

Abstract
The effects of probenecid and cimetidine on the pharmacokinetics of valaciclovir and its metabolite acyclovir have been investigated. Twelve healthy male volunteers participated in this open single-dose study with a four-way-crossover randomized and balanced design. At the first of four administrations, volunteers in four groups received 1 g of valaciclovir alone, valaciclovir with 1 g of probenecid, valaciclovir with 800 mg of cimetidine, or valaciclovir with a combination of probenecid and cimetidine. At three subsequent administrations, drug regimens were alternated among groups so that each group received each regimen. Probenecid and cimetidine increased the mean maximum concentrations in serum (Cmax) of valaciclovir by 23 and 53% and the areas under the concentration-time curves (AUC) for valaciclovir by 22 and 73%, respectively; probenecid and cimetidine also increased the mean acyclovir Cmax by 22 and 8% and its AUC by 48 and 27%, respectively. The combination had a greater effect than either drug alone. Their effects may be due to competitive inhibition of membrane transport of valaciclovir and acyclovir in the liver and kidney. Neither cimetidine nor probenecid affected the absorption of valaciclovir. Both probe drugs reduced the rate of valaciclovir metabolism but not its extent. These pharmacokinetic modifications did not affect the tolerability of valaciclovir.
Valaciclovir (Valtrex) is the l-valine ester of acyclovir and is extensively converted to the antiherpetic compound acyclovir by hepatic first-pass metabolism following oral administration. Its bioavailability as valaciclovir is three- to fivefold greater than acyclovir's oral bioavailability (13). The active metabolite acyclovir is excreted 85% unchanged in the urine, with the rate of renal clearance (CLR) being three times that of the glomerular filtration rate, indicating that renal excretion has a significant tubular-secretion component. Valaciclovir and acyclovir, which have anionic and cationic forms in plasma, are secreted by organic anion and cation transporters. Acyclovir CLR is reduced by probenecid (6), which was thought to be due to inhibition of the renal tubular secretion of acyclovir by the anionic pathway.
We investigated the effects of probenecid and cimetidine on valaciclovir pharmacokinetics, as these drugs have been reported to inhibit the metabolism of some compounds and the active membrane transport of a number of organic anions and cations (4, 6, 8, 9, 12). Additionally, we examined the effects of probenecid and cimetidine on the pharmacokinetics of acyclovir. The drug interactions at the renal level were modeled as a function of the concentrations of the interaction drugs in plasma in order to characterize more precisely their mechanisms and potential consequences.
MATERIALS AND METHODS
Study design.
We employed an open, randomized, balanced, crossover study design with four drug treatments separated by intervals of at least 1 week. Twelve healthy male volunteers (age range, 22 to 43 years; weight range, 54 to 111 kg) participated in the study. They gave written informed consent for participation before enrollment. The fasting subjects were given 1 g of valaciclovir (two 500-mg tablets) with either (i) 1 g of probenecid (Benemid, two 500-mg tablets; Merck Sharp and Dome) 2 h before valaciclovir dosing (probenecid), (ii) 800 mg of cimetidine (Tagamet, 800-mg tablets; Smith Kline and French Laboratory) 10 and 1 h before valaciclovir dosing (cimetidine), (iii) a combination of the treatments noted in sections i and ii (combination), or (iv) no concomitant treatment (control).
Cimetidine was administered 10 h prior to valaciclovir dosing to increase gastric pH to examine possible effects of altered gastric pH on valaciclovir absorption. Cimetidine (second dose) and probenecid were administered 1 and 2 h prior to valaciclovir dosing, respectively, which corresponded to their peak activities as inhibitors of renal secretion in previous studies (2, 7). Tablets were administered with 200 ml of squash. On study occasions, volunteers received a fixed fluid regimen allowing frequent urine collections. They also received standard low-protein meals to minimize the effects of protein loading on renal function.
Blood sampling and urine collection.
