

Abstract
A single-dose study and a multiple-dose study of the safety and pharmacokinetics of ruprintrivir, a new selective irreversible inhibitor of human rhinovirus 3C protease, were conducted with healthy adult volunteers. Both studies were double-blind, randomized, placebo-controlled, parallel-group investigations of ruprintrivir administered intranasally at two dose levels. The parent drug and its acid metabolite, AG7185, were measured in plasma samples and nasal washings, and the safety of the treatments was monitored. Intranasal ruprintrivir, administered as single doses of 4 and 8 mg or every 3 h, six times per day, for 7 days was safe and well tolerated. Adverse events were mild, short-lived, and confined to the upper respiratory tract (i.e., nose and throat, taste and smell perceptions). Adverse events were similar after placebo and after single or multiple doses of active drug. Systemic exposure to ruprintrivir was rarely detectable with the highest measured concentration of ≤0.52 ng/ml; the assay had a lower limit of quantification of 0.2 ng/ml. Systemic exposure to the metabolite was also low, with a highest measured concentration of 3.25 ng/ml. Concentrations of AG7185 observed during multiple dosing were higher than those observed after the first dose but were no more than predicted from the single-dose study. Substantial amounts of ruprintrivir were observed intranasally for at least 9 h after multiple doses of ruprintrivir.
The most common illness in humans is viral infection of the upper respiratory tract. Rhinoviruses are the predominant pathogens causing common colds, accounting for 30 to 40% of all colds, and young adults have close to one rhinoviral cold per year (1). Common colds associated with these infections are a worldwide leading cause of acute infectious morbidity. In subjects with underlying lung diseases, rhinovirus infection can lead to severe consequences that require acute care and hospitalization. Symptoms associated with the underlying lung disease can be magnified in severity and duration by viral infections (3, 7). Several agents currently used for the symptomatic treatment of colds include antihistamines and anticholinergics for rhinorrhea and sneezing, analgesics for headache and myalgias, and antitussives and decongestants for coughs and nasal congestion. Antibiotics are frequently prescribed despite their lack of effectiveness.
Ruprintrivir (trans-(4 S,2"R,5"S,3‴S)-4-{2"-4-(4-fluorobenzyl)-6"-methyl-5′-[(5"-methylisoxazole)-3"-carbonylamino]-4-oxoheptanoylamino}-5-(2‴-oxopyrrolidin-3‴-yl)pent-2-enoicacid ethyl ester) is an irreversible 3C protease inhibitor in clinical development for use against human rhinoviral infections. The design of ruprintrivir was based on the three-dimensional structure of the active site of rhinovirus 3C protease (2, 4, 5), which is responsible for the cleavage of viral precursor polyproteins into structural and enzymatic proteins essential for viral replication. Potent antiviral activity for the viral 3C protease has been demonstrated in vitro against all 48 different human rhinovirus (HRV) serotypes tested with a mean 90% effective concentration (EC90) of 82 nM or 49 ng/ml (6). Antiviral activity of ruprintrivir has also been demonstrated against HRV replication in a normal human bronchial epithelial cell line (BEAS-2B), along with reduced release of inflammatory cytokines into the cell supernatant (8).
In preclinical animal studies, hydrolysis of ruprintrivir (the ethyl ester) produced ethanol and the carboxylic acid metabolite, AG7185, which was 400-fold less active than ruprintrivir and was the predominant biotransformation pathway. In vitro studies with human microsomal preparations suggested that nonspecific esterases in cytosol and the liver cytochrome P450 enzymes were responsible for the hydrolysis of ruprintrivir. However, no change of ruprintrivir concentration was observed for 2 h after incubation with human plasma or nasal wash. In rabbits, high levels of radioactivity remained at the intranasal dosing site for 24 h after administration of radiolabeled ruprintrivir, and the systemic exposure was found to be low. Studies of the clinical formulation indicated that acute and chronic intranasal administration was well tolerated in rats and dogs.
