


Abstract
We recently reported that the antitumor triazoloacridone, compound C-1305, is a topoisomerase II poison with unusual properties. In this study we characterize the DNA interactions of C-1305 in vitro, in comparison with other topoisomerase II inhibitors. Our results show that C-1305 binds to DNA by intercalation and possesses higher affinity for GC- than AT-DNA as revealed by surface plasmon resonance studies. Chemical probing with DEPC indicated that C-1305 induces structural perturbations in DNA regions with three adjacent guanine residues. Importantly, this effect was highly specific for C-1305 since none of the other 22 DNA interacting drugs tested was able to induce similar structural changes in DNA. Compound C-1305 induced stronger structural changes in guanine triplets at higher pH which suggested that protonation/deprotonation of the drug is important for this drug-specific effect. Molecular modeling analysis predicts that the zwitterionic form of C-1305 intercalates within the guanine triplet, resulting in widening of both DNA grooves and aligning of the triazole ring with the N7 atoms of guanines. Our results show that C-1305 binds to DNA and induces very specific and unusual structural changes in guanine triplets which likely plays an important role in the cytotoxic and antitumor activity of this unique compound.
INTRODUCTION
Triazoloacridone C-1305 belongs to a large group of acridine derivatives that have been synthesized at Gdansk University of Technology in a search for new antitumor compounds (1). This compound showed potent antitumor activity toward a wide range of different experimental tumors in vitro and in vivo including both murine and human colon carcinomas (2). The mechanism of action of C-1305 is currently under extensive investigation. Our recent studies show that C-1305 is a DNA topoisomerase II inhibitor which stabilizes covalent complexes between DNA and the enzyme (3). Interestingly, at cytotoxic doses C-1305 produces only low levels of unusually toxic cleavable complexes compared to classic topoisomerase II inhibitors such as amsacrine (m-AMSA). Another unusual feature of C-1305 is its potent cytotoxic activity toward PARP1-deficient cells in striking contrast to other topoisomerase II inhibitors that are less cytotoxic toward cells with compromised PARP1 function (4).
DNA topoisomerase II is an essential nuclear enzyme that regulates DNA topology and chromatin organization in living cells (5). The enzyme relieves torsional stress in DNA by controlled breakage-reunion of both DNA strands and passage of a DNA segment through the strand breaks. All topoisomerase II directed agents interfere with at least one step of the catalytic cycle and thus classify as topoisomerase inhibitors. Compounds that stabilize the covalent enzyme intermediate (cleavable complex) between DNA and topoisomerase II are traditionally called topoisomerase poisons (6), while agents that act on other steps of the catalytic cycle are called catalytic inhibitors (7). The majority of clinically used topoisomerase II inhibitors are topoisomerase poisons.
Although topoisomerase II inhibitors are among the most effective antitumor drugs that are used in the treatment of human tumors (8), the mechanistic basis for their activity is not fully elucidated. Inhibition of DNA topoisomerase II by antitumor drugs leads to many different cellular effects including DNA strand breaks through the conversion of cleavable complexes into direct DNA lesions during transcription and replication. Although the formation of DNA damage is considered as a crucial event in the cytotoxic activity of topoisomerase II inhibitors, perturbations of DNA topology and chromosome condensation during G2 and mitosis may also participate in the cytotoxic and antitumor effects of these agents (9). Recent results suggest that topoisomerase II inhibitors might work as transcriptional modulators, in particular with respect to the expression of genes responding to environmental changes (10).
It has long been debated whether topoisomerase II inhibitors induce enzyme-mediated DNA damage in specific DNA regions such as MAR sequences (11). Many factors could influence the drug-induced topoisomerase II-mediated sequence-specific DNA cleavage including the enzyme's binding site preference, the sequence preference for drug–DNA-binding, the binding site accessibility for topoisomerase II or drug-induced changes in DNA structure/topology (12). Specific cleavage at defined DNA sites by topoisomerase II in the presence of its inhibitors could be important from at least two perspectives. First, it could explain the chromosome band-specific DNA damage that is observed in patients treated with the topoisomerase II inhibitor etoposide (13). Second, different topoisomerase II inhibitors could have distinct effect on the DNA transcription pattern of drug-treated cells, depending on where and to which extent region-specific DNA cleavage is induced. Topoisomerase II inhibition principally interferes with DNA transcription due to the presence of immobilized cleavable complexes that arrest the progression of the transcription machinery (14). It may also result from accumulation of superhelical tension ahead of the transcription complex that prevents further progression of the RNA polymerase (15). We would predict such region-specific transcriptional modifications to be particularly important for topoisomerase II inhibitors with strong DNA-binding.
We speculated that the pronounced toxicity of triazoloacridone-induced DNA damage might be due to stimulation of cleavable complexes in specific DNA regions. In this study, we show that C-1305 binds to DNA by intercalation and induces unusual structural changes in DNA regions with guanine triplets. Comparison with other topoisomerase inhibitors and different DNA interacting agents showed that the structural perturbations in guanine-rich regions were specific for C-1305. We also characterized the structural elements of the triazoloacridones that are responsible for this effect and built a model to illustrate this feature.
MATERIALS AND METHODS
Drugs and chemicals
All triazoloacridone derivatives and m-AMSA were synthesized by Dr Barbara Horowska at the Department of Pharmacological Technology and Biochemistry, Gdansk University of Technology, Poland (see Figure 1 and Table 1 for chemical structures). Triazoloacridones (free bases) were used from 10 mM stock solutions prepared in 0.2% lactic acid in water (v/v), m-AMSA was used from 10 mM stock solutions prepared in water and kept at −20°C until use. Radiolabeled [α-32P]-dATP (3000 Ci/mmol), was purchased from Amersham Biosciences AB (Uppsala, Sweden). Calf thymus DNA, double-stranded polymers [poly(dA–dT)]2 and [poly(dG–dC)]2 were purchased from Sigma Chemicals Co. (St Louis, MO). All other reagents were of analytical grade and obtained either from Sigma or from the local purchaser.
Figure 1.

Chemical structures of triazoloacridones C-1305 and C-1533.
Table 1.
Chemical structures of selected triazolo- and imidazoacridone derivatives, and their effect on stabilization of the DNA secondary structure
 |
Ten different imidazo- and triazoloacridone derivatives were chosen for structure–activity relationship studies using chemical probing and melting temperature measurements. Bold face indicates the lead compound which induces unusual structural changes at guianine-triplets.
aMelting temperatures determined for calf thymus DNA in the presence of the studied compounds, the drug–DNA (nucleotide) ratio was 0.5, results are presented as means ± SD.
bMelting temperatures for the [poly(dA–dT)]2 DNA polymer at a drug–DNA (nucleotide) ratio of 0.5. Results are presented as means ± SD.
Absorption spectroscopy and melting temperature studies
Absorption spectra and melting curves were determined using an Uvikon 943 spectrophotometer (Kontron) coupled to a Neslab RTE111 cryostat or CaryBio 300 spectrophotometer coupled to a Peltier system (Varian). Titrations of the drug with DNA covering a large range of DNA-phosphate/drug ratios, were performed with fixed drug concentration (20 µM) and increasing concentrations of DNA substrates (in base pairs): for ctDNA 7–140 µM, 3–60 µM for poly(dA–dT) and poly(dG–dC). The experiments were conducted at 25°C in BPE buffer pH 7.1 (6 mM Na2HPO4, 2 mM NaH2PO4 and 1 mM EDTA). DNA substrates were included in reference samples.
For each series of Tm measurements, 12 samples were placed in a thermostatically controlled cell-holder, and the quartz cuvettes (10 mm pathlength) were heated by circulating water. The measurements were performed in BPE buffer. The temperature inside the cuvette was controlled with a platinum probe and increased over the range 20–100°C with a heating rate of 1°C/min. The ‘melting’ temperature Tm was calculated as the mid-point of the hyperchromic transition curves.