Venous blood samples were taken just before valaciclovir dosing and then at 15, 30, 45, 60, 75, and 90 min and at 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 16, and 24 h after administration. Samples taken up to 3 h after valaciclovir dosing were assayed for valaciclovir. All samples were assayed for acyclovir. Probenecid and cimetidine were assayed in samples taken just before valaciclovir administration and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, and 24 h after valaciclovir administration. Urine was collected before valaciclovir administration and then in 12 fractions over 24 h following acyclovir administration (0 to 0.5, 0.5 to 1, 1 to 1.5, 1.5 to 2, 2 to 3, 3 to 4, 4 to 6, 6 to 8, 8 to 10, 10 to 12, 12 to 16, and 16 to 24 h). For each urine collection, an aliquot acidified with trichloroacetic acid was kept at −20°C until analysis. All urine collections were assayed for acyclovir.
Valaciclovir assay.
Valaciclovir was assayed in plasma by liquid chromatography as specified by Weller et al. (13).
Acyclovir assay.
Acyclovir concentrations in plasma and urine were determined by a specific competitive radioimmunoassay. The limits of quantification were 0.04 μM in plasma and 0.80 μM in urine. The quantifiable range was 0.04 to 0.5 μM in plasma, with inter- and intra-assay precision ranging from negligible to 10% and accuracy (bias) ranging from −3.5 to +12.3%. In urine, the quantifiable range was 0.80 to 9.80 μM, with inter- and intra-assay precision ranging from negligible to 10.5% and accuracy ranging from −5.4 to +5.8%.
Cimetidine assay.
Cimetidine concentrations in plasma were determined by liquid chromatography and UV detection with SKF92374 (cimetidine structural analogue) as an internal standard (IS). Cimetidine and the IS were extracted from alkaline medium with octanol and back extracted by using acetonitrile in dilute acid medium. The supernatants were analyzed on a Lichrosorb Si60 column (250 by 4.6 mm; granulometry, 5 μm) eluted with a mobile phase (acetonitrile-methanol-water-20% ammonia in a ratio of 425:60:12:3) at a flow rate of 1.5 ml/min. The UV absorbance of the column effluent was monitored at a wavelength of 226 nm. The retention times were approximately 6 min for cimetidine and 8 min for the IS. The quantifiable range was 40 to 5,000 mg/liter. Mean recovery over the calibration range was 84% for cimetidine and 77% for the IS. The intra- and interassay precision ranged from 2.09 to 13.51%, and the accuracy (bias) ranged from −17 to +7.5%.
Probenecid assay.
Probenecid concentrations in plasma were determined by liquid chromatography and UV detection by using indoprofene as an IS. Briefly, probenecid and the IS were extracted by diethyl ether in acid medium. The ether phase was evaporated to dryness, and the extract was reconstituted in 50 mM ammonium acetate buffer (pH 5.0) containing 30% acetonitrile. The extracts were analyzed on a Chromasil C18 column (100 by 4.6 mm, 5 μm) eluted with a mobile phase (500 mM acetate buffer [pH 5.0]-water-acetonitrile in a ratio of 10:65:25) at a flow rate of 2 ml/min. The UV absorbance of the column effluent was monitored at a wavelength of 250 nm. The retention times were approximately 6 min for probenecid and 4.5 min for the IS. The quantifiable range was 0.5 to 50 mg/liter. The mean recovery was 81% for probenecid and 97% for the IS. The intra- and interassay precision ranged from 2.4 to 9.5%, and the accuracy (bias) ranged from −7.9 to +0.5%.
Pharmacokinetics.
Pharmacokinetic parameters were determined for all drugs using noncompartmental methods. The AUC were calculated by use of the linear trapezoidal rule, and extrapolation to infinity was obtained by adding the ratio of the last measured concentration (Ct) to the slope of the elimination phase (λ) on a semilogarithmic scale. Cmax and times to maximum concentration of drug in serum (Tmax) were taken directly from the concentrations in plasma. Elimination half-lives (t1/2) were calculated as ln2/λ. The elimination rate constant for valaciclovir could not be reliably determined. Hence, only Cmax, Tmax, and AUC from 0 to 3 h (AUC0-3) were reported.