The projected regimen of ruprintrivir in humans was intranasal administration several times per day for several days for the treatment and possible prophylaxis of rhinoviral infections. Ruprintrivir was the first protease inhibitor to be tested for in vivo anti-HRV activity. Prior to the testing of efficacy in phase II trials, a single-dose study and a multiple-dose study were conducted in healthy adult volunteers to determine the safety, tolerability, and pharmacokinetics of ruprintrivir. Since there is no efficacy animal model for the common cold and ruprintrivir is very well tolerated in animals, the 8-mg dose was selected in the first in human testing to deliver the maximal amount of drug to the human nose. A lower dose of 4 mg of ruprintrivir was also studied to investigate safety and pharmacokinetics related to dose.
MATERIALS AND METHODS
The single-dose study and the multiple-dose study were double-blind, randomized, placebo-controlled, parallel-group investigations. Each protocol was approved by an independent review board before the start of the study. All subjects were healthy male volunteers, 18 to 50 years of age, who provided written informed consent before participation in the studies. They were selected from the clinical research unit's panel of volunteers and were enrolled in either the single-dose or multiple-dose study. Smokers were excluded. Strenuous exercise, alcohol, and caffeine consumption were prohibited, and the subjects remained at the clinical facility for the entire duration of each study. On acceptance into the study, subjects were randomized to receive ruprintrivir or placebo. For both studies, the 4-mg dose was administered first, and the blinded safety data were reviewed and evaluated before the 8-mg dose was administered.
The single-dose treatments were 4 and 8 mg of ruprintrivir from a 2% suspension or placebo supplied with an intranasal delivery device intended to administer 100 μl (2 mg) per actuation. The vehicle alone was used as the placebo treatment. Six subjects were studied in each treatment group: four subjects were given ruprintrivir, and two subjects were given placebo. Each subject was administered two 100-μl actuations in each nostril. For the placebo treatment, both instillations in each nostril were placebo. For the 4-mg treatment, one instillation in each nostril was ruprintrivir and one was placebo. For the 8-mg treatment, both instillations in each nostril were ruprintrivir. Treatments were administered while subjects were seated, and they remained supine or seated for 1 h after administration. Blood samples for the assessment of the plasma pharmacokinetics of ruprintrivir and its acid metabolite, AG7185, were obtained prior to dosing and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 h after dosing.
The multiple-dose treatments were 4 and 8 mg every 3 h, given six times from 7 a.m. to 10 p.m. per day, for 7 days. The formulation was the same as that used for the single-dose study. Twelve subjects were studied in each treatment group: six subjects were given ruprintrivir, and six subjects were given placebo. Each subject in the 4-mg group was administered one 100-μl actuation in each nostril. Each subject in the 8-mg group was administered two 100-μl actuations in each nostril. The treatments were administered while subjects were seated, and they remained supine or seated for 30 min after administration. Blood samples for assessment of the plasma pharmacokinetics of ruprintrivir and its acid metabolite were obtained prior to dosing and at 0.25, 0.5, 1, 1.5, 2, 2.5, and 3 h after the first dose and the last (i.e., the 42nd) dose. Nasal washing samples for assessment of the levels of ruprintrivir and AG7185 in the nasal cavity after intranasal dosing were obtained 0.5 and 3 h after the second dose on the first day of dosing and immediately before and 0.5 and 3 h after the first dose on the second day of dosing. Half of the subjects were studied at 0.5 h, and half were studied at 3 h. Nasal washings were obtained after 5 ml of 0.9% saline solution was infused into each nostril (total volume, 10 ml) and drained into a cup after a few seconds.