Circular dichroism (CD)
The CD spectra were obtained with a J-810 Jasco spectropolarimeter at 20°C controlled by a PTC-424S/L Peltier type cell changer (Jasco). A quartz cell of 10 mm path length was used to obtain spectra from 500 to 230 nm with a resolution of 0.2 nm. Titrations of the drug with DNA, covering a large range of P/D, were performed with fixed drug concentration (50 µM) and increasing concentrations of DNA substrates: for ct DNA 14–270 µM, for poly(dA–dT) and poly(dG–dC) 6–80 µM, until no further changes in drug–DNA spectra were observed. All the measurements were performed in 1 ml of BPE buffer at 25°C.
Electric linear dichroism (ELD)
ELD measurements were performed with a computerized optical measurement system using the procedures described previously (16,17). All experiments were conducted with a 10 mm pathlength Kerr cell having 1.5 mm electrode separation. The samples were oriented under an electric field strength varying from 1 to 14 kV/cm. The drug under test was present at 10 µM concentration together with the DNA at 200 µM concentration unless stated otherwise. This electro-optical method has proved particularly useful to determine the orientation of the drugs bound to DNA. It has the additional advantage that it senses only the orientation of the polymer-bound ligand since the free ligand is isotropic and does not contribute to the signal.
Surface plasmon resonance
Binding and kinetics measurements were performed with BIAcore 3000 system and streptavidin coated sensor chips (CM-5 chips from BIAcore) as described previously (18). Two 5′-biotin-labeled DNA hairpins (PAGE purified; Eurogentec, Belgium) were used (hairpin loop underlined): d(biotin-CATATATATCCCCATATATATG) and d(biotin-CGCGCGCGTTTTCGCGCGCG). All procedures for binding studies were automated as methods using repetitive cycles of sample injection and regeneration. Steady-state binding analysis was performed with multiple injections of different compound concentrations over the immobilized DNA surfaces for a 9 min period at a flow rate of 20 µl min−1 at 25°C. Drug solutions at concentrations ranging from 0.01 to 25 µM were prepared in filtered and degassed buffer by serial dilutions from stock solution and were injected from 7 mm plastic vials with pierceable plastic crimp caps (BIAcore Inc.). In all cases, the DNA surface was regenerated by buffer flow during 30 min without additional regeneration agents.
Average fitting of the sensorgrams at the steady-state level was performed with the BIAevaluation 3.0 program by fitting data from the steady-state regions to a multiple equivalent site model using Kaleidagraph with the following equation.
where K, the microscopic binding constant, is one variable to fit, r represents mol bound compound per mol DNA hairpin duplex, Cfree is the concentration of the compound in equilibrium with the complex and is fixed by the concentration in the flow solution and n is the number of compound binding sites on the DNA duplex. The r values were calculated by the ratio RU/RUmax, where RU is the steady-state response at each concentration and RUmax is the predicted RU for binding of a compound to the DNA in a flow cell. Global kinetic fitted to the sensorgrams to obtain association and dissociation kinetics constants were done using BIAevaluation software and an equivalent site interaction model.
Purification and radiolabeling of DNA restriction fragments
The 176 bp DNA fragment was prepared by 3′ end 32P-labeling of a EcoRI–PvuII (Promega) double digest of the pBS plasmid (Stratagene, La Jolla, CA) using [α-32P]dATP (Amersham) and avian myeloblastosis virus reverse transcriptase (Roche). Digestion products were separated on a 6% polyacrylamide gel under native conditions in Tris–borate/EDTA-buffered solution [89 mM Tris–borate (pH 8.3) and 1 mM EDTA). After autoradiography, DNA bands were excised, crushed and eluted overnight in 500 mM ammonium acetate and 10 mM magnesium acetate at 37°C. This suspension was filtered through a 0.22 µm filter (Millipore Corporation, Bedford, MA) and the DNA was precipitated with ethanol. After washing with 70% ethanol and vacuum drying of the precipitate, the labeled DNA was dissolved in 10 mM Tris–HCl (pH 7.0) containing 10 mM NaCl.
DNase I footprinting
Bovine pancreatic deoxyribonuclease I (DNase I, Sigma Chemical Co.) was stored as a 7200 U/ml solution in 20 mM NaCl, 2 mM MgCl2, 2 mM MnCl2 (pH 8.0). The stock solutions of DNase I were kept at −20°C and freshly diluted to the desired concentration immediately before use. Footprinting experiments were performed essentially as described previously (19,20). Briefly, reactions were conducted in a total volume of 10 µl. For each sample, ∼3000 c.p.m. of radiolabeled DNA was used which corresponds to ∼1 ng DNA. Samples (2 µl) of the labeled DNA fragment were incubated with 6 µl of the buffered solution containing the ligand at appropriate concentration. After 15 min incubation at room temperature to ensure equilibration of the binding reaction, the digestion was initiated by the addition of 2 µl of a DNase I solution whose concentration was adjusted to yield a final enzyme concentration of ∼0.01 U/ml in the reaction mixture. The reaction was stopped by freeze-drying after 3 min. Samples were lyophilized and dissolved in 5 µl of an 80% formamide solution containing tracking dyes. The DNA samples were then heated at 90°C for 3 min and chilled in ice for 3 min before electrophoresis.
DEPC chemical probing
Chemical probing experiments were performed essentially as described previously (21,22). Briefly, DEPC (diethylpyrocarbonate, Sigma) reactions were conducted in a total volume of 20 µl. Samples (2 µl) of the labeled DNA fragment were incubated with 18 µl of ligand in 10 mM Tris–HCl/NaCl (pH 7.5). After 10 min incubation at 4°C to ensure equilibration of the binding reaction, the reaction was initiated by the addition of 1 µl of DEPC solution and continued on ice with intermittent mixing. After 15 min the reaction was stopped by addition of 2 µl of 3 M sodium acetate and DNA precipitation with 70% ethanol. Samples were lyophilized and applied to DNA digestion with piperidine (30 µl of 1M solution in water) at 90°C for 10 min. Samples were then lyophilized and re-suspended in 5 µl of 80% formamide solution containing tracking dyes, heated and resolved by PAGE under denaturating conditions (0.3 mm thick gels and 8% acrylamide containing 8 M urea). After electrophoresis (∼1.5 h at 55 W/1600 V in Tris–Borate-EDTA-buffered solution and BRL sequencing apparatus model S2), gels were soaked in 10% acetic acid for 10 min, transferred onto Whatman 3 MM paper, and dried under vacuum at 80°C. A Molecular Dynamics 425E PhosphorImager system was used to collect data from the storage screens exposed to dried gels overnight at room temperature. Base line-corrected scans were analyzed by integrating all the densities between two selected boundaries using ImageQuant software (version 3.3, Molecular Dynamics). Each resolved band was assigned to a particular band within the DNA fragments by comparison of its position relative to sequencing standards generated by treatment of the DNA with dimethyl sulphate followed by piperidine-induced cleavage at the modified guanine bases in DNA (G-track).
Competition dialysis assay
The binding affinity of C-1305 to different DNA substrates was determined as described (23). For each competition dialysis assay, 250 ml of low salt (10 mM NaCl) BPE buffer pH 7.1 (6 mM Na2HPO4, 2 mM NaH2PO4 and 1 mM EDTA) containing 5 µM C-1305 was used. Oligonucleotides containing comparable GC content (50%) were purchased from ThermoElectron (see Table 2 for sequences). All substrates were high-performance liquid chromatography (HPLC)-purified, annealed in high salt BPE buffer, and dialyzed into BPES buffer for 24 h at room temperature. All measurements were performed using constant oligonucleotide concentration (75 µM per bp), in a 100 µl Slide-A-Lyzer MINI dialyzer unit with a 3500 Da molecular weight cut-off membranes (Pierce, Rockford, IL) and continuous stirring for 24 h at room temperature. After equilibration, SDS was added to each sample to 1% final concentration and the drug concentrations were determined by measuring the absorbance with a CaryBio 300 spectrophotometer (Varian) at 421 nm for both compounds. The fraction of DNA-bound drug was calculated as the difference between total drug concentration and unbound (free) drug concentration.
Table 2.