Acyclovir's CLR from time 1 to time 2 [CLR(t1-t2)], its total CLR, and the percentage of the dose recovered as acyclovir in the urine were also calculated. Acyclovir's CLR(t1-t2)was calculated as the ratio of the amount of acyclovir excreted in urine between t1 and t2 to the corresponding acyclovir AUC. The CLR of acyclovir was analyzed for each urine collection as a function of the mean concentrations of probenecid and cimetidine in plasma observed during the urine collection period.
AUC for cimetidine and probenecid were determined only for volunteers administered valaciclovir for up to 24 h.
Statistical analysis.
Due to the log-normal nature of the data, AUC and Cmax were log transformed prior to analysis. According to the crossover design and as specified in the protocol, pharmacokinetics parameters were subjected to analysis of variance, taking into account sources of variation due to subject, period, and concomitant treatment. The effect of carryover from each drug was also examined, but if not significant, it was removed from the model.
Geometric means were calculated for each probenecid-cimetidine combination. The point estimates and 95 and 90% confidence intervals produced were dependent on the presence or absence of an interaction between the effects of the two probe drugs (cimetidine and probenecid).
If the effects of cimetidine and probenecid on a pharmacokinetic variable were not independent, i.e., if an interaction between the effects of probe drugs was significant (5% F-ratio test), then point estimates and 95 and 90% confidence intervals were calculated for the ratios of the values for valaciclovir administered with each additional treatment to the values for valaciclovir administered alone (the control).
If the effects of the two probe drugs were independent, i.e., if an interaction between the effects of the probe drugs was not significant (5% F-ratio test), then point estimates and 95 and 90% confidence intervals were calculated for the ratios of the overall effects of the probe drugs individually (comparing the results for the two treatments in which a drug was present to the results for the two treatments in which that drug was absent).
Further results presenting 90% confidence intervals were calculated in order to comply with the latest regulatory requirements (Note for Guidance on the Investigation of Drug Interactions, Committee for Proprietary Medicinal Products, European Commission document CPMP/EWP/560/95).
For Tmax, the medians were calculated for each treatment limb. The interaction of the two drugs was examined. Differences in Tmax medians were compared using a Wilcoxon signed rank test. Similar to results of the analysis discussed above for AUC, the 95% confidence intervals produced were dependent on the presence or absence of a significant interaction between the effects of the probe drugs.
Analyses of variance were performed on the t1/2 and AUC of each probe drug to check whether the presence of the other probe drug had an effect on its pharmacokinetics.
Modeling of the interactions.
The pharmacokinetic model of the interactions of cimetidine and probenecid on acyclovir elimination was built as follows. By definition, the urinary elimination rate of acyclovir, dU/dt, is given by the following equation (10):
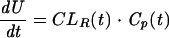 |
(1) |
where CLR(t) is the CLR of acyclovir at time t and Cp(t) is the concentration of acyclovir at time t in plasma. Since acyclovir is assumed to be eliminated by glomerular filtration, tubular secretion by the organic cation transporter, and tubular secretion by the organic anion transporter [CLGF, CLT1, and CLT2, respectively], we have
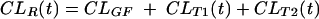 |
(2) |
In this equation, CLGF is assumed to be constant over time but CLT1 and CLT2 may change as the respective concentrations of acyclovir, probenecid, and cimetidine vary. Basically, the aim of the model was to describe CLR(t), since the acyclovir elimination rate during each time interval and the concentration-versus-time profile of each drug were known. When acyclovir alone is present (control treatment), each tubular-secretion process can be described according to a Michaelis-Menten model (10) as follows:
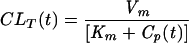 |
(3) |
