. Bañeras, C. Pedrós-Alió, and G. Muyzer. 2000. Identification of and spatio-temporal differences between microbial assemblages from two neighboring sulfurous lakes: comparison by microscopy and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 66:499-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferris, M. J., G. Muyzer, and D. M. Ward. 1996. Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl. Environ. Microbiol. 62:340-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillan, D. C., A. G. C. L. Speksnijder, G. Zwart, and C. De Ridder. 1998. Genetic diversity of the biofilm coverning Montacuta ferruginosa (Mollusca, Bivalvia) as evaluated by denaturing gradient gel electrophoresis analysis and cloning of PCR-amplified gene fragments coding for 16S rRNA. Appl. Environ. Microbiol. 64:3464-3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes, M. S., L.-A. Beck, and R. A. Skuce. 1994. Identification and elimination of DNA sequences in Taq DNA polymerase. J. Clin. Microbiol. 32:2007-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwamoto, T., K. Tani, K. Nakamura, Y. Suzuki, M. Kitagawa, M. Eguchi, and M. Nasu. 2000. Monitoring impact of in situ biostimulation treatment on groundwater bacterial community by DGGE. FEMS Microbiol. Ecol. 32:129-141. [DOI] [PubMed] [Google Scholar]
- 8.Kawai, M., N. Yamaguchi, and M. Nasu. 1999. Rapid enumeration of physiologically active bacteria in purified water used in the pharmaceutical manufacturing process. J. Appl. Microbiol. 86:496-504. [DOI] [PubMed] [Google Scholar]
- 9.Meier, A., D. H. Persing, M. Finken, and E. C. Böttger. 1993. Elimination of contaminating DNA within polymerase chain reaction reagents: implications for a general approach to detection of uncultured pathogens. J. Clin. Microbiol. 31:646-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muyzer, G., E. C. de Waal, and A. G. Uitterlinden. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 59:695-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nielsen, A. T., W.-T. Liu, C. Filipe, L. Grady, Jr., S. Molin, and D. A. Stahl. 1999. Identification of a novel group of bacteria in sludge from a deteriorated biological phosphorus removal reactor. Appl. Environ. Microbiol. 65:1251-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver, J. D. 1995. The viable but non-culturable state in the human pathogen Vibrio vulnificus. FEMS Microbiol. Lett. 133:203-208. [DOI] [PubMed] [Google Scholar]
- 13.Reasoner, D. J., and E. E. Geldreich. 1985. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ. Microbiol. 49:1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rölleke, S., G. Muyzer, C. Wawer, G. Wanner, and W. Lubitz. 1996. Identification of bacteria in a biodegraded wall painting by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl. Environ. Microbiol. 62:2059-2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roszak, D. B., and R. R. Colwell. 1987. Survival strategies of bacteria in the natural environment. Microbiol. Rev. 51:365-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmidt, T. M., E. F. DeLong, and N. R. Pace. 1991. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J. Bacteriol. 173:4371-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanaka, Y., N. Yamaguchi, and M. Nasu. 2000. Viability of Escherichia coli O157:H7 in natural river water determined by the use of flow cytometry. J. Appl. Microbiol. 88:228-236. [DOI] [PubMed] [Google Scholar]
- 18.Ward, D. M., R. Weller, and M. M. Bateson. 1990. 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 345:63-65. [DOI] [PubMed] [Google Scholar]
- 19.Wilson, I. G. 1997. Inhibition and facilitation of nucleic acid amplification. Appl. Environ. Microbiol. 63:3741-3751. [DOI] [PMC free article] [PubMed] [Google Scholar]