

Abstract
Single-nucleotide polymorphisms (SNPs) are rapidly replacing microsatellites as the markers of choice for genetic linkage studies and many other studies of human pedigrees. Here, we describe an efficient approach for modeling linkage disequilibrium (LD) between markers during multipoint analysis of human pedigrees. Using a gene-counting algorithm suitable for pedigree data, our approach enables rapid estimation of allele and haplotype frequencies within clusters of tightly linked markers. In addition, with the use of a hidden Markov model, our approach allows for multipoint pedigree analysis with large numbers of SNP markers organized into clusters of markers in LD. Simulation results show that our approach resolves previously described biases in multipoint linkage analysis with SNPs that are in LD. An updated version of the freely available Merlin software package uses the approach described here to perform many common pedigree analyses, including haplotyping and haplotype frequency estimation, parametric and nonparametric multipoint linkage analysis of discrete traits, variance-components and regression-based analysis of quantitative traits, calculation of identity-by-descent or kinship coefficients, and case selection for follow-up association studies. To illustrate the possibilities, we examine a data set that provides evidence of linkage of psoriasis to chromosome 17.
Introduction
Until recently, most genomewide linkage scans and other studies of human pedigrees relied on highly polymorphic microsatellite markers to track inheritance of chromosomal regions (Weber and Broman 2001). Microsatellites are highly informative, so that linkage scans using microsatellites require fewer markers (typically, ∼400–800 are used to cover the genome) than scans using less polymorphic markers, such as SNPs (Kruglyak 1997). Nevertheless, the role of microsatellites as the markers of choice for genomewide studies is changing. Technical advances have made rapid, accurate, and automated genotyping of very large numbers of SNP markers practical and inexpensive (Kwok 2001; Kennedy et al. 2003), and very large collections of SNP markers are now available (Sachidanandam et al. 2001), including some designed specifically for linkage studies (Matise et al. 2003; Shaw et al. 2004). These advances have been so substantial that it is now faster and more cost-effective to perform genomewide linkage scans with SNPs rather than with microsatellite markers (John et al. 2004; Middleton et al. 2004; Schaid et al. 2004), even after replacing each microsatellite with multiple SNPs.
These are welcome developments, since inexpensive genotyping technologies are necessary for detailed examination of the large data sets required for the identification of many complex-disease genes (Hirschhorn and Daly 2005). Extracting the maximum benefit from these new SNP data sets is likely to require that current tools for the analysis of human pedigrees be updated. For example, many of the proposed SNP linkage panels will include markers that are in linkage disequilibrium (LD) (Goddard and Wijsman 2002; Matise et al. 2003), but current linkage-analysis tools assume linkage equilibrium between markers. This assumption can lead to inaccurate results, especially when parental genotypes are missing (Schaid et al. 2002, 2004; Broman and Feingold 2004; Huang et al. 2004).
LD can be readily incorporated into the Elston-Stewart algorithm (Elston and Stewart 1971; Lathrop et al. 1984; Ott 1991; O’Connell 2000), but that algorithm is limited to the analysis of a relatively small number of genetic markers. Here, we describe a practical approach for incorporating marker-marker LD into multipoint analyses by use of the Lander-Green algorithm (Lander and Green 1987), which is commonly used for pedigree analyses with tens to thousands of markers. Our algorithm clusters tightly linked markers and uses haplotype frequencies to model LD within each cluster. A key issue in the use of our approach for the analysis of real data is the estimation of these haplotype frequencies. To address this issue, we describe a complementary gene-counting algorithm for efficiently estimating maximum-likelihood allele and haplotype frequencies in family data. Together, the methods described here are computationally efficient and allow marker-marker LD to be incorporated into many common pedigree analyses, including haplotyping and haplotype frequency estimation, parametric and nonparametric multipoint linkage analysis, variance-components and regression-based analysis of quantitative traits, calculation of identical-by-descent (IBD) or kinship coefficients, case selection for follow-up association studies (Fingerlin et al. 2004), and relationship inference.