where Vm is the maximal velocity of transport and Km is the concentration at which half the maximal velocity is reached. Cimetidine and probenecid were assumed to be competitive inhibitors. When a competitive inhibitor is present, the CLT is decreased according to the following equation (11):
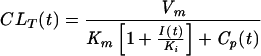 |
(4) |
where I(t) is the concentration of the inhibitor in plasma at time t and Ki is its inhibition constant. In the control group, the CLR of acyclovir was nearly constant over time. Hence, Cp(t) was negligible compared to the Km of each transporter and even more negligible in CLT expressions when probenecid and/or cimetidine were coadministered. This finding allowed a simplification of the CLT formula as follows:
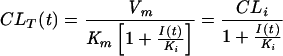 |
(5) |
where CLi is the intrinsic clearance of acyclovir. The elimination rate of acyclovir from the urine of each subject was finally described as follows. In the control administration (valaciclovir alone),
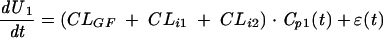 |
(6) |
In the administration of valaciclovir plus cimetidine,
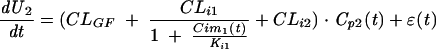 |
(7) |
Similar equations were used for the other treatments. In these equations, Cp1, C2, Cp3, and Cp4 are the concentrations of acyclovir in plasma after administrations 1 to 4, respectively; CLi1 and CLi2 are the CLis of acyclovir for the cationic and anionic transports, respectively; Ki1 and Ki2 are the Kis of cimetidine and probenecid, respectively; and Cim1(t) and Cim2(t) are the concentrations of cimetidine in plasma after its first and second administrations, respectively. The concentrations of probenecid in plasma could be calculated by the same equation. The error term ɛ(t) was assumed to be random, with normal distribution, a zero mean, and the standard deviation (ς) being linearly related to the elimination rate. Provided that the concentration-versus-time profile of each drug is described adequately, the five parameters for estimation by nonlinear regression for each individual are CLGF, CLi1, CLi2, Ki1, and Ki2. The model is identifiable if the four equations are fitted simultaneously to the data gained on the four occasions of drug administration.
A model of the pharmacokinetic profile of each drug is required. The acyclovir profile was best described by use of a bicompartmental model with a zero order absorption rate and a lag time before onset of absorption. Cimetidine and probenecid kinetics were described by a one-compartment model with first-order absorption and elimination rate and a lag time before absorption. The analysis was carried out in two steps. In the first step, all the individual parameters of these pharmacokinetic models were estimated by nonlinear regression by weighted least-squares analysis. In the second step, equations 6 and 7 were fitted to the data (acyclovir renal elimination rates versus time) from each individual by weighted least-squares analysis to determine CLGF, CLi1, CLi2, Ki1, and Ki2, with the values of the other kinetic parameters being fixed to the values determined in the first step. All these estimations were made by using ADAPT II software (ADAPT II users guide, Biomedical Simulation Resource, Los Angeles, Calif.).
RESULTS
The study medications were well tolerated over the whole study. No evidence was found of any carryover between drug administrations for either valaciclovir or acyclovir parameters.
Valaciclovir pharmacokinetics.
Plasma valaciclovir concentrations were usually not detectable by 3 h postdose except in four subject profiles. A summary of valaciclovir pharmacokinetic parameters for each treatment is presented in Table 1. Point estimates and 95 and 90% confidence intervals for changes in pharmacokinetic parameters relative to those of the control group are presented in Table 2. Mean concentrations of valaciclovir in plasma after each treatment are shown in Fig. 1.Cimetidine had a greater effect than probenecid on valaciclovir's Cmax and AUC0-3. With cimetidine and probenecid combined, valaciclovir's Cmax increased with an additive effect (134%) while valaciclovir's AUC increased with a greater-than-additive effect (196%).
TABLE 1.
Valaciclovir pharmacokinetic parameters for each treatment
| Treatment | Cmax (μM)a | Median Tmax (h) | AUC0-3 (μM · h)a |
|---|---|---|---|
| None | 0.81 ± 0.31 | 1.00 | 0.83 ± 0.32 |
| Cimetidine | 1.24 ± 0.42 | 1.00 | 1.44 ± 0.34 |
| Probenecid | 1.00 ± 0.37 | 1.00 | 1.01 ± 0.40 |
| Combined | 1.90 ± 0.84 | 1.13 | 2.46 ± 1.01 |
Values are means ± SD.