Sequence of double-stranded DNA oligonucleotides used in melting temperature studies and competition dialysis assays (only the upper strand sequence is shown)
| Oligonucleotides | Sequence | Tm (°C) |
|---|---|---|
| cGGGc | 5′-ATTCCGGGCTATATACGGGCCTAA-3′ | 53.4 ± 0.1 |
| aGGGa | 5′-GCAAAGGGAAACTGAAGGGAAACG-3′ | 52.8 ± 0.2 |
| tGGGt | 5′-GCAATGGGTTACTGATGGGTTACG-3′ | 52.7 ± 0.5 |
| cGGGa | 5′-AGTGCGGGATATATGCGGGATTGA-3′ | 54.5 ± 0.4 |
| GcGcG | 5′-TAATGCGCGCTATTACGCGCGATA-3′ | 59.3 ± 0.5 |
| cGaGc | 5′-AGTACGAGCATCCTACGAGCATTG-3′ | 55.5 ± 0.6 |
| CGtGc | 5′-AGTACGTGCATCCTACGTGCATTG-3′ | 56.0 ± 0.1 |
Tm values correspond to empirically determined melting temperatures of studied oligonucleotides in BPES buffer, pH 7.1. Bold face indicates potential DNA binding/interaction sites for C-1305.
Molecular modeling
Atomic coordinates of a 10mer double-stranded DNA sequence: 5′-GCGTGGGACC-3′, containing a G-triplet, were obtained from its X-ray structure (Nucleic Acid Database, accession no. pdt055). The structure was fixed for missing atoms (both heavy and hydrogen atoms) and the terminal nucleotides were patched with hydroxyl groups to obtain integer charge model (i.e. −18). The resulting DNA substrate was neutralized with sodium counterions and solvated with water molecules in a 15 Å box. The complete system was optimized through molecular mechanics minimization of the global energy function and then molecular dynamics simulation was performed for the total time of 10 ns. The NPT ensemble with isotropic pressure and thermal coupling was implemented. The CHARMM 27 (24) force field within NAMD (25) molecular dynamics engine was used with 11 Å cut-off distance for non-bonding interactions and Particle Mesh Ewald summation. The integrator was set to 2 fs and data were collected every 1 ps. The relaxed DNA structure obtained after 3 ns of MD simulation was then used to build C-1305:DNA complex. The structure of C-1305 was constructed using Quanta molecular modeling package (Accelrys, San Diego, CA), where partial charges were obtained by Merz–Kollman electrostatic fitting procedure from optimized B3LYP/6-31+G(df,p) ab initio calculations within Gaussian 03 program. We also calculated macroscopic pKa values of triazoloacridones in a broad range of pH. For this purpose, we submitted all data to the SPARC (Predictive Modeling System) online server at the U.S. Environmental Protection Agency (Research Triangle Park, NC) which previously has enabled correct estimation of pKa values for more than 4000 organic compounds (26). Calculated macroscopic pKa values were in agreement with the values obtained from analyses of drug protonation based on changes in UV-VIS spectra (Zofia Mazerska, unpublished data). Based on all gathered data, we found that the zwitterionic form of C-1305, with a protonated terminal nitrogen atom in the side chain and a deprotonated hydroxyl oxygen in the planar chromophore, was the most abundant form at pH close to physiological and higher (pH > 7.5).
In the next step, two systems containing DNA with intercalated compound in two different positions, were prepared in the same manner as for DNA alone. Resulting trajectories became a source for further analysis and calculations of static properties for studied systems. To evaluate which of the intercalation sites is favored by the compound, an Molecular Mechanics–Poisson Boltzmann Solvent Accessible (MM–PBSA) approach was used, which was described in detail previously (27). In this procedure, MM–PBSA is the sum of energy contributions of bonds, angles, dihedrals, Coulomb and Van der Waals interactions (MM part), the solvation energy obtained from continuum solvent calculations (PB part) and non-polar hydrophobic energy correlated with Solvent Accessible surface area (SA part). The MM–PBSA energy should be regarded only as the approximation of free energy since it does not include explicit entropy terms. However, such approach gives a good estimate of the differences in binding constants of the two systems, where all parts either do not change their conformations or change in a similar way. In both cases, entropic contributions can be neglected and MM–PBSA energy is correlated with binding constants, according to the Equation 1.
| 1 |
where ΔEMM–PBSA is a difference between total MM–PBSA energy of two ligand–DNA complexes with different positions of the drug, ΔΔG is a difference between free energy of two ligand–DNA complexes with different positions of the drug in drug–DNA complex, and ΔK is a difference between ligand–DNA-binding constants with two different intercalation sites.
Our own protocols to calculate MM–PBSA energy were used as described previously (28). Each selected simulation frame from MD was minimized with GB continuum model (20 steps of steepest descent and 200 steps of conjugate gradients algorithm) to avoid steric hindrances that could occur during MD simulations. The MM contribution was calculated using CHARMM 27 software and force field (29), solvation and non-polar energy was calculated using the UHBD program (30). Changes in molecular electrostatic potential (MEP) of the DNA upon drug intercalation were calculated with APBS software package (31). Final analyses and plots were done by Visual Molecular Dynamics software package (32).
RESULTS
UV-VIS absorption spectral changes upon triazoloacridone–DNA-binding
The absorption spectra of C-1305 and the structurally related biologically inactive C-1533 compound are shown on Figures 2A and 3A. Binding of C-1305 and C-1533 to different DNA substrates (calf thymus DNA, [poly(dA–dT)]2 and [poly(dG–dC)]2) revealed a marked hypochromic shifts in absorption for triazoloacridone derivatives with maxima at 407 and 421 nm for C-1305, and 419 nm for C-1533. Bathochromic shift was also observed for C-1305 spectra with a 3 nm shift for both peaks (407 and 421 nm) with ctDNA and [poly(dG–dC)]2 whereas a 3 nm shift could be observed only for 421 nm peak. No bathochromic shift was obtained for C-1305 with [poly(dA–dT)]2. These results show that the two compounds clearly bind to DNA and form stable complexes with both AT and GC polymers.
Figure 2.

Absorption (A) and CD spectra (B) of C-1305 in the presence of increasing concentrations of DNA (calf thymus DNA, [poly(dA–dT)]2 and [poly(dG–dC)]2). DNA titration of the drug was performed in BPE buffer at pH 7.1. To 1 ml of drug solution of 20 µM for absorption measurement and 50 µM for CD spectra, aliquots of a concentrated DNA solution were added. Vertical arrows indicate the increase of phosphate-DNA/drug ratio, horizontal arrows correspond to bathochromic shifts.
Figure 3.

Absorption (A) and CD spectra (B) of C-1533 in the presence of increasing concentration of DNA (calf thymus DNA, [poly(dA–dT)]2 and [poly(dG–dC)]2). DNA titration of the drug were performed in BPE buffer at pH 7.1. To 1 ml of drug solution at 20 µM for absorption measurement and 50 µM for CD spectra, aliquots of a concentrated DNA solution were added. Vertical arrows indicate the increase of phosphate-DNA/drug ratio.
CD
CD spectra were recorded at fixed drug concentrations of C-1305 and C-1533 and increasing concentrations of DNA and are shown in Figures 2B and 3B. These spectra cover a wide range of drug–DNA nucleotide ratios (ranging from 7:1 to 0.2:1). These results reveal a very distinct behavior of C-1305 to different types of DNA (Figure 2B) compared with C-1533 (Figure 3B). Compound C-1533 gave very weak CD signals at 420–450 nm for ctDNA with no major differences in CD signals for [poly(dA–dT)]2 and [poly(dG–dC)]2 homopolymers. In striking contrast, for the biologically active C-1305 derivative we observed a strong increase of the CD signal at 305–365 nm, a decrease at 375–415 nm and an increase at 430–470 nm. It is also possible to distinguish three isodichroic crossovers closely positioned to isosbestic points for absorption measurements. The CD amplitude of these bands differs between DNA substrates, the strongest signal was observed for calf thymus DNA that contains comparable proportions of AT (58%) and GC (42%) base pairs while [poly(dA–dT)]2 gave the weakest signal. Although the exact origin of the induced CD signals is not known, it is possible that the negative band centered around 400 nm reflects the intercalation of the drug between base pairs. Intercalating agents frequently, but not systematically, generate negative induced CD signals in their respective absorption band (33). The magnitude of the negative CD bands is roughly identical for ctDNA, [poly(dA–dT)]2 and [poly(dG–dC)]2, suggesting that C-1305 can intercalate in all three sequence contexts, which is in agreement with the absorption and ELD measurements. This explanation is insufficient when one considers the behavior of C-1533. This inactive compound was also capable of intercalating into DNA (see ELD data below) but did not give rise to a negative CD band at 400 nm (Figure 3B). Therefore, we are inclined to believe that the CD signals observed with C-1305 in this region reflect the specific structural distortion of DNA induced by this compound (see DEPC data below), rather than the intercalative binding process per se. The intense positive CD bands centered at 330 and 450 nm seen with ctDNA very likely reflect the unique capacity of this compound to alter the DNA structure around intercalation sites. The positive bands were very pronounced with ctDNA and the GC polymer and much weaker for the AT homopolymer.