Methods
Linkage Analysis with Clustered Markers
We consider the problem of accurately extracting multipoint inheritance information from an arbitrary pedigree. Following convention (Cannings et al. 1978; Kruglyak et al. 1996; Lange 1997), we divide individuals in a pedigree into a set of f founders, whose parents are not observed, and their n descendants. Our objective is to extract inheritance information by use of genotype data collected at a series of genetic markers for one or more of the individuals in the pedigree, even when some of the markers are in LD.
Our method assumes that markers can be organized into nonoverlapping clusters of consecutive markers, so that (1) markers in the same cluster may be in LD, (2) markers in different clusters will exhibit only low levels of LD, and (3) the recombination rate is extremely low within each cluster. To construct a computationally tractable solution, our model uses haplotype frequencies within each cluster to describe patterns of LD and makes two approximations: we ignore LD between markers in different clusters, and we assume that the recombination rate within each cluster is zero. Consequences of these approximations are examined in the “Results” and “Discussion” sections. In the following sections, we review the Lander-Green algorithm and provide further details of our approach.
The Lander-Green Algorithm
The first step of the Lander-Green algorithm is the enumeration of all possible inheritance vectors in a pedigree. Each inheritance vector denotes a possible pattern of segregation for founder alleles in the pedigree. Since there are 2n meiosis events in the pedigree, each with two possible outcomes (transmission of the maternal or the paternal allele), there will be up to 22n inheritance vectors (Lander and Green 1987). Typically, many of these will be indistinguishable and can be grouped to simplify calculations (Kruglyak et al. 1996; Gudbjartsson et al. 2000; Abecasis et al. 2002). Our algorithm for the analysis of clustered markers leaves this step unchanged.
The second step involves iterating over inheritance vectors and markers and calculating the probability of the observed genotypes for each marker conditional on a particular inheritance vector (Lander and Green 1987). Typically, this step of the calculation is performed by first identifying groups of connected founder alleles, then enumerating possible states for the founder alleles in each group, and, finally, calculating the probability of drawing each group of founder alleles from the population. A good description of the procedure is given by Sobel and Lange (1996), who use the term “genetic descent graph” rather than “inheritance vector.” Our algorithm affects this portion of the calculation: rather than iterate over markers, we iterate over clusters of markers in LD. Then, for each inheritance vector, we calculate the conditional probability of observed genotypes for all markers within the cluster conditional on estimated haplotype frequencies, which are used to model LD. Details are given in the next section.
The final step of the Lander-Green algorithm uses a Markov process to describe the joint distribution of inheritance vectors along a chromosome (Lander and Green 1987). This step relies on the observation that, under the assumption of no genetic interference, inheritance vectors form a hidden Markov chain. The matrices of transition probabilities between inheritance vectors at consecutive markers are a function of recombination fractions between markers. The Markov-chain calculations can be performed efficiently with either a divide-and-conquer algorithm (Idury and Elston 1997) or fast Fourier transform (Kruglyak and Lander 1998). This portion of the calculation is also left unchanged by our algorithm.
Figures 1 and 2 illustrate the various components of the likelihood calculation, with use of either the standard approach (fig. 1) or our proposed approach with clustering of neighboring markers that are in LD (fig. 2). In figure 2, all clusters include exactly two markers, but our implementation has no such restriction. Rather than iterating over inheritance vectors at each marker (v1…vK in fig. 1), we iterate over inheritance vectors at each cluster (vcluster1…vclusterN in fig. 2). In both approaches, the distribution of inheritance vectors along the chromosome is modeled with a hidden Markov model, with transition probabilities defined by the distance between markers in the standard approach and the distance between clusters in our approach. In either approach, probabilities of observed genotypes must be calculated conditional on a particular inheritance vector.
Figure 1.

Schematic representation of the standard Lander-Green algorithm.
Figure 2.

Schematic representation of the Lander-Green algorithm, with clustering of neighboring markers.