The concentrations of ruprintrivir and AG7185 in plasma were measured by use of validated liquid-liquid extraction and liquid chromatography with tandem mass-spectrometric detection at Covance Laboratories, Ltd. (Harrogate, England). Plasma samples (0.2 ml) containing various concentrations of ruprintrivir and AG7185 were mixed with internal standards (ruprintrivir-D5 for ruprintrivir and AG7214, an chemical analogue of AG7185, for AG7185) in solution (0.025 ml). Each sample was mixed with 0.2 ml of 0.1 N HCl solution and then extracted by using 2.0 ml of ethyl acetate. The mixture was rotated on rotary mixer for 15 min, and the organic layer was transferred to a new tube and evaporated to dryness under a nitrogen stream at 30°C. The residue was reconstituted with 0.1 ml of methanol-0.01% trifluoroacetic acid (50:50 [vol/vol]) solution and transferred to a high-pressure liquid chromatography-tandem mass spectrometer. The separation was achieved on a Perkin-Elmer LC-MS/MS System with a Micro LC pump and a Hypersil BDS C8 column with a flow rate of 1.0 ml of the mobile phase (water-methanol-acetonitrile-10 mMammonium acetate buffer [pH 4.0]-trifluoroacetic acid [20/35/25/20/0.01, by volume]), followed by detection of ruprintrivir and AG7185 at the following ions: 599.3 to 373.3 for ruprintrivir and 571.2 to 373.1 for AG7185. This method was validated within the concentration range of 0.2 to 200 ng/ml for AG&088 and AG7185. The standard curves were linear with R 2 values of >0.980. The precision (percent coefficient of variation) levels of the assay for ruprintrivir and AG7185 were ≤10.2 and 17.1%, respectively. The accuracy levels (i.e., the deviation from the nominal concentrations) of the assay for ruprintrivir and AG7185 were within −8.7 to 1.7 and −3.3 to −5.0%, respectively.
Nasal wash samples were analyzed by using a similar method except that nasal wash samples were diluted before analysis with normal saline to the assay range (1 to 1,000 ng/ml).
The maximum concentration in plasma (Cmax) and the time at which it occurred (Tmax) were the observed parameters. The area under the concentration-time curve from 0 h to the time of the last measurable concentration (AUC0-last) was calculated by linear interpolation up to Cmax and exponential interpolation for the remainder of the data. AUC0-3 was calculated for the first and last doses in the multiple-dose study. The half-life (t1/2) and elimination rate constant (kel) were estimated by linear regression of the logarithm of the terminal concentration versus time data provided that at least three points from the terminal elimination phase were available. The R2 values for these linear regression curves ranged from 0.62 to 1.0. The AUC from 0 h to infinity (AUC0-∞) after a single dose was computed by the summation of AUC0-last and AUCextrap, the latter parameter estimated from the ratio of the last measurable concentration and the elimination rate constant, kel. The extrapolated AUC ranged from 8 to 44% of the total AUC. All pharmacokinetic parameters were estimated by using WinNonLin, professional version 2.1, (Scientific Consulting, Inc., Apex, N.C.) and were summarized with descriptive statistics (mean and standard deviation or median) by using SAS version 6.12, (SAS Institute, Inc., Cary, N.C.).
The total amounts of ruprintrivir and AG7185 recovered in the nasal washes were calculated from the product of the measured concentrations and the volume of the wash.
In both studies, adverse events, vital signs, 12-lead electrocardiography (ECG) data, clinical laboratory evaluations, and physical evaluations were assessed at preselected time points to ascertain the safety and tolerability of ruprintrivir. Blood pressure, heart rate, respiratory rate, and ECG with a 10-s rhythm strip data were obtained after individual volunteers had been in a supine position for at least 5 min. Predose blood pressure and heart rate were measured three times, and the median was used as baseline.
Because of the limited number of subjects and few safety observations, no formal statistical analysis of the safety observations or pharmacokinetic data was performed.
RESULTS
All volunteers satisfactorily completed the studies. All subjects were male nonsmokers that were between 19 and 49 years of age, 58.2 to 99.6 kg in weight, and 162 to 197 cm in height. All were Caucasians except for one Asian volunteer in the single-dose study. The mean age of the subjects administered the 8-mg ruprintrivir treatment in the multiple-dose study was lower (23 ± 4.5 years) than in the other groups (Table 1). Since subjects were randomly assigned to the treatment groups, this difference in age was a random event. It is unlikely to affect the outcome of the study. There were no other apparent differences among the treatment groups within both studies.