TABLE 2.
Point estimates and confidence intervals for effects of cimetidine and probenecid (relative to no drug treatment) on valaciclovir and acyclovir pharmacokinetic parameters
| Analyte | Param- eter | Value | Result with probe drug
|
||
|---|---|---|---|---|---|
| Cimetidine | Probenecid | Combination | |||
| Valaciclovir | Cmax | Mean | 1.71 | 1.36 | |
| 95% CI | (1.50-1.94) | (1.19-1.54) | |||
| 90% CI | (1.53-1.92) | (1.21-1.53) | |||
| AUC0-3 | Mean | 1.82 | 1.22 | 2.95 | |
| 95% CI | (1.54-2.16) | (1.03-1.44) | (2.49-3.50) | ||
| 90% CI | (1.58-2.10) | (1.06-1.41) | (2.55-3.41) | ||
| Tmax (h)a | Median | 0.13 | 0.03 | ||
| 95% CI | (−0.06-0.25) | (−0.13-0.25) | |||
| Acyclovir | Cmax | Mean | 1.05 | 1.20 | |
| 95% CI | (0.96-1.16) | (1.09-1.32) | |||
| 90% CI | (0.96-1.14) | (1.11-1.30) | |||
| AUC0-24 | Mean | 1.23 | 1.42 | ||
| 95% CI | (1.15-1.32) | (1.32-1.52) | |||
| 90% CI | (1.16-1.31) | (1.33-1.51) | |||
| t1/2 | Mean | 1.03 | 1.08 | ||
| 95% CI | (0.99-1.08) | (1.04-1.13) | |||
| 90% CI | (0.99-1.07) | (1.04-1.12) | |||
| CLR | Mean | 0.78 | 0.67 | ||
| 95% CI | (0.72-0.85) | (0.61-0.72) | |||
| 90% CI | (0.72-0.84) | (0.62-0.73) | |||
| Tmax (h)a | Median | 0.33 | 0.13 | ||
| 95% CI | (0.06-0.50) | (−0.13-0.31) | |||
Estimated median differences between concomitant medication present and absent.
FIG. 1.

Mean concentrations (+ 2 standard errors) of valaciclovir in plasma.
Valaciclovir's Tmax was not significantly affected by cimetidine and probenecid. Therefore, point estimates and 95 and 90% confidence intervals were calculated for the overall effect of probe drugs individually and not for the effect of each treatment in comparison with that of the control treatment.
Acyclovir pharmacokinetics.
Acyclovir pharmacokinetic parameters for each treatment are summarized in Table 3. Ratios of point estimates and 95 and 90% confidence intervals for changes in pharmacokinetic parameters to those of the control group are presented in Table 2. Mean concentrations of acyclovir in plasma for each treatment are shown in Fig. 2.The overall effects of cimetidine and probenecid on acyclovir pharmacokinetics were independent: with combined treatments, acyclovir's Cmax and AUC from 0 to infinity (AUC0-∞) were increased with an additive effect. Probenecid had a greater effect than cimetidine on the acyclovir Cmax and AUC0-∞. The effects of the probe drugs on acyclovir pharmacokinetics were much less strong than those of valaciclovir.
TABLE 3.