Probability of Observed Genotypes within a Cluster
Since our algorithm leaves unchanged the first step (enumeration of inheritance vectors) and the last step (hidden–Markov-chain calculation along a chromosome) of the Lander-Green algorithm, here we describe our strategy for implementing the second step, in which the probability of observed genotypes for each pedigree, conditional on estimated haplotype frequencies and an inheritance vector, is calculated for a cluster of markers in LD. For a cluster with M markers, let G1…GM denote the observed genotypes for each marker. Further, let h be the number of distinct haplotypes in the population and p1…ph denote their respective frequencies. Finally, for i=1…2f, let Hi denote the state of founder haplotype i. For each inheritance vector v, we wish to calculate the probability of observed genotypes at a particular cluster of markers, Pr(G1…GM|p1…pf,v), conditional on population haplotype frequencies.
One straightforward way to calculate this quantity is to iterate over founder haplotype sets and take the product of Pr(H1…H2f|p1…ph), the prior probability of each haplotype set, and Pr(G1…GM|H1…H2f,v), the conditional probability of observed genotypes, given a founder haplotype set and inheritance vector v. Pr(H1…H2f|p1…ph) is a simple product of haplotype frequencies. Since the inheritance vector v specifies the founder haplotypes carried by each individual, Pr(G1…GM|H1…H2f,v) is equal to 1 if the implied haplotypes for each individual are compatible with the observed genotypes and is zero otherwise. Thus:
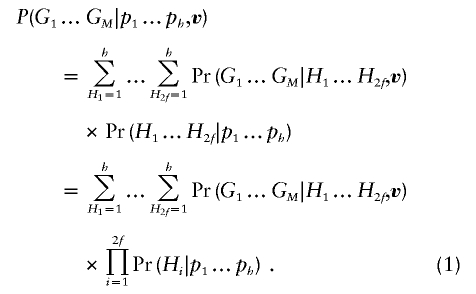 |
Although this implementation is straightforward, it is also extremely inefficient, since most founder haplotype sets typically will be incompatible with the observed genotype data and, therefore, most terms in the summation will be zero. A better way is to identify the set S(G1…GM,v) of founder haplotype configurations compatible with inheritance vector v and observed genotype data G1…GM. By definition, Pr(G1…GM|H1…H2f,v)=1 for all configurations in this set. Then, equation (1) can be replaced with a smaller sum of products of haplotype frequencies:
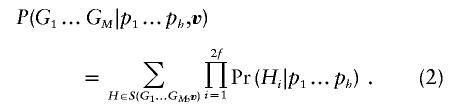 |
Identifying the Set of Compatible Haplotype Configurations
For any particular inheritance vector v and set of observed genotypes G1…GM, the list of compatible founder haplotypes can be quickly calculated as the Cartesian product of possible founder allele states at each marker. An effective procedure for listing possible founder allele states at a marker has been described in detail elsewhere (Sobel and Lange 1996), and here we provide only a short review for the sake of completeness. For each marker, proceed as follows: (1) using the inheritance vector v, assign two founder alleles to each individual; (2) identify connected components in the resulting genetic descent graph, where each founder allele is a vertex and an edge is drawn between pairs of founder alleles that are transmitted to the same genotyped individual; (3) generate a list of the zero, one, or two possible allele states for each of the connected components. When executing the final step, it is important to note that, although connected components can include any number of founder alleles, there will never be more than two possible allelic states for a set of connected founder alleles. In fact, there will be either (a) no compatible founder allele states if the genotype data are incompatible with the proposed inheritance vector and, therefore, P(G1…GM|p1…ph,v)=0; (b) one possible state if the component includes at least one homozygous individual or two individuals with different heterozygous genotypes; or (c) two possible states if all individuals connected by this component have the same heterozygous genotype. Alleles in components that do not include any genotyped individuals can be in any state. Once possible allele states for each component are determined, it is straightforward to identify compatible founder haplotype sets. Specifically, picking one of the possible states for each component will fix all founder allele states and produce one potential founder haplotype set. Furthermore, the Cartesian product of these sets is S(G1…GM,v), the set of all compatible founder haplotypes.