TABLE 1.
Summary of subject demographics
| Dose of ruprintrivir (n)a | Mean ± SD
|
||
|---|---|---|---|
| Age (yr) | Body wt (kg) | Ht (cm) | |
| Single-dose placebo (4) | 30 ± 5.4 | 74.6 ± 4.86 | 179 ± 6.6 |
| Single dose, 4 mg (4) | 33 ± 9.7 | 73.1 ± 2.42 | 179 ± 2.9 |
| Single dose, 8 mg (4) | 37 ± 10.0 | 84.3 ± 10.56 | 181 ± 6.4 |
| Multiple-dose placebo, 4 mg (6) | 36 ± 7.9 | 73.7 ± 8.27 | 172 ± 6.2 |
| Multiple-dose placebo, 8 mg (6) | 33 ± 4.6 | 79.2 ± 13.90 | 185 ± 8.7 |
| Multiple dose, 4 mg (6) | 35 ± 10.1 | 76.8 ± 10.14 | 177 ± 4.0 |
| Multiple dose, 8 mg (6) | 23 ± 4.5 | 72.7 ± 9.99 | 178 ± 8.5 |
n, number of subjects.
In the single-dose study, the concentrations of ruprintrivir in plasma at both dose levels were below or barely above the lower limit of quantitation of the assay (0.2 ng/ml), so it was not possible to compute pharmacokinetic parameters for ruprintrivir between the two treatment groups. After the 4-mg dose, ruprintrivir was not measurable in the plasma samples from any subject. After the 8-mg dose, ruprintrivir was measurable in three plasma samples from two subjects, and the concentrations ranged from 0.21 to 0.28 ng/ml.
The concentrations of the metabolite (AG7185) in plasma were measurable in seven of the eight subjects administered ruprintrivir (Fig. 1).In these subjects, AG7185 could be measured in the first sample, which had been obtained 15 min after dosing. In most subjects, AG7185 could be measured up to 8 h but not by 12 h after dosing. The pharmacokinetics of AG7185 are summarized in Table 2.
FIG. 1.

Concentrations of the metabolite AG7185 observed in plasma in individual subjects after a single intranasal dose of 4 or 8 mg of ruprintrivir (note the difference in the ordinate scales).
TABLE 2.
Pharmacokinetic parameters of metabolite AG7185 in plasma after a single intranasal dose of ruprintrivir
| Dose of ruprintrivir (mg) | Median (n),a range
|
||||
|---|---|---|---|---|---|
| Cmax (ng/ml) | Tmax (h) |
t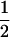 (h) (h) |
AUC0-last (ng ml−1 h−1) | AUC0-∞ (ng ml−1 h−1) | |
| 4 | 0.46 (4), 0-1.01 | 1.0 (3), 0.5-4.0 | 4.54 (2), 2.56-6.52 | 3.27 (3), 0.73-4.39 | 5.94 (2), 4.01-7.87 |
| 8 | 2.75 (4), 2.48-3.21 | 3.0 (4), 1.0-4.0 | 3.77 (4), 1.40-5.61 | 7.81 (4), 7.12-5.26 | 10.09 (4), 9.92-10.74 |
n, number of subjects.
In the multiple-dose study, all concentrations of ruprintrivir in plasma at both dose levels were <0.2 ng/ml after the first dose on the first day of treatment. Concentrations were detectable after the last dose on the seventh day in one subject treated with 4 mg and in three subjects treated with 8 mg. In these four subjects the Cmax was observed within 1 h of dosing and ranged from 0.26 to 0.52 ng/ml. The AUC0-3 ranged from 0.27 to 1.04 ng ml−1 h−1. The elimination half-life of ruprintrivir was estimated for two subjects, at 2.45 and 1.45 h, respectively.