ELD
To determine the binding mode and orientation of the bound drug molecule to the long axis of DNA, we have used ELD. The ELD spectra of studied drugs bound to ctDNA are shown in Figure 4. These measurements were performed in a low ionic strength buffer (1 mM sodium cacodylate), as required for the ELD experiments due to the high electric field applied to the solution) at a high DNA–drug ratio (P/D = 20) to ensure that all drug molecules are bound to DNA. The intensity of the ELD signal is a function of the degree of alignment of the DNA molecules in the electric field and for this reason, the reduced dichroism (ΔA/A) values were measured at increasing field strength at 430 nm for C-1305 and C-1533 and 440 nm for m-AMSA (used as a reference acridine intercalator) are comparable to the one obtained for different types of DNA at 260 nm (Figure 4A). In all three cases, the reduced dichroism ΔA/A was strongly negative in the drug absorption band, which indicates an orientation of the chromophore perpendicular to the helix axis (Figure 4B). The higher ΔA/A values measured with the drug–DNA complexes compared to naked DNA reflects drug-induced stiffening effects which would favor the orientation of the DNA molecules in the electric field. This phenomenon is commonly seen with intercalating agents (34). Altogether, the ELD data suggest that the studied compounds are oriented parallel to the base pairs and strongly supports the intercalative binding mode, independent of the particular DNA substrate used (ct DNA, and both alternating homopolymers).
Figure 4.

ELD data for drug binding to DNA. Dependence of the reduced dichroism ΔA/A on the wavelength (A) and electric field strength (B) for DNA (open square), m-AMSA (closed square) and triazoloacridone derivatives C-1305 (open circle) and C-1533 (closed circle). Conditions: 13.6 kV/cm, P/D = 20 (200 µM DNA and 10 µM drug) (A) and ΔA/A was measured in absorption band of 440 nm for the DNA–amsacrine complexes (P/D = 20), 430 nm for the DNA–triazoloacridone derivative complexes (P/D = 20) and 260 nm for the DNA alone (B). All measurements were performed in 1 mM sodium cacodylate buffer, pH 7.0.
DNA-binding affinity
To evaluate the DNA-binding affinity of C-1305 a quantitative analysis of the drug–DNA interaction was performed by SPR (Figure 5) at constant temperature. Two 5′ biotin-labeled hairpin oligomers containing a [AT]4 or [GC]4 tract were immobilized on the sensor surface through streptavidin–biotin coupling and blank flow cell was used as a control (32). Representative sensogram at different concentrations of C-1305 binding to the GC duplex is shown in Figure 5A. In the association phase of the experiment, a solution containing increasing concentrations of the triazoloacridone derivatives (ranging from 0.01 to 25 µM) was introduced, and the progress of drug–DNA-binding was continuously monitored. The number of C-1305 molecules bound to the [AT]4 oligonucleotide is much lower than what is obtained for the [GC]4 oligonucleotide, and the RU value for the [AT]4 oligonucleotide is almost three times lower than that observed for the [GC]4 oligonucleotide (data not shown). Furthermore, C-1305 rapidly associated with DNA and formed stable drug–DNA complexes. After addition of the buffer, the drug–DNA complex rapidly dissociated. Compound C-1533 associated even more rapidly with DNA, formed less stable drug–DNA complexes that dissociated instantly (data not shown). The steady-state RU values obtained for each compound were fitted to the equations as described in Materials and Methods. The binding constants (K) are shown in the table on Figure 5B. The binding constants obtained for C-1305 clearly show a higher preference of C-1305 toward GC sequences. The binding constant for [GC]4 is almost 10 times higher than that measured for [AT]4 with 3.0 × 105 versus 4.9 × 104 (M−1). In contrast, the C-1533 derivative exhibits much weaker binding ability, with almost 10 times lower binding affinity constant for both [AT]4 and [GC]4 although some preference toward GC sequences could be observed (Figure 5B). Data obtained with SPR are in agreement with the results obtained with other techniques, where C-1305 displayed higher DNA-binding ability compared to C-1533.
Figure 5.

SPR sensograms (A) for binding of C-1305 to the [GC]4 DNA hairpin oligomer in HBS-EP buffer at 25°C. Table (B) represents equilibrium binding constants of C-1305 and C-1533 to [AT]4 and [GC]4 DNA.
DNase I footprinting for sequence selectivity of drug binding
DNase I footprinting is one of the classic methods to investigate the in vitro sequence-specific interaction of drug with DNA (35). We have compared the sequence selectivity of triazoloacridone derivatives with compound Hoechst 33258, a well-defined DNA minor groove binder with high specificity for AT sequences (36). In this study, a 176 bp DNA fragment from pBS plasmid DNA was used, covering a wide range of possible base pair combinations. For Hoechst 33258, we observed two main regions of DNA cleavage inhibition, mainly in AT-rich region (5′-GTTAA-3′ and 5′-CTTTTG-3′) (see Supplementary Figure 1). In contrast, both studied triazoloacridones gave no sequence-specific DNase I footprint. We could only observe a non-specific inhibition of DNase I activity with increasing concentrations of C-1305 (10–50 µM), whereas compound C-1533 had practically no effect.
Chemical probing
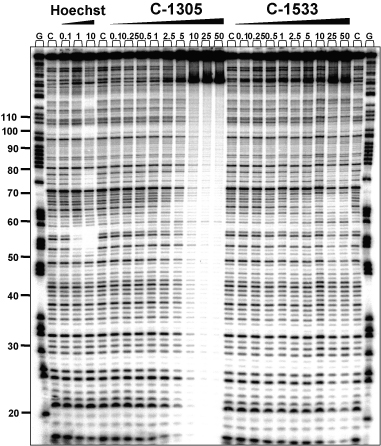
To further elucidate the potential sequence-specific interaction of C-1305 with purine-rich DNA regions, we carried out a series of chemical probing experiments with DEPC. Chemical probing of DNA with DEPC is based on diethylpyrocarbonate alkylation of the N7 residue of purines in the major groove of DNA, resulting in destabilization of phosphodiester bonds in the presence of piperidine (22). With increasing concentrations of C-1305, we observed a markedly increased DEPC reactivity in drug-containing lanes compared to drug-free lanes (Figure 6). At drug concentration as low as 100 nM, we could observe DEPC-associated DNA cleavage at DNA sites containing stretches of three consecutive guanines (positions from 44 to 65, modification sites are indicated in Figure 6 with arrows). In some cases, the drug-mediated enhanced reactivity of DEPC was observed at GG doublets (e.g. positions 25 and 33) but at these sites the band intensity is considerably lower than at the GGG-triplets. The effect was highly specific for C-1305 since the C-1533 triazoloacridone derivative did not produce similar changes in DNA. Higher DEPC reactivity in G-rich DNA regions induced by C-1305 was dose-dependent, and the maximum effect was observed at 50 µM. Comparative studies with a series of 12 other DNA topoisomerase II inhibitors revealed that none of the tested compounds were able to induce DNA cleavage in guanine triplets (Figure 7). We identified a group of compounds with the ability to specifically disturb DNA (bisantrene, TAS-103, echinomycin and to a lesser extent, mitoxantrone), but the DNA cleavage profile (see schematic below the gel in Figure 6) was completely different from the one obtained for C-1305.