An example is given in figure 3. Figure 3A lists the observed genotypes for a hypothetical pedigree, whereas figure 3B gives the genetic descent graph corresponding to one potential inheritance vector. Figure 3C gives the founder allele graph resulting from the genotypes observed in 3A and the inheritance pattern specified in 3B. In this case, the pattern of missing data is the same at both markers, and the founder allele graph has two components, one with alleles A, C, E, and F and another with alleles B and D, whichever marker is considered. Let Bi denote the state of founder allele B for marker i. Then, for marker 1, there is only one set of possible states for the first component, (A1=1, C1=2, E1=1, F1=2), and two possible states for the second component, (B1=1, D1=2) and (B1=2, D1=1). For the second marker, there is again only one set of possible states for the first component, (A2=2, C2=2, E2=2, F2=2), but two possible states for the second component, (B2=1, D2=2) and (B2=2, D2=1). The Cartesian product of these sets {(A1=1, C1=2, E1=1, F1=2)} × {(B1=1, D1=2), (B1=2, D1=1)} × {(A2=2, C2=2, E2=2, F2=2)} × {(B2=1, D2=2), (B2=2, D2=1)} gives the list of four possible founder haplotype sets in figure 3D.
Figure 3.

Founder allele graphs used to identify possible haplotype states for a pedigree. A, Summary of the observed genotype data for two markers. B, Possible inheritance vector or genetic descent graph. Genotyped alleles are shown in gray, and the connections they induce between founder alleles are denoted with dashed lines. C, Representation of the founder allele graph corresponding to panels A and B. D, Possible haplotype states, calculated as the Cartesian product of possible states for each founder allele graph component.
Two additional savings are possible. If some haplotype frequency estimates are zero, then one can trim the list of founder haplotype sets to exclude configurations where at least one haplotype has zero frequency. This trimming can be done either after evaluating the full Cartesian product or, preferably, by evaluating the product gradually and removing partial configurations that require a haplotype with zero frequency after performing each multiplication. We have implemented the latter, more efficient approach. Since only configurations that include haplotypes with zero frequency are removed, this computational savings does not affect the result of likelihood calculations.
A second savings is possible when iterating over many distinct inheritance vectors, as in the Lander-Green algorithm. This additional savings reduces the number of times that the summation in equation (2) must be used to evaluate P(G1…GM|p1…ph,v). For each inheritance vector vj, we check whether the set of compatible founder haplotypes is identical to that identified for one of the previously evaluated inheritance vectors—that is, whether there is a k < j for which S(G1…GM,vk)≡S(G1…GM,vj). If a match is found, we reuse the previously calculated value P(G1…GM|p1…ph,vk) instead of re-evaluating equation (2). The check is computationally inexpensive and takes <2% of the time required to list possible founder haplotype configurations and calculate their probabilities. Even when many inheritance vectors are compatible with the observed genotype data for a cluster of markers, we find that the number of distinct founder haplotypes sets is much smaller and that the savings resulting from this simple check can be quite useful.
Estimation of Haplotype Frequencies in General Pedigrees
The approach described in the previous section requires founder haplotype frequencies for each cluster as input. Although estimating haplotype frequencies in a set of unrelated individuals is now straightforward (Excoffier and Slatkin 1995; Stephens et al. 2001), estimating maximum-likelihood haplotype frequencies in pedigrees is more challenging. Here, we outline a gene-counting–based expectation-maximization (EM) algorithm (Ceppellini et al. 1955; Dempster et al. 1977) that can be used to estimate haplotype frequencies within each cluster. The approach is applicable to nuclear families and other small pedigrees. The exact constraints on the algorithm depend not only on pedigree size but also on the number of markers being considered and the informativeness of the marker data. The algorithm performs fastest on pedigrees for which founder haplotype configurations have little uncertainty (for example, when founder genotypes are available) and performs slower when founder haplotype configurations are more uncertain (for example, when founder genotypes are missing and sibships are small). We find that our algorithm can typically handle ∼20 markers per cluster in pedigrees with ∼20 individuals. Our algorithm also allows for allele frequency estimation and, for pedigrees of modest size, executes much faster than standard algorithms based on numerical optimization of the likelihood (Boehnke 1991).