Acyclovir pharmacokinetic parameters for each treatmenta
| Treatment | Cmax (μM) | Tmax (h) (median) | AUC0-∞ (μM · h) | t1/2 (h) | CLR (ml/min) | Ae (%)b |
|---|---|---|---|---|---|---|
| Control | 23.77 ± 5.94 | 1.25 | 69.50 ± 12.63 | 2.82 ± 0.14 | 349 ± 68 | 46.4 ± 8.6 |
| Cimetidine | 25.70 ± 8.73 | 1.50 | 88.78 ± 18.45 | 2.98 ± 0.20 | 273 ± 51 | 45.9 ± 7.0 |
| Probenecid | 29.12 ± 9.40 | 1.50 | 103.31 ± 25.65 | 3.13 ± 0.23 | 234 ± 59 | 44.8 ± 6.4 |
| Combined | 30.61 ± 9.48 | 1.50 | 121.90 ± 27.01 | 3.16 ± 0.28 | 187 ± 63 | 42.4 ± 9.7 |
Values are means ± SD unless otherwise noted.
Ae, percentage of the dose recovered as acyclovir in the urine.
FIG. 2.

Mean concentrations (+ 2 standard errors) of acyclovir in plasma.
The mean increases in acyclovir AUC observed with probenecid (48%), cimetidine (27%), and both drugs combined (75%) compared to the acyclovir AUC observed in the control group were consistent with the reductions in CLR (33, 22, and 46%, respectively). The changes in the values for acyclovir CLR versus time interval from values for the control treatment showed the inhibition to be greatest at the interval of 0 to 0.5 h (Fig. 3).The plasma elimination t1/2 was similar to that of the control group when valaciclovir was combined with cimetidine and increased by 8% when valaciclovir and probenecid were administered concomitantly. The amount of acyclovir recovered in the urine was almost constant after all treatments (42.4 to 46.4% of the dose).
FIG. 3.

Change in acyclovir's CLR from values for the control treatment versus time.
Cimetidine and probenecid pharmacokinetics.
The AUC0-∞ (mean ± standard deviation [SD]) for cimetidine given with valaciclovir alone or combined with probenecid was 58.5 ± 12.7 or 63.4 ± 15.2 μM · h, respectively, but the increase with the valaciclovir-probenecid combination was not significant (P = 0.15). Likewise, the AUC0-∞ of probenecid given with valaciclovir alone or combined with cimetidine was 2,701 ± 964 or 2,899 ± 820 μM · h, respectively, but the increase was not significant (P = 0.10).
Modeling of the drug interactions.
The pharmacokinetic parameters of the interaction model of cimetidine and probenecid on acyclovir renal elimination are summarized in Table 4. Based on an examination of the residual plots, the adequacy of the individual plasma drug concentration-versus-time curves or acyclovir elimination-rate-versus-time curves with respect to the experimental data was very good (data not shown). Acyclovir's CLR (mean ± SD) estimated using the interaction model (equation 2) was 384 ± 86 ml/min, while the corresponding value determined using the noncompartmental method was 349 ± 68 ml/min.
TABLE 4.
Values for the pharmacokinetic parameters of the cimetidine-probenecid interaction model for acyclovir renal elimination
| Parameter | Mean (SD) | Median (range) |
|---|---|---|
| CLGF (ml/min) | 146 (46) | 151 (84-223) |
| CLi1 (ml/min) | 102 (61) | 87 (21-213) |
| CLi2 (ml/min) | 136 (41) | 130 (63-203) |
| Ki1 (μM) | 5.86 (7.43) | 2.84 (0.63-18.6) |
| Ki2 (μM) | 62.8 (32.2) | 66.4 (26.0-92.2) |
| CLR (ml/min)a | 385 (71) | 378 (265-468) |
Determined for the control group according to equation 2.
DISCUSSION
Valaciclovir pharmacokinetics.
Valaciclovir's AUC increased with all concomitant treatments. This increase may be due to either increased absorption or decreased first-pass metabolism or clearance. However, a significant change in absorption is not likely, because the overall recovery of acyclovir in the urine did not change with concomitant treatment. Thus, it appears that suppression of gastric acid by cimetidine does not affect the absorption of valaciclovir. The increase in valaciclovir's AUC with probenecid and cimetidine is likely to be due to a reduced clearance upon conversion to acyclovir. This may be due to reduced hepatic uptake or metabolism.