Figure 6.

DEPC reactivity on the 176 bp DNA fragment. Experiments were done in the presence of increasing concentrations of C-1305 and C-1533. Digestion products were resolved on 8% polyacrylamide gel containing 7 M urea. Control tracks contained no drug. Numbers on the left side of the gel refer to the standard numbering scheme for the nucleotide sequence of the DNA fragment based on comparison to the position of the guanines on guanine-specific track. Arrows indicate dose-dependent sequence-specific interaction of C-1305 in guanine-rich regions of DNA. G, guanine track; D, DNA unmodified; C, control DNA.
Figure 7.

Effect of DNA topoisomerase inhibitors on DEPC reactivity toward the 176 bp DNA fragment. Digestion products were resolved on 8% polyacrylamide gel containing 7 M urea. Control tracks contained no drug. Numbers on the left side of the gel refer to the standard numbering scheme for the nucleotide sequence of the DNA fragment based on comparison to the position of the guanines on guanine-specific track.
We also performed chemical probing with osmium tetraoxide/pyridine that cleaves DNA specifically at thymines and detects changes in DNA structure at AT-rich sequences. We did not observe any enhanced reactivity of the osmium tetraoxide/pyridine complex toward DNA for either C-1305 or C-1533 (data not shown).
Affinity of C-1305 to different DNA substrates
To clarify whether induction of structural changes in DNA structure by C-1305 is associated with its higher affinity to DNA containing G-triplets, we performed melting temperature studies and equilibrium microdialysis experiments with different DNA substrates. We specifically designed 7 oligonucleotides (24 bp long), with comparable GC content of ∼50%, which either contained G-triplets with different neighboring nucleotides or ‘mutated’ G-triplets (see Table 2 for oligonucleotide sequences). Both melting temperature studies as well as microdialysis measurements showed no major differences in DNA affinity of C-1305 to studied oligonucleotides (Figure 8). It follows that the induction of structural changes in guanine triplets is not directly associated with higher affinity of C-1305 to guanine-rich DNA.
Figure 8.

(A) Sequence-dependent stabilization of the DNA secondary structure in the presence of C-1305. The graph represents variation of the melting temperature differences ΔTm (, in degrees) determined for complexes between C-1305 and different DNA substrates (see Table 2 for oligonucleotide sequences) in BPES buffer pH 7.1. Gray and black bars correspond to drug–DNA ratios of 0.5:1 and 1:1, respectively. (B) Sequence-dependent binding affinity of C-1305 obtained by the competition microdialysis assay in BPES buffer. All experiments were carried out in duplicates at least twice. Results are presented as means ± SD.
Structure–activity relationship studies for a series of acridones
We then wanted to define the structural elements that are responsible for the unusual ability of C-1305 to induce structural changes in DNA at guanine triplets. To this end, we selected a set of nine acridone derivatives that differ in their chemical structures. There were four major differences in the chemical structures of the studied compounds: (i) changes in the aminoalkylamino side chain (a number of methylene groups and substituent R1); (ii) substituent R2 in position 8 of the acridone ring; (iii) presence or absence of the methyl group at the nitrogen atom in position 5 of the acridone ring (R3); and (iv) conversion of triazolo to imidazo ring condensed with the acridone chromophore (X). The chemical structures of all studied compounds are shown in Table 1.
We compared the sequence-specific interaction of triazolo- and imidazoacridone compounds with the aforementioned 176 bp DNA fragment by chemical probing with DEPC. As could be seen in Figure 9A, only the C-1305 derivative effectively increased the reactivity of DNA with DEPC at guanine triplets compared to drug-free lanes. We also compared the relative effect induced by different compounds by densitometry analysis of band intensity corresponding to guanine 46 which is the central guanine of the guanine triplet (indicated with an arrow in Figure 9B). This analysis showed ∼20-fold increase in DNA cleavage induced by C-1305 and only minor effect of three other tested compounds. These compounds include two triazoloacridones C-1297, C-1234 and imidazoacridone C-1371. These findings suggest that a hydroxyl group in position 8 of the acridone chromophore is crucial for the induction of specific changes in the DNA structure by C-1305, that result in increased reactivity with DEPC.
Figure 9.

Effect of triazolo- and imidazoacridone derivatives on DEPC reactivity toward a 176 bp DNA fragment (A) Optical density analysis of specific drug–DNA interaction sites. (B) Arrow indicates the position of the band on which the cleavage intensity for each compound was measured by densitometry. The graph represents an increase of the cleavage at guanine sites compared to control/untreated DNA.
A substantial fraction of compound C-1305 at pH > 7 is present as zwitterion, with a protonated terminal nitrogen atom in the side chain and a deprotonated hydroxyl group at position 8 of the chromophore (see Supplementary Figure 2). Therefore, we studied the influence of pH on drug-induced DEPC reactivity. At acidic pH (pH < 7), we could observe only very low DEPC reactivity at guanine triplets in the presence of C-1305 (Figure 10). In contrast, at pH 7 and higher, we observed increasing DEPC reactivity at guanine triplets of DNA incubated with C-1305. This could not be explained by altered DNA-binding affinity of C-1305 at different pH since we observed no apparent change in stabilization of the secondary DNA structure by C-1305 with increasing pH as revealed by DNA melting temperature studies (data not shown). DNA alone did not show increased DEPC reactivity at different pH since DNA cleavage was observed in the absence of drug.
Figure 10.

Influence of pH on DEPC reactivity toward a 176 bp DNA fragment incubated in the presence of C-1305 (A) Experiments were carried out in 10 mM Tris–HCl/NaCl buffer with increasing pH value (ranging from 5.55 to 9.0). Control tracks with DNA (D) were added for each pH value and contained no drug. G, guanine track; C, DNA unmodified; D, control DNA. 1, 20–1 and 20 µM C-1305. Optical density analysis of specific drug–DNA interaction sites. (B) Arrow indicates the position of the band on which the cleavage intensity for each compound was measured by densitometry. The graph represents an increase of the cleavage at guanine site over control/untreated DNA.
Molecular modeling
Based on our experimental data, we concluded that C-1305 binds to DNA and induces specific structural changes within guanine triplets. However, the exact position of C-1305 in the drug–DNA complex as well as the way the DNA structure is perturbed upon ligand binding is not known. Formally, the compound can intercalate outside (‘on top of’) the G-triplet or it can be localized inside the G-triplet. In addition, in both situations there are two possible orientations of the drug due to the asymmetry of the planar chromophore. We applied theoretical chemistry and molecular modeling analysis in order to: (i) determine the position of C-1305 inside the drug–DNA complex that is energetically favorable, (ii) analyze the structural changes that occur upon drug–DNA complex formation, and (iii) investigate the molecular basis of the unusual DEPC reactivity at N7 atom of the middle guanine of the G-triplet.
To compare the energetics of C-1305 binding at the two distinct drug–DNA interaction sites, the MM–PBSA free energy calculations were performed on time evolving data (MD). Calculations revealed a difference of ∼5 kcal/mol between the two studied complexes and in the more energetically favorable model, the drug was positioned inside the G-triplet. For this drug location, we analyzed structural changes in DNA induced by the intercalated drug within a G-triplet. Compound C-1305 induced stronger structural changes in guanine triplets at higher pH suggesting that drug protonation/deprotonation might be important for this drug-specific effect. Therefore, we compared structural changes induced by C-1305 with protonated terminal nitrogen atom of the side chain and the zwitterionic form of the drug, with both a protonated nitrogen in the side chain and a deprotonated hydroxyl group of the chromophore. Comparison of the structure of drug–DNA complexes for protonated and zwitterionic forms of C-1305 revealed significant differences. For C-1305, which is only protonated at the terminal nitrogen atom of the side chain, the hydroxyl group in the chromophore is able to form hydrogen bond with O4′ atom of deoxyribose in the ‘upper’ cytidine of the G-triplet (Figure 11a, arrow). In striking contrast, this is not possible for the zwitterionic form of C-1305 where the very same O4′ atom now forms hydrogen bond with the protonated terminal nitrogen atom of the side chain of C-1305 (Figure 11b, arrow). This hydrogen bonding leads to immobilization of the side chain in the minor groove and produces a substantial distortion in base pairing between ‘upper’ guanine and cytosine. Deprotonation of the hydroxyl group allows the drug chromophore to re-position in such a way that the nitrogen atoms of the triazole ring are aligned with the N7 atoms of the three guanines. Simulation with C-1305, which had protonated hydroxyl oxygen showed that the hydrogen bonding between OH group and O4′ atom of deoxyribose in cytidine, prevents the ligand to adopt the same position as the zwitterionic form of the compound (compare structures B and C in Figure 11). In both cases, drug intercalation leads to significant distortions of the phosphate-sugar backbone and widening of both the major and the minor DNA grooves in order to minimize conformational hindrances. Interestingly, for the zwitterionic form of C-1305 intercalated into DNA we observed a strong exposure to the solvent of the N7 atom of the middle guanine in the G-triplet (Supplementary Figure 3, structure B, arrow).