The pharmacokinetics of AG7185 after multiple dosing are summarized in Table 3. AG7185 was measurable after the first and forty-second doses in all six subjects given 4 mg of active drug. AG7185 concentrations in plasma were not measurable in two subjects after the first 8-mg dose but were measurable in all plasma samples obtained just prior to and for 3 h after the forty-second 8-mg dose. The concentrations observed after the 42nd dose are plotted in Fig. 2.The highest measured concentration was 3.25 ng/ml. The elimination half-lives of AG7185 could not be determined because the pharmacokinetic blood sampling was limited to the 3-h dosing interval and there was inconsistent evidence of concentration decay over that interval (Fig. 2).
TABLE 3.
Pharmacokinetic parameters of metabolite AG7185 in plasma after the first and forty-second intranasal doses of ruprintrivir
| Dose of ruprintrivir (n)a | Dose no. | Median (range)
|
||
|---|---|---|---|---|
| Cmax (ng/ml) | Tmax (h) | AUC0-3 (ng ml−1 h−1) | ||
| 4 mg (6)a | 1 | 0.44 (0.20-0.69) | 2.25 (0.25-3.0) | 0.58 (0.05-1.18) |
| 8 mg (6) | 1 | 0.67 (0-1.46) | 3.0b (1.5-3.0) | 1.30 (0-2.34) |
| 4 mg (6) | 42 | 1.46 (0.71-3.25) | 0.5 (0-2.5) | 3.46 (1.57-7.28) |
| 8 mg (6) | 42 | 2.18 (1.26-3.13) | 1.75 (0-3.0) | 5.48 (2.61-6.85) |
n, number of subjects.
n = 4 (for two subjects who had no detectable AG7185 concentration in plasma; Cmax and AUC0-3 were assigned a value of 0, whereas Tmax was assigned as missing).
FIG. 2.

Concentrations of the metabolite AG7185 observed in plasma in individual subjects after the intranasal administration of 4 or 8 mg of ruprintrivir every 3 h, six times daily, for 7 days.
The amounts of ruprintrivir recovered from the nasal wash varied widely from 5.1 to 326.1 μg for the 4-mg dosage group and 2.3 to 79.1 μg for the 8-mg dosage group. The median amounts recovered prior to the first dose of the second day, 9 h after the preceding dose, were 25 and 10 μg for the 4- and 8-mg dosage groups, respectively (Table 4). The amounts of the metabolite, AG7185, recovered in nasal washings ranged from 0.22 to 1.09 μg for the 4-mg dosage group to 0.29 to 1.56 μg for the 8-mg dosage group. The median amounts recovered prior to the first dose of the second day, 9 h after the preceding dose, were 0.70 and 0.66 μg for the 4- and 8-mg dosage groups, respectively (Table 4). There was no apparent difference between the 4- and 8-mg dosage groups.
TABLE 4.
Nasal amount of ruprintrivir and its metabolite AG7185 after intranasal 4- and 8-mg doses of ruprintrivir
| Dose (mg) | Median [μg] (range)a
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ruprintrivir
|
AG7185
|
||||||||||
| Day 1
|
Day 2
|
Day 1
|
Day 2
|
||||||||
| 0.5 h | 3 h | Predose | 0.5 h | 3 h | 0.5 h | 3 h | Predose | 0.5 h | 3 h | ||
| 4 | 60 (39-87) | 34 (9-326) | 25 (5-57) | 41 (35-48) | 30 (17-31) | 0.8 (0.2-0.8) | 0.4 (0.3-0.7) | 0.7 (0.3-1.1) | 0.7 (0.2-1.2) | 0.50 (0.5-0.5) | |
| 8 | 50 (47-53) | 16 (13-28) | 10 (2-51) | 43 (37-45) | 33 (12-79) | 0.5 (0.4-0.6) | 0.3 (0.2-0.5) | 0.7 (0.4-1.6) | 0.5 (0.5-0.6) | 0.8 (0.4-1.5) | |
Predose, that is, 9 h from the dose of the previous evening.