As regards metabolism, valaciclovir is converted mainly to acyclovir by a specific mitochondrial hydrolase (1). The effect of cimetidine or probenecid on this enzyme is unknown. As regards hepatic uptake, cimetidine and probenecid might reduce the hepatic uptake of cations and anions, respectively (2, 3, 4, 6, 7). Approximately 50% of valaciclovir in plasma can be predicted to be in a cationic form, and only 1% is predicted to be in an ionic form (pKa, 1.7 to 7.47 to 9.41). The hypothesis of cimetidine and probenecid inhibiting the hepatic uptake of valaciclovir is consistent with the more profound effect of cimetidine on valaciclovir's AUC. However, the extent of conversion of valaciclovir to acyclovir, as assessed by urinary recovery of acyclovir, was not altered.
No adverse effects could be attributed to the higher levels of valaciclovir in plasma occurring after some treatments. This result is consistent with the small amount of pharmacological activity attributable to valaciclovir.
Acyclovir pharmacokinetics.
The increases in acyclovir's AUC with administration of the probe drugs, either separately or in combination, were entirely accounted for by the reduction in CLR of acyclovir. This reduction is consistent with the low contribution (15%) of metabolic clearance to acyclovir elimination. The comparable recoveries in urine after each treatment suggest that the bioavailability of acyclovir had not been altered.
Acyclovir CLR is almost threefold greater than the glomerular-filtration rate in healthy subjects. Comparison of CLGF, CLi1, and CLi2 shows that each mechanism of elimination accounts for roughly one-third of CLR. Hence, the complete inhibition of one mechanism by a drug interaction cannot have great consequences on acyclovir mean concentrations, since CLR would be at most decreased by one-third.
The estimate of the CLGF of acyclovir is in fact the part of CLR that is not subject to interaction with cimetidine or probenecid. It amounted to 146 ± 46 ml/min, which is higher than the creatinine clearance of the healthy volunteers (108 ± 25 ml/min). The difference might represent the clearance of another renal secretion process, involving a different carrier. Among the known transporters, the nucleoside carrier might be a good candidate (8), owing to the chemical structure of acyclovir. Alternatively, the discrepancy may result from a rapid diffusion of acyclovir from blood cells, because the model estimate does not take this phenomenon into account.
Some acyclovir in plasma is present as an anion (about 3%) and as a cation (traces) (pKa, 2.27 to 9.25). The reduction in acyclovir CLR with probenecid (33%) was similar to that reported in a previous study in which a 32% decline was observed (6). This reduction was thought to be due to inhibition of the renal tubular secretion of acyclovir by the anionic pathway. However, inhibition of renal secretion of neutral and basic compounds by probenecid has also been reported (2). Thus, probenecid can inhibit acyclovir CLR for all of its ionic species.
Since the equilibrium among anionic, cationic, and neutral species of a compound in solution is dynamic and the rate of conversion is very high, species could be cleared by a transporter in a larger amount than could be expected from the proportions at equilibrium. It has been suggested that acyclovir may inhibit the transport of creatinine, which is excreted by the renal tubular pathway for cations (5). Hence, acyclovir may be transported by this transporter and cimetidine may compete with the renal tubular secretion of acyclovir via the cationic pathway.
As regards the kinetics of the interactions, acyclovir CLR from each urine sample collected was most reduced by probenecid and cimetidine at the first collection period, which was 2 h after the probenecid dose and 1 h after the cimetidine dose (Fig. 3). This period corresponds to the peak actions of these drugs as inhibitors of drug CLR reported in previous studies (2, 7). According to the kinetic model, the cimetidine Ki is 5.9 μM, while its peak concentration is in the range of 10 to 20 μM and its t1/2 is about 2 h. Hence, the time required for the Ki of a cimetidine concentration to become negligible (i.e., less than 0.2 Ki) is about 3 to 4 t1/2, i.e., 6 to 8 h. Cimetidine cannot interact with acyclovir secretion beyond this limit. Analogous calculations for probenecid (peak concentration, 150 to 250 μM; t1/2, 5 h; Ki, 60 μM) show that probenecid can interact with acyclovir during 3 to 4 t1/2, i.e., 15 to 20 h.