Figure 11.

Formation of the hydrogen bond between the O4′ atom of the ribose in the ‘upper’ cytidine (arrow) and deprotonated hydroxyl group at position 8 of C-1305 of the zwitterionic form of the drug (a) and protonated terminal nitrogen atom in the side chain of C-1305 (b). Arrow marks O4′ atom of deoxyribose in the ‘upper’ cytosine of the G-triplet. Structures of the DNA fragment without drug and C-1305:DNA complexes (A) where the compound is intercalated inside the G-triplet as a zwitterionic form (B) and with protonated terminal nitrogen of the side chain only (C). Guanine residues from the G-triplet are colored by name, nitrogen atoms N7 of guanines in blue. Compound C-1305 is shown in yellow with nitrogens from triazole ring are marked blue while hydroxyl oxygen is marked red, other nucleotides of the DNA fragment are indicated in cyan. The hydroxyl group at position 8 of the drug chromophore is marked with an arrow. The distribution of MEP on equipotential surfaces for the DNA (A′) and both studied C-1305:DNA complexes (B′ and C′). Colors correspond to potential values, negative potential at −5 kcal/mol is shown in red, positive potential at +5 kcal/mol is shown in blue.
Analysis of the MEP distribution in the major DNA groove upon drug intercalation showed that the N7 atoms of three consecutive guanines in the G-triplet generate a long patch of the negative potential (structure A′, Figure 11). Since the intercalated zwitterionic form of C-1305 locates its triazole ring in close proximity to these atoms, this leads to the production of even stronger local negative electrostatic potential within the major groove, which is not observed for C-1305 with a protonated hydroxyl oxygen at position 8 (compare structures A′, B′ and C′, Figure 11).
DISCUSSION
Among the antitumor drugs used in the treatment of human cancers, a significant portion is comprised of DNA interacting compounds. Search for new low molecular weight ligands that could specifically bind to particular DNA sequences has been carried out for years, in the hope that new therapeutics could be found that would selectively modulate aberrant gene expression. However, despite great advances in our understanding of ligand-DNA recognition, these efforts have so far been only partially successful and only relatively few compounds show high DNA sequence-specificity. Notable exceptions are at least some bis-intercalators and DNA alkylators [for recent review see (37)]. Compounds that bind selectively to a particular DNA sequence of three nucleotides or longer are sandramycin and luzopeptins, that recognize 5′-CATG sequence (38,39), as well as ditercalinium that shows specificity to 5′-CGCG sequence (40). Interestingly, aflatoxin has been shown to intercalate within G-triplets and to bind covalently at the middle guanine of the G-triplet (41).
Our previous studies have shown that C-1305 is an unusual inhibitor of DNA topoisomerase II in vitro and in tumor cells (3). At cytotoxic doses, C-1305 produces only low levels of unusually toxic cleavable complexes compared to classic topoisomerase II inhibitors such as m-AMSA. One of the possible explanations for this phenomenon could be that C-1305 stimulates formation of cleavable complexes in specific DNA regions resulting in the formation of DNA damage that is very toxic to cells. In the present study, we systematically characterized the interactions of C-1305 and other triazoloacridones with DNA in vitro. We were particularly interested in a possible sequence-specific DNA-binding of C-1305 or production of structural changes in DNA, that could explain the unusual biological properties of this compound. We here show that C-1305 binds to DNA with limited sequence-specificity but induces very unusual structural changes in DNA sequences containing guanine triplets. Comparison with 22 other topoisomerase inhibitors and DNA interacting agents showed that this structural perturbation was specific for the C-1305 compound. This finding is particularly interesting, since to the best of our knowledge, this property is unique among anticancer compounds that bind to DNA.
DEPC probing showed prominent changes in the secondary DNA structure within regions containing guanine triplets, but had no effect on adenine-rich sequences. DEPC reacts with the N7 atoms of purines in the major groove (21). The structural changes in DNA induced by C-1305 binding principally perturb the major groove geometry in DNA regions containing guanine triplets. It is also worth noting, that C-1305 did not produce significant DEPC reactivity within a run of 12 consecutive GC pairs or GAGG sequence, that are present in the studied DNA fragment. These findings strongly suggest that regions with consecutive guanine residues and not just purine-rich triplets are absolutely required for the induction of structural changes in DNA by C-1305. However, the most pronounced structural DNA perturbations induced by C-1305 occurred in GGG-triplets.
Based on molecular modeling analysis, we predict that C-1305 intercalates between the first or the last guanine in the stretch of three guanines, which leads to local unwinding of DNA and widening of both the minor and the major DNA grooves. These structural changes are associated with changes in the structure of the phosphate backbone and the positions of the guanine residues within the G-triplet, resulting in increased reactivity of the N7 guanine atoms with the chemical probe DEPC. Our model predicts that the most prominent change in the position of the N7 atom occurs at the middle guanine of the G-triplet. This is in full agreement with our experimental data where densitometric analysis indicated that the middle guanine is the most reactive to DEPC, compared to other guanine residues of the G-triplet. It is also possible that the strong and localized negative electrostatic potential, that is present in the major groove once the drug is intercalated within guanine triplet, may facilitate chemical reaction of carbethoxylation by DEPC.
A fundamental question is why C-1305, which is a relatively small molecular weight DNA intercalator, is able to produce such a specific effect on the secondary DNA structure within very defined regions i.e. guanine triplets. Mixed-function DNA intercalators, such as the studied triazoloacridone C-1305, with flat chromophore moiety combined with an aminoalkyl side chain, intercalate into DNA in the minor groove and cover a maximum of 2–3 nt with the side chain located in the minor groove. It appears that the specific effect of C-1305 within guanine triplets is not directly linked to a higher affinity of the drug to DNA, as revealed by DNA melting temperature studies and microdialysis experiments with DNA substrates containing three consecutive guanine residues. Based on the structure–activity relationship for a group of acridones used in our studies as well as molecular modeling studies we propose that unique properties of C-1305 result from its ability to combine specific structural features of a typical DNA intercalator with the synergistic contribution of electrostatic interactions of the charged (zwitterionic) form of C-1305.
We were able to define three structural features that are absolutely required for C-1305 to induce the unusual structural perturbation in guanine triplets. These include: (i) triazole ring in the chromophore; (ii) hydroxyl group at position 8 of the chromophore; and (iii) aminoalkyl side chain with three methylene groups and methyl groups at the terminal nitrogen atom. It should be noted that among all studied triazoloacridone derivatives this unique combination of structural features is only present in compound C-1305. Based on our results, we propose a molecular basis to explain the contribution of each of these three. First, when the drug chromophore intercalates between base pairs a sufficiently long side chain (with three methylene groups in the aminoalkyl chain) is needed to allow the chromophore to penetrate DNA helix and the triazole ring of the chromophore can interact within the major groove of DNA. Second, at physiological pH our compound possesses a fully protonated terminal nitrogen atom on the side chain and a substantial fraction of the drug (more than 50% of the total drug molecules at pH 7.9) is present as a zwitterion i.e. with a deprotonated hydroxyl group at position 8 of the chromophore. The presence of deprotonated hydroxyl group allows re-positioning of the chromophore within the drug–DNA complex and, when C-1305 is located within G-triplet, the triazole ring can become aligned with three N7 atoms of guanines. Third, the protonated terminal nitrogen of the side chain forms a hydrogen bond with O4′ atom of the ribose and relatively non-bulky substitutents, such as methyl groups at the terminal nitrogen of the side chain, do not obstruct this interaction. There is a very interesting question as to whether zwitterionic form of C-1305 can intercalate into DNA or if the deprotonation of the hydroxyl group occurs only after C-1305 becomes intercalated into DNA. We would expect that the presence of the negative charge on the chromophore in the zwitterionic form could lead to electrostatic repulsion during initial stages of DNA intercalation. Further studies are required to fully clarify the role of electrostatic interactions in DNA-binding by C-1305.