As with other gene-counting strategies for allele and haplotype frequency estimation, our algorithm involves two basic steps. First, conditional on current haplotype frequency estimates, the expected number of copies of each haplotype in the sample is calculated. Next, these expected counts are used to generate a new set of haplotype frequency estimates. After updating haplotype frequencies and estimated counts in turn several times, the process converges to maximum-likelihood estimates of haplotype frequencies. Adequate convergence can be verified by repeating the process with different initial guesses for haplotype frequencies.
The key step in implementing this gene-counting–based EM algorithm is evaluating the expected number of copies of each haplotype in each pedigree, conditional on current haplotype frequency estimates. For each haplotype k=1…h, let 0⩽nk(H1…H2f)⩽2f be the number of copies of haplotype k among founder haplotypes H1…H2f. Typically, founder haplotypes are not observed directly, so that calculating the expected number of copies of haplotype k, conditional on observed genotypes G1…GM and allele frequencies, requires summing over all configurations that are compatible with the observed genotype data and weighting each configuration by its probability. Thus,
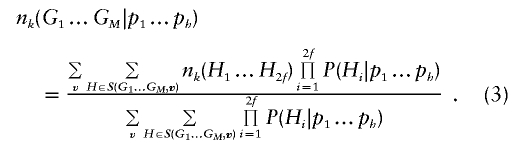 |
Although this formulation is sufficient for implementing an EM algorithm for haplotype frequency estimation in family data, we again note that many inheritance vectors will produce identical sets of compatible founder haplotypes. Thus, it is advantageous to group these vectors and reduce the number of terms in the sums above. Thus, we define a nonredundant subset of inheritance vectors U={j:∀i<j,S(G1…GM,vj)≠S(G1…GM,vi)}, so that each vector in U produces a different list of compatible founder haplotypes. For each of these vectors we define a list of equivalent inheritance vectors Ej={i:S(G1…GM,vj)≡S(G1…GM,vi)} and a weight wj=|Ej|, which is simply the number of equivalent inheritance vectors. Equation (3) now becomes
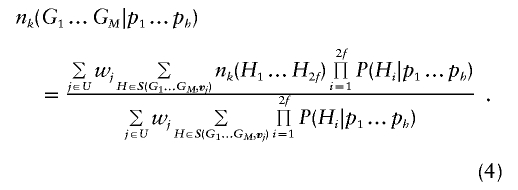 |
With this formula, the expected number of copies of a particular haplotype in any family can be calculated quickly. This quantity can be summed over all families and divided by the total number of founder haplotypes in the sample to update estimated haplotype frequencies and proceed with the gene-counting–based EM algorithm.
Implementation
We have implemented our methods for multipoint analysis with clustered markers and for haplotype frequency estimation in the Merlin package (Abecasis et al. 2002). Since these methods enhance the underlying hidden Markov model for multipoint analysis, they naturally extend to all the analyses performed by Merlin, including haplotyping and haplotype frequency estimation, parametric and nonparametric multipoint linkage analysis, variance-components and regression-based analysis of quantitative traits (Sham et al. 2002), calculation of IBD or kinship coefficients, and case selection for follow-up association studies (Fingerlin et al. 2004).
Our implementation can handle user-specified clusters or, alternatively, can automatically identify clusters with a simple criteria based on pairwise r2 or intermarker distance thresholds. The r2 criterion groups markers for which pairwise r2 exceeds a predefined threshold, together with intervening markers, into a cluster. The distance criterion groups markers that are close together into a cluster, without taking marker-marker LD into account. By default, allele frequencies within each cluster are calculated with a single run of the EM algorithm as described in the previous section, but user-defined haplotype frequencies can also be accommodated. Although our method assumes that the recombination fraction within clusters is zero, real data will sometimes include obligate recombinants within a cluster of markers in LD. For families in which an obligate recombinant is observed within a cluster, our implementation automatically flags the “problematic” genotypes and treats them as missing. Since recombination events between markers in LD should be extremely rare, only a very small fraction of the data should be treated in this manner. Our approach is not appropriate when there is substantial recombination between markers in LD within the available pedigrees.