There were no treatment-related trends or clinically significant changes from baseline with regard to vital signs, physical examination, clinical laboratory values, or ECG results in the single-dose study or in the multiple-dose study. All adverse events that may have been judged related to treatment were mild in intensity. They are summarized in Table 5. In the single-dose study adverse events were reported by two of the four subjects receiving placebo, 4 mg of ruprintrivir, or 8 mg of ruprintrivir. Rhinitis occurred between 1 and 5 min after dosing and lasted for 1 min to 1 h. Taste perversions started between 1 and 5 min after dosing and lasted 5 min and 1 h. In the multiple-dose study, adverse events were reported by three of of the six subjects who received placebo in both the 4- and the 8-mg treatment groups or 8 mg of ruprintrivir and by all six of the subjects who received 4 mg of ruprintrivir. Adverse events were reported evenly across the dosing days, with no evidence of greater prevalence at the start or end of treatment. Of the 14 episodes of pharyngitis, 11 were reported by two subjects receiving 4 mg of ruprintrivir. The onset of pharyngitis ranged from immediately after dosing to ca. 7 h after dosing and lasted between 30 min and ca. 5 days. The median duration of pharyngitis was 2 h. Rhinitis, reported by five subjects, started between 1 min and 7 h 20 min after dosing and lasted between less than 1 min and 10 days. The median duration of rhinitis was 1 min. Taste perversion commenced between 4 and 10 min after dosing and lasted 1 to 45 min, with a median duration of 3 min. All adverse events resolved without treatment except for a headache reported by one subject receiving placebo.
TABLE 5.
Frequency of drug-related adverse events
| Adverse event | No. of adverse event experiences (no. of subjects with adverse event)a
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Single-dose study
|
Multiple-dose study
|
|||||||
| Placebo (n = 4) | 4 mg (n = 4) | 8 mg (n = 4) | Placebo (4 mg) (n = 6) | Placebo (8 mg) (n = 6) | 4 mg (n = 6) | 8 mg (n = 6) | ||
| Pharyngitis | 0 | 0 | 0 | 1 (1) | 0 | 12 (3) | 1 (1) | |
| Rhinitis | 2 (2) | 1 (1) | 3 (2) | 1 (1) | 3 (1) | 5 (3) | 2 (1) | |
| Epistaxis | 0 | 0 | 0 | 0 | 3 (2) | 0 | 1 (1) | |
| Cough increased | 1 (1) | 0 | 0 | 0 | 0 | 1 (1) | 0 | |
| Taste perversion | 2 (2) | 0 | 0 | 0 | 0 | 4 (3) | 1 (1) | |
| Parosmia | 0 | 1 (1) | 0 | 0 | 0 | 3 (1) | 0 | |
| Headache | 0 | 1 (1) | 0 | 2 (2) | 0 | 0 | 1 (1) | |
| Dizziness | 0 | 0 | 0 | 0 | 0 | 1 (1) | 0 | |
| Nausea | 0 | 1 (1) | 0 | 0 | 0 | 0 | 0 | |
| Dehydration | 0 | 1 (1) | 0 | 0 | 0 | 0 | 0 | |
| Total | 5 (2) | 5 (2) | 3 (2) | 4 (3) | 6 (3) | 26 (6) | 6 (3) | |
n, number of subjects.
DISCUSSION
The pharmacokinetic data in both studies indicated minimal systemic exposure to ruprintrivir after intranasal administration. Most plasma samples had no detectable ruprintrivir concentrations. Only low concentrations (≤0.52 ng/ml) in plasma were occasionally detected during the seventh day of dosing.