The ability of C-1305 to induce structural perturbations in guanine triplets could have important biological consequences. Many functional DNA regions in the human genome contain guanine triplets or guanine-rich sequences. These include telomeres, centromeres and GC-rich gene promoter regions. Therefore, it is possible that the structural changes induced upon C-1305 binding might interfere with functionality of these DNA regions. Vertebrate telomeric sequences contain guanine triplets and may fold into higher order structures, called G-quadruplexes (42). It is possible that compound C-1305 could specifically bind telomeric DNA and studies are under way in our laboratory to evaluate the affinity of the drug for telomeric sequences or G-quartet structures. Another possibility is that the affinity of regulatory proteins, such as transcription factors, may be altered when C-1305 is bound to promoter regions that contain guanine triplets due to changes both in the local DNA structure and distribution of MEP. This could lead to changes in gene expression profiles which would most likely be limited to those genes that are regulated by guanine-rich promoters. Finally, production of structural changes by C-1305 in specific DNA regions with guanine triplets might be associated with the inhibition of topoisomerase II activity by this compound. Structural changes in guanine triplets might lead to preferential stabilization of cleavable complexes in these regions which, in turn, could result in region-specific DNA cleavage in telomeres, centromeres or guanine-rich promoters. Interestingly, etoposide has been shown to induce DNA damage in both telomeric and centromeric DNA (43,44).
Together, here we report that the antitumor triazoloacridone compound C-1305 binds to DNA and induces unusual structural changes specifically in DNA sequences containing guanine triplets. Comparison with 22 topoisomerase inhibitors and other DNA interacting agents, including a series of structurally related triazoloacridones showed that the structural perturbation of guanine-rich regions was specific for C-1305. We suggest that the structural perturbations induced by C-1305 play an important role in the cytotoxic and antitumor effects of this unique compound.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Supplementary Material
Acknowledgments
This work was supported by the Foundation for the Advancement of Polish Pharmacy and Medicine, the Ministry of Scientific Research and Information Technology, Poland, grant # 2P05F 015 27 and Association pour la Recherche sur le Cancer (ARC) Villejuif, France, grant no 4659, and the Institut de Recherches sur le Cancer de Lille (IRCL). K.L. was supported by a Marie Curie Fellowship from the European Community. Support by Academic Computer Center TASK in Gdansk (CPU time) and the ‘Actions intégrées Franco-Belge, Programme Tournesol’ is acknowledged. The authors are grateful to Dr Zofia Mazerska for communicating of data before publication and Jakub Olewniak for help with microdialysis measurements. We would also like to thank to an anonymous reviewer for prompting us to perform additional experiments which addressed the role of protonation of C-1305 in its DNA-binding. Funding to pay the Open Access publication charges for this article was provided by Ministry of Science and Information Society Technologies (Poland).
Conflict of interest statement. None declared.
REFERENCES
- 1.Cholody W.M., Martelli S., Konopa J. 8-Substituted 5-[(aminoalkyl)amino]-6H-v-triazolo[4,5,1-de]acridin-6-ones as potential antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 1990;33:2852–2856. doi: 10.1021/jm00172a028. [DOI] [PubMed] [Google Scholar]
- 2.Kusnierczyk H., Cholody W.M., Paradziej-Lukowicz J., Radzikowski C., Konopa J. Experimental antitumor activity and toxicity of the selected triazolo- and imidazoacridinones. Arch. Immunol. Ther. Exp. (Warsz.) 1994;42:415–423. [PubMed] [Google Scholar]
- 3.Lemke K., Poindessous V., Skladanowski A., Larsen A.K. The antitumor triazoloacridone C-1305 is a topoisomerase II poison with unusual properties. Mol. Pharmacol. 2004;66:1035–1042. doi: 10.1124/mol.104.000703. [DOI] [PubMed] [Google Scholar]
- 4.Wesierska-Gadek J., Schloffer D., Gueorguieva M., Uhl M., Skladanowski A. Increased susceptibility of poly(ADP-ribose) polymerase-1 knockout cells to antitumor triazoloacridone C-1305 is associated with permanent G2 cell cycle arrest. Cancer Res. 2004;64:4487–4497. doi: 10.1158/0008-5472.CAN-03-3410. [DOI] [PubMed] [Google Scholar]
- 5.Wang J.C. Cellular roles of DNA topoisomerases: a molecular perspective. Nature Rev. Mol. Cell. Biol. 2002;3:430–440. doi: 10.1038/nrm831. [DOI] [PubMed] [Google Scholar]
- 6.Wilstermann A.M., Osheroff N. Stabilization of eukaryotic topoisomerase II-DNA cleavage complexes. Curr. Top. Med. Chem. 2003;3:321–338. doi: 10.2174/1568026033452519. [DOI] [PubMed] [Google Scholar]
- 7.Larsen A.K., Escargueil A.E., Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 2003;99:167–181. doi: 10.1016/s0163-7258(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 8.Hande K.R. Clinical applications of anticancer drugs targeted to topoisomerase II. Biochim. Biophys. Acta. 1998;1400:173–84. doi: 10.1016/s0167-4781(98)00134-1. [DOI] [PubMed] [Google Scholar]
- 9.Larsen A.K., Skladanowski A. Cellular resistance to topoisomerase-targeted drugs: from drug uptake to cell death. Biochim. Biophys. Acta. 1998;1400:257–274. doi: 10.1016/s0167-4781(98)00140-7. [DOI] [PubMed] [Google Scholar]
- 10.Larsen A.K., Lemke K., Oestergaard V., Andersen A.H., Vekris A., Haaz M.C., Robert J., Escargueil A.E., Skladanowski A. Topoisomerases and transcriptional regulation. Eur. J. Cancer. 2002;38(suppl):S71. [Google Scholar]
- 11.Miassod R., Razin S.V., Hancock R. Distribution of topoisomerase II-mediated cleavage sites and relation to structural and functional landmarks in 830 kb of Drosophila DNA. Nucleic Acids Res. 1997;25:2041–2046. doi: 10.1093/nar/25.11.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binaschi M., Farinosi R., Borgnetto M.E., Capranico G. In vivo site specificity and human isoenzyme selectivity of two topoisomerase II-poisoning anthracyclines. Cancer Res. 2000;60:3770–3776. [PubMed] [Google Scholar]
- 13.Lovett B.D., Strumberg D., Blair I.A., Pang S., Burden D.A., Megonigal M.D., Rappaport E.F., Rebbeck T.R., Osheroff N., Pommier Y.G., et al. Etoposide metabolites enhance DNA topoisomerase II cleavage near leukemia-associated MLL translocation breakpoints. Biochemistry. 2001;40:1159–1170. doi: 10.1021/bi002361x. [DOI] [PubMed] [Google Scholar]
- 14.Collins I., Weber A., Levens D. Transcriptional consequences of topoisomerase inhibition. Mol. Cell. Biol. 2001;21:8437–8451. doi: 10.1128/MCB.21.24.8437-8451.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mondal N., Parvin J.D. DNA topoisomerase IIalpha is required for RNA polymerase II transcription on chromatin templates. Nature. 2001;413:435–438. doi: 10.1038/35096590. [DOI] [PubMed] [Google Scholar]