Conclusion.
Cimetidine and probenecid, separately and together, reduced the rate but not the extent of the conversion of valaciclovir to acyclovir and reduced the CLR of acyclovir. The effects were independent of each other. The pattern of inhibition of the CLR of acyclovir is consistent with competitive inhibition. Drugs which substantially reduce the renal tubular secretion of cations or anions may reduce the CLR of acyclovir, with the effect being greater for anionic inhibitors. These interactions are not expected to have clinical consequences regarding the safety of valaciclovir.
Acknowledgments
We thank Steve Jeal, Lewis Kanics, and Mira Doig for analytical support and Garry Layton for statistical analysis.
REFERENCES
- 1.Burnette, T. C., J. A. Harrington, J. E. Reardon, B. M. Merrill, and P. De Miranda. 1995. Purification and characterization of a rat liver enzyme that hydrolyzes valaciclovir, the l-valyl ester prodrug of acyclovir. J. Biol. Chem. 270:15827-15831. [DOI] [PubMed] [Google Scholar]
- 2.Cunningham, R. F., Z. H. Israili, and P. G. Dayton. 1981. Clinical pharmacokinetics of probenecid. Clin. Pharmacokinet. 6:135-151. [DOI] [PubMed] [Google Scholar]
- 3.Hedaya, M. H., and R. J. Sawchuk. 1989. Effect of probenecid on the renal and nonrenal clearance of zidovudine and its distribution in cerebrospinal fluid in the rabbit. J. Pharm. Sci. 78:716-722. [DOI] [PubMed] [Google Scholar]
- 4.Kenwright, S., and A. J. Levi. 1973. Impairment of hepatic uptake of rifamycin antibiotics by probenecid, and its therapeutic implications. Lancet 2:1401-1405. [DOI] [PubMed] [Google Scholar]
- 5.Laskin, O. L. 1983. Clinical pharmacokinetics of aciclovir. Clin. Pharmacokinet. 8:187-201. [DOI] [PubMed] [Google Scholar]
- 6.Laskin, O. L., P. De Miranda, and D. H. King. 1982. Effects of probenecid on the pharmacokinetics and elimination of aciclovir in humans. Antimicrob. Agents Chemother. 21:804-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muirhead, M. R., A. A. Somogyi, P. E. Rolan, and F. Bochner. 1986. Effect of cimetidine on renal and hepatic drug elimination: studies with triamterene. Clin. Pharmacol. Ther. 40:400-407. [DOI] [PubMed] [Google Scholar]
- 8.Sadée, W., V. Drübbish, and G. L. Amidon. 1995. Biology of membrane transport proteins. Pharm. Res. 12:1823-1837. [DOI] [PubMed] [Google Scholar]
- 9.Somogyi, A. A., C. Stockley, J. Keal, P. Rolan, and F. Bockner. 1987. Reduction of metformin renal tubular secretion by cimetidine in man. Br. J. Clin. Pharmacol. 23:545-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker, G. T. 1981. Measurement of the renal clearance of drugs. Br. J. Clin. Pharmacol. 12:761-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Ginneken, C. A., and F. G. Russel. 1989. Saturable pharmacokinetics in the renal excretion of drugs. Clin. Pharmacokinet. 16:38-54. [DOI] [PubMed] [Google Scholar]
- 12.Van Grugten, J., F. Bochner, J. Keal, and A. Somogyi. 1986. Selectivity of the cimetidine-induced alterations of the renal handling of organic substrates in humans. Studies with anionic, cationic and zwitterionic drugs. J. Pharmacol. Exp. Ther. 236:481-487. [PubMed] [Google Scholar]
- 13.Weller, S., M. R. Blum, and M. Doucette. 1993. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clin. Pharmacol. Ther. 54:595-605. [DOI] [PubMed] [Google Scholar]