The amount of ruprintrivir in the nose appeared to decrease quickly. Within half an hour of drug administration, <2% of the drug was recovered from the nose by nasal wash. Nevertheless, substantial amounts of ruprintrivir (25 and 10 μg after the 4- and 8-mg doses, respectively) were recovered from the nose even 9 h after intranasal administration. The median calculated nasal wash concentrations of ruprintrivir were 2.5 and 1.0 μg/ml after the 4- and 8-mg doses, respectively, which was at least 20-fold higher than the EC90 of 49 ng/ml. The actual ruprintrivir concentrations in the nasal surface fluid were likely even higher than the nasal wash concentrations since significant dilutions by the nasal washing occurred during the collection of nasal wash samples. Although the nasal therapeutic amount of ruprintrivir is not established, the concentrations of ruprintrivir observed in the studies were probably sufficient to have an antirhinoviral effect in the nose after both doses during the intranasal administration of ruprintrivir. Therefore, dosing less frequently than every 3 h, six times per day may be feasible.
The nasal amounts of ruprintrivir after the 4- and 8-mg administrations were more variable (Table 4) and did not appear to be different between the two dosing groups. It is not clear whether there was a lack of dose relationship or the variability was too great and the number of samples was too small to detect any differences between the two dosing groups. Further investigation is needed in future studies.
Although the metabolite was detected in the nose, the amounts were much lower than for ruprintrivir. This suggests that very little of ruprintrivir was hydrolyzed to AG7185 at that site. Since AG7185 does not have significant antirhinoviral activity, the systemic exposure of AG7185 may be more relevant to potential systemic side effects, which were essentially absent in this small, short-duration study. All measures of systemic exposure to AG7185 (Cmax, AUC0-last, and AUC0-∞) indicated that it was greater after a single 8-mg dose of ruprintrivir than after a single 4-mg dose. The Cmax values appeared to increase less than proportional, whereas the AUC values were roughly proportional to dose in the single-dose study. This appeared to be caused by the flatter shape of plasma concentration-time curves after the 4-mg dose (Fig. 1). During the multiple-dosing study, a greater systemic exposure at the higher dose was also observed. Because of the small number of subjects and the variability of the pharmacokinetics of AG7185, dose proportionality could not be accurately estimated. However, it appeared that at steady state, the values of Cmax increased more than proportionally and the AUC0-3 values increased approximately proportionally (Table 3). Because of the erratic concentration-time profiles of AG7185 observed in both studies, AUC might be a better indicator of systemic exposure. The low systemic concentrations of ruprintrivir and AG7185 might explain the minimal systemic side effects of the doses of ruprintrivir.
The variability in the pharmacokinetics of the metabolite, AG7185, was high (with a coefficient of variation of >40%) except for the 8-mg single dose and the last 8-mg dose of the multiple-dose study. This may be an artifact of the presence of the very low concentrations observed after the lower doses. In the first 8 h after the single doses, AG7185 concentrations were below the lower limit of quantitation in none of the samples obtained after 8 mg and in 35% of the samples obtained after the 4-mg dose. Similarly, AG7185 concentrations were below the lower limit of quantitation in 33 and 44% of the samples after the first 4- and 8-mg doses, respectively, but in none of the samples obtained after the 42nd doses. Thus, variability in the pharmacokinetics of AG7185 tends to be low to moderate when concentrations are well above the lower quantification limits of the analytical method.
Based upon the median half-life of ca. 4 h for AG7185, it was anticipated that the concentrations observed in plasma during multiple dosing at 3-h intervals six times per day would be higher than after a single dose. This was the case in that median measures of systemic exposure for 3 h after the forty-second dose were four to six times greater than for 3 h after the first dose. However, the systemic exposure during a 3-h dosing interval (AUC0-3) after the 42nd dose (last dose on day 7) was not greater than the systemic exposure observed in the single-dose study (AUC0-∞), indicating no more accumulation during multiple dosing than expected based on linear pharmacokinetics. At the end of 7 days of multiple dosing, the systemic exposure to ruprintrivir and its metabolite AG7185 were low. The highest single concentrations of ruprintrivir and AG7185 observed in these studies were 0.52 and 3.25 ng/ml, respectively. The highest average concentration of AG7185 measured over a 3-h dosing interval was 2.43 ng/ml.
In both the single-dose and the multiple-dose studies the 4- and 8-mg doses of ruprintrivir were safe and well tolerated. No subject discontinued the study prematurely, and no serious adverse events were reported. There were no treatment-related trends or clinically significant changes. The adverse event profile during the multiple-dose study was similar to that observed in the single-dose study. In general, adverse events in both studies were mild and confined to the upper respiratory tract. Pharyngitis and rhinitis were the most frequently reported adverse events, and most incidences started shortly after dosing and were short-lived. Similar adverse events were reported by subjects receiving either placebo or ruprintrivir. These adverse events are likely related to intranasal dosing rather than to the study drug. The occurrence of an adverse event largely at the site of administration is consistent with the very low systemic exposure to ruprintrivir and AG7185 observed in these studies.
In conclusion, single and multiple intranasal administrations of 4- and 8-mg doses of ruprintrivir the treatments were safe and well tolerated, the systemic exposure of ruprintrivir and AG7185 was low, and a substantial amount of ruprintrivir was observed in the nose for at least 9 h after multiple doses of ruprintrivir were administered.
REFERENCES
- 1.Arruda, E., A. Pitkaranta, T. J. Witek, Jr., C. A. Doyle, and F. G. Hayden. 1997. Frequency and natural history of rhinovirus infections in adults during autumn. J. Clin. Microbiol. 35:2864-2868. [DOI] [PMC free article] [PubMed]
- 2.Dragovich, P. S., T. J. Prins, R. Zhou, S. E. Webber, J. T. Marakovits, S. A. Fuhrman, A. K. Patick, D. A. Matthews, C. A. Lee, C. E. Ford, B. J. Burke, P. A. Rejto, T. F. Hendrickson, T. Tuntland, E. L. Brown, J. W. Meador III, R. A. Ferre, J. E. V. Harr, M. B. Kosa, and S. T. Worland. 1999. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitor. 4. Incorporation of P1 lactam moieties as l-glutamine replacements. J. Med. Chem. 42:1213-1224. [DOI] [PubMed] [Google Scholar]
- 3.Gwaltney, J. M., Jr., and R. R. Rueckert. 1997. Rhinovirus, p. 1025-1047. In D. D. Richman, R. J. Whitley, and F. G. Hayden (ed.), Clinical virology. Churchill Livingstone, New York, N.Y.
- 4.Matthews, D. A., P. S. Dragovich, S. E. Webber, S. A. Fuhrman, A. K. Patick, L. S. Zalman, T. Hendrickson, T. J. Prins, J. T. Marakovits, R. Zhou, J. Tikhe, C. E. Ford, J. W. Meador III, R. A. Ferre, E. L. Brown, S. L. Binford, D. M. Delisle, and S. T. Worland. 1999. Structure-assisted design of mechanism based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA96:11000-11007. [DOI] [PMC free article] [PubMed]
- 5.Matthews, D. A., W. W. Smith, R. A. Ferre, B. Condon, G. Budahazi, W. Sisson, J. E. Villafranca, C. A. Janson, H. E. McElrov, C. L. Gribskov, and S. Worland. 1994. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 77:761-771. [DOI] [PubMed] [Google Scholar]
- 6.Patick, A. K., S. L. Binford, M. A. Brothers, R. L. Jackson, C. E. Ford, M. D. Diem, F. Maldonado, P. S. Dragovich, R. Zhou, T. J. Prins, S. A. Fuhrman, J. W. Meador, L. S. Zalman, D. A. Matthews, and S. T. Worland. 1999. In vitro antiviral activity of ruprintrivir, a potent inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 43:2444-2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pitkaranta, A., and F. G. Hayden. 1998. What's new with common colds? Complications and management. Infect. Med. 15:117-128. [Google Scholar]
- 8.Zalman, L. S., M. A. Brothers, P. S. Dragovich, R. Zhou, T. J. Prins, S. T. Worland, and A. K. Patick. 2000. Inhibition of human rhinovirus-induced cytokine production by AG&088, a human rhinovirus 3C protease inhibitor. 44:1236-1241. [DOI] [PMC free article] [PubMed]