Simulations
To evaluate the performance of our approach, we analyzed a series of simulated data sets. Each data set consisted of a series of affected sibships, each with two, three, or four affected siblings and zero, one, or two genotyped parents (for a total of nine different family configurations). We fixed the total number of genotyped individuals at 2,000 in each data set and adjusted the number of simulated sibships accordingly. For example, data sets with two affected sibs and no genotyped parents included 1,000 affected sibships each, whereas those with four affected sibs and two genotyped parents included only 333 sibships. We simulated genotypes for a SNP linkage mapping panel covering chromosome 13 (∼100 cM) and used HapMap information to determine realistic levels of marker-marker LD for nearby markers (The International HapMap Consortium 2003). Briefly, we first selected 100,000-bp windows along the heterochromatic portion of chromosome 13, with window centers separated by 5,000,000 bp. Next, within each window, we defined parameters for a cluster of SNPs in LD by using HapMap data for the CEPH Utah panel to select three SNPs with minor-allele frequency >5% and to estimate their corresponding haplotype frequencies. This resulted in a total of 20 clusters and 59 SNPs (one window included only 2 SNPs, and 3 SNPs were selected from each of the remaining 19 windows). Each cluster included an average of 3.7 common haplotypes (frequency >5%). Average pairwise r2 was 0.227 for markers within the same cluster, with 9% of within-cluster marker pairs in complete LD (r2=1.0) and 14%, 17%, and 26% of within-cluster marker pairs exceeding r2 thresholds of 0.80, 0.50, and 0.20, respectively. We then used the estimated haplotype frequencies to generate founder haplotypes for use in gene-dropping simulations (Ott 1991). Note that this simulation does not generate LD between clusters. When segregating SNPs through the pedigree, we assumed a recombination rate of 10−8 per bp (corresponding to the genomewide average of ∼1 cM per 1,000,000 bp [Yu et al. 2001]). For comparison purposes, we also repeated our simulations with the within-cluster recombination rate set to zero and/or with a larger window size of 500,000 bp. We performed simulations both under the null, with no linked genetic effect simulated, and under the alternative, with use of a multiplicative model corresponding to sibling recurrence risk λs=1.08 (disease-allele frequency of 0.10; penetrances of 0.01, 0.02, and 0.04 for genotypes with 0, 1, and 2 copies of the susceptibility allele, respectively). We calculated Kong and Cox LOD scores for each data set with the use of the linear model (Kong and Cox 1997) and the Spairs statistic (Whittemore and Halpern 1994). We considered three analysis strategies: (1) modeling LD for clusters of tightly linked markers, as described in the previous sections; (2) using a naive approach that ignores LD between markers; or (3) selecting one SNP from each cluster for analysis, thereby focusing on a set of “independent” SNPs that are in linkage equilibrium. To evaluate the informativeness of these SNP linkage maps, we also simulated microsatellite data for markers separated by recombination fractions of 0.05 and 0.10 (∼5 cM and 10 cM) for comparison. Each simulated microsatellite had four alleles of equal frequency (heterozygosity of 75%).
Applied Example: Analysis of a Map Including SNPs and Microsatellites
Our exemplary data set consists of the psoriasis data of Stuart et al. (2005). The data were collected to examine evidence for linkage and association between psoriasis and chromosome 17q (a locus originally suggested by Tomfohrde et al. [1994]). The data consist of 3,158 individuals in 274 families recruited in Germany and the United States, each with ⩾2 individuals with psoriasis. Genotypes were available for up to 21 individuals per family, corresponding to a total of 2,598 genotyped individuals. Genotype data were available for a total of 32 microsatellites and six SNPs, including data from an initial linkage scan and markers selected for fine mapping. Clusters were defined such that markers in a cluster have a standardized multiallelic disequilibrium coefficient D′ (Hedrick 1987) of at least 0.3 and a nominal χ2 contingency table P value of <.001. Disequilibrium coefficients were calculated with GOLD (Abecasis and Cookson 2000).