- 16.Bailly C., Helbecque N., Hénichart J.P., Colson P., Houssier C., Rao K.E., Shea R.G., Lown J.W. Molecular recognition between oligopeptides and nucleic acids. DNA sequence specificity and binding properties of an acridine-linked netropsin hybrid ligand. J. Mol. Recognit. 1990;3:26–35. doi: 10.1002/jmr.300030103. [DOI] [PubMed] [Google Scholar]
- 17.Colson P., Bailly C., Houssier C. Electric linear dichroism as a new tool to study sequence preference in drug binding to DNA. Biophys. Chem. 1996;58:125–140. doi: 10.1016/0301-4622(95)00092-5. [DOI] [PubMed] [Google Scholar]
- 18.Bailly C., Arafa R.K., Tanious F.A., Laine W., Tardy C., Lansiaux A., Colson P., Boykin D.W., Wilson W.D. Molecular determinants for DNA minor groove recognition: design of a bis-guanidinium derivative of ethidium that is highly selective for AT-rich DNA sequences. Biochemistry. 2005;44:1941–1952. doi: 10.1021/bi047983n. [DOI] [PubMed] [Google Scholar]
- 19.Bailly C., Waring M.J. Comparison of different footprinting methodologies for detecting binding sites for a small ligand on DNA. J. Biomol. Struct. Dyn. 1995;12:869–898. doi: 10.1080/07391102.1995.10508782. [DOI] [PubMed] [Google Scholar]
- 20.Bailly C., Kluza J., Martin C., Ellis T., Waring M.J. DNase I footprinting of small molecule binding sites on DNA. In: Herdewijn P., editor. Methods in Molecular Biology—Oligonucleotide Synthesis: Methods and Applications. Vol. 288. Totowa, NY: Humana Press Inc.; 2004. pp. 319–342. [DOI] [PubMed] [Google Scholar]
- 21.Bailly C., Gentle D., Hamy F., Purcell M., Waring M.J. Localized chemical reactivity in DNA associated with the sequence-specific bisintercalation of echinomycin. Biochem. J. 1994;300:165–73. doi: 10.1042/bj3000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailly C., Waring M.J. Diethylpyrocarbonate and osmium tetroxide as probes for drug-induced changes in DNA conformation in vitro. In: Fox K.R., editor. Drug–DNA Interaction Protocols. Vol. 90. NJ: Humana Press; 1997. pp. 51–79. [DOI] [PubMed] [Google Scholar]
- 23.Ren J., Chaires J.B. Sequence and structural selectivity of nucleic acid binding ligands. Biochemistry. 1999;38:16067–16075. doi: 10.1021/bi992070s. [DOI] [PubMed] [Google Scholar]
- 24.MacKerell A.D., Jr, Bashford D., Bellott M., Dunbrack R.L., Jr, Evanseck J.D., Field M.J., Fischer S., Gao J., Guo H., Ha S., et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 25.Kalé L., Skeel R., Bhandarkar M., Brunner R., Gursoy A., Krawetz N., Phillips J., Shinozaki A., Varadarajan K., Schulten K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 26.Hilal S.H., Karickhoff S.W., Carreira L.A. Athens, GA: U.S. Environmental Protection Agency; 2003. Verification and Validation of the SPARC Model. publication No. EPA/600/R-03/033. [Google Scholar]
- 27.Fogolari F., Tosatto S.C. Application of MM/PBSA colony free energy to loop decoy discrimination: towards correlation between energy and root mean square deviation. Protein Sci. 2005;14:889–901. doi: 10.1110/ps.041004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fogolari F., Moroni E., Wojciechowski M., Baginski M., Ragona L., Molinari H. MM/PBSA analysis of molecular dynamics simulations of bovine b-lactoglobulin: free energy gradients in conformational transitions? Proteins. 2005;59:91–103. doi: 10.1002/prot.20384. [DOI] [PubMed] [Google Scholar]
- 29.Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S., Karplus M. CHARMM: A program for macromolecular energy, minimization and dynamics calculations. J. Comput. Chem. 1983;4:187–217. [Google Scholar]
- 30.Madura J.D., Briggs J.M., Wade R.C., Davis M.E., Luty B.A., Ilin A., Antosiewicz J., Gilson M.K., Bagheri B., Scott L.R., et al. Electrostatics and diffusion of molecules in solution: Simulations with the University of Houston Brownian Dynamics Program. Comput. Phys. Commun. 1995;91:57–95. [Google Scholar]
- 31.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl Acad. Sci. USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Humphrey W., Dalke A., Schulten K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 33.Wilson W.D., Tanious F.A., Ding D., Kumar A., Boykin D.W., Colson P., Houssier C., Bailly C. Nucleic acid interactions of unfused aromatic cations: Evaluation of proposed minor-groove, major-groove and intercalation binding modes. J. Am. Chem. Soc. 1998;120:10310–10321. [Google Scholar]
- 34.Bonjean K., De Pauw-Gillet M.C., Defresne M.P., Colson P., Houssier C., Dassonneville L., Bailly C., Greimers R., Wright C., Quentin-Leclerq J., et al. The DNA intercalating alkaloid cryptolepine interferes with topoisomerase II and inhibits primarily DNA synthesis in B16 melanoma cells. Biochemistry. 1998;37:5136–5146. doi: 10.1021/bi972927q. [DOI] [PubMed] [Google Scholar]
- 35.Fox K.R. DNase I footprinting. In: Fox K.R., editor. Drug–DNA Interaction Protocols. vol. 90. NJ: Humana Press; 1997. pp. 1–22. [Google Scholar]
- 36.Brown P.M., Fox K.R. Minor groove binding ligands alter the rotational positioning of DNA fragments on nucleosome core particles. J. Mol. Biol. 1996;262:671–685. doi: 10.1006/jmbi.1996.0544. [DOI] [PubMed] [Google Scholar]
- 37.Tse W.C., Boger D.L. Sequence-selective DNA recognition: natural products and nature's lessons. Chem. Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Boger D.L., Ledeboer M.W., Kume M., Searcey M., Jin Q. Total synthesis and comparative evaluation of luzopeptin A–C and quinoxapeptin A–C. J. Am. Chem. Soc. 1999;121:11375–11383. [Google Scholar]
- 39.Boger D.L., Saionz K.W. DNA-binding properties of key sandramycin analogues: systematic examination of the intercalation chromophore. Bioorg. Med. Chem. 1999;7:315–321. doi: 10.1016/s0968-0896(98)00206-5. [DOI] [PubMed] [Google Scholar]
- 40.Pelaprat D., Delbarre A., Le Guen I., Roques B.P., Le Pecq J.B. DNA intercalating compounds as potential antitumor agents. 2. Preparation and properties of 7H-pyridocarbazole dimers. J. Med. Chem. 1980;23:1336–143. doi: 10.1021/jm00186a010. [DOI] [PubMed] [Google Scholar]
- 41.Shimada T., Guengerich F.P. Evidence for cytochrome P-450NF, the nifedipine oxidase, being the principal enzyme involved in the bioactivation of aflatoxins in human liver. Proc. Natl Acad. Sci. USA. 1989;86:462–465. doi: 10.1073/pnas.86.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williamson J.R. G-quartet structures in telomeric DNA. Annu. Rev. Biophys. Biomol. Struct. 1994;23:703–730. doi: 10.1146/annurev.bb.23.060194.003415. [DOI] [PubMed] [Google Scholar]
- 43.Yoon H.J., Choi I.Y., Kang M.R., Kim S.S., Muller M.T., Spitzner J.R., Chung I.K. DNA topoisomerase II cleavage of telomeres in vitro and in vivo. Biochim. Biophys. Acta. 1998;1395:110–120. doi: 10.1016/s0167-4781(97)00139-5. [DOI] [PubMed] [Google Scholar]
- 44.Floridia G., Zatterale A., Zuffardi O., Tyler-Smith C. Mapping of a human centromere onto the DNA by topoisomerase II cleavage. EMBO Rep. 2000;1:489–493. doi: 10.1093/embo-reports/kvd110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Facompre M., Carrasco C., Colson P., Houssier C., Chisholm J.D., Van Vranken D.L., Bailly C. DNA-binding and topoisomerase I poisoning activities of novel disaccharide indolocarbazoles. Mol. Pharmacol. 2002;62:1215–1227. doi: 10.1124/mol.62.5.1215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}