Results
Simulated Data
In our simulations, we evaluated three alternative strategies for analyzing the simulated SNP linkage data: (1) we ignored LD and used a standard implementation of the Lander-Green algorithm; (2) we modeled marker-marker LD within clusters with the strategy described in the “Methods” section; or (3) we selected a subset of “independent” SNPs for analysis, retaining a single SNP from each cluster and discarding the others. Tables 1 and 2 summarize the performance of the three strategies for 5,000 simulated data sets generated under the null hypothesis (i.e., when no linked genetic effect was simulated and the analyzed sibships exhibited only random sharing). Table 1 presents results with no missing data among genotyped individuals, and table 2 includes 5% missing data among genotyped individuals.
Table 1.
Comparison of Different Analysis Strategies under the Null Hypothesis, without Missing Data[Note]
|
Average LOD |
Information Content |
Significance Threshold for α=.05a |
|||||||
| Analysis Strategy | Ignore LD | Model LD | Independent SNPs | Ignore LD | Model LD | Independent SNPs | Ignore LD | Model LD | Independent SNPs |
| No parents genotyped: | |||||||||
| 2 sibs per family | 1.762 | −.005 | −.004 | .413 | .394 | .247 | 13.65 | 1.33 | 1.22 |
| 3 sibs per family | 2.971 | .003 | .002 | .547 | .537 | .358 | 20.16 | 1.34 | 1.27 |
| 4 sibs per family | 2.470 | −.007 | −.008 | .646 | .641 | .452 | 16.30 | 1.26 | 1.20 |
| One parent genotyped: | |||||||||
| 2 sibs per family | .586 | −.002 | −.004 | .710 | .705 | .470 | 6.06 | 1.40 | 1.27 |
| 3 sibs per family | .686 | −.004 | −.006 | .786 | .785 | .568 | 6.34 | 1.31 | 1.22 |
| 4 sibs per family | .484 | −.002 | −.002 | .832 | .833 | .635 | 4.51 | 1.37 | 1.31 |
| Two parents genotyped: | |||||||||
| 2 sibs per family | −.001 | −.001 | −.001 | .804 | .806 | .608 | 1.46 | 1.45 | 1.32 |
| 3 sibs per family | −.003 | −.003 | −.002 | .837 | .838 | .654 | 1.44 | 1.44 | 1.36 |
| 4 sibs per family | .003 | .003 | .004 | .858 | .859 | .687 | 1.41 | 1.42 | 1.32 |
Note.— We fixed the number of genotyped individuals at 2,000. When neither parent was genotyped, this resulted in 1,000, 666, and 500 families with 2, 3, and 4 affected siblings, respectively. When one parent was genotyped, this resulted in 666, 500, and 400 families with 2, 3, and 4 affected siblings, respectively. When both parents were genotyped, this resulted in 500, 400, and 333 families, with 2, 3, and 4 affected siblings, respectively.
In simulated ∼100-cM chromosome.
Table 2.
Comparison of Different Analysis Strategies under the Null Hypothesis, with 5% Missing Data
|
Average LOD |
Information Content |
Significance Threshold for α=.05a |
|||||||
| Analysis Strategy | Ignore LD | Model LD | Independent SNPs | Ignore LD | Model LD | Independent SNPs | Ignore LD | Model LD | Independent SNPs |
| No parents genotyped: | |||||||||
| 2 sibs per family | 1.539 | −.004 | −.004 | .397 | .378 | .230 | 11.81 | 1.29 | 1.23 |
| 3 sibs per family | 2.699 | .003 | .002 | .531 | .521 | .336 | 18.11 | 1.34 | 1.23 |
| 4 sibs per family | 2.380 | −.006 | −.007 | .631 | .627 | .426 | 15.54 | 1.26 | 1.20 |
| One parent genotyped: | |||||||||
| 2 sibs per family | .549 | −.003 | −.004 | .683 | .680 | .432 | 5.40 | 1.40 | 1.29 |
| 3 sibs per family | .682 | −.004 | −.007 | .769 | .769 | .534 | 6.18 | 1.31 | 1.23 |
| 4 sibs per family | .508 | −.001 | −.001 | .820 | .821 | .603 | 4.62 | 1.38 | 1.25 |
| Two parents genotyped: | |||||||||
| 2 sibs per family | .032 | −.001 | −.001 | .781 | .784 | .561 | 1.54 | 1.43 | 1.32 |
| 3 sibs per family | .027 | −.002 | −.004 | .823 | .825 | .619 | 1.52 | 1.41 | 1.35 |
| 4 sibs per family | .024 | .003 | .003 | .849 | .850 | .659 | 1.49 | 1.40 | 1.32 |
In simulated ∼100-cM chromosome.
In each table, the first three columns summarize average estimated Kong and Cox (1997) LOD scores, calculated by use of the linear model (Kong and Cox 1997). Limited analyses with the exponential model produced similar results (data not shown). The expected LOD under the null is zero because a negative sign was arbitrarily assigned to the statistics when less-than-expected sharing was observed. It is clear that, when parental genotypes are not available, ignoring marker-marker LD produces noticeable biases. The bias is severe when both parents are missing (average LOD > 1.5 for sibships with 2, 3, or 4 affected siblings) and is still large when only one parent is genotyped (average LOD > 0.5 for sibships with 2, 3, or 4 affected siblings). In contrast, both our clustering strategy for modeling LD and selection of independent SNPs perform correctly and produce average LOD scores of ∼0, whether or not parental genotypes are available.
The next three columns of tables 1 and 2 summarize information content (calculated with use of the entropy of the inheritance vector distribution [Kruglyak et al. 1996]). It appears that ignoring marker-marker LD produces slightly inflated estimates of information content, whereas discarding genotypes for two SNPs per cluster and retaining only independent markers significantly lowers information content. As expected, information content is higher when genotypes for one parent are available and is even higher when genotypes for both parents are available.
The final set of three columns summarizes thresholds corresponding to a 5% significance level in these simulation experiments (i.e., exceeded in only 5% of simulated chromosomes). When parental genotypes are not available and marker-marker LD is ignored, very large LOD scores can occur in the absence of linkage (Huang et al. 2004), and very high significance thresholds must be employed (e.g., >11 when both parents are missing). If parental genotypes are available, modeling LD between markers is less important, but even 5% missing data among genotyped individuals can produce inflated LOD scores and significance thresholds (table 2). When marker-marker LD is modeled, or when independent markers are selected, significance thresholds are much lower and vary only slightly with sibship size and the proportion of parental genotypes available. As expected, significance thresholds for multipoint analysis in each family configuration increase slightly with information content (for a discussion of related issues, see Kruglyak and Daly [1998]), and, thus, estimated thresholds are slightly higher (1) when data are complete for genotyped individuals than when some genotypes are missing at random; (2) when parental genotypes are available than when they are missing; and, finally, (3) when LD is modeled than when independent markers are selected for analysis. In all three cases, the more informative setting requires slightly higher thresholds.
Next, using the empirical significance thresholds discussed above and listed in the final three columns of tables 1 and 2, we evaluated power for the three analysis strategies. Again, we simulated data sets with 2,000 genotyped individuals and a trait locus with disease-allele frequency 0.10 and penetrances 0.01, 0.02, and 0.04 (corresponding to λsib≈1.08). The trait locus was simulated at 62.5 cM and in linkage equilibrium with genotypes for the two flanking clusters (one at ∼60.0 cM and another at ∼65.0 cM). The relative performance of the three analysis strategies was similar when larger effect sizes were simulated (data not shown). Figure 4 gives representative results for one of the simulated data sets. Note the two extremely high LOD score peaks that result when LD between markers is ignored (LOD of 6.75 at ∼20 cM and LOD of 4.72 at ∼70 cM). When LD between markers is modeled, the higher peak completely disappears. A single peak persists (LOD of 2.92 at 75 cM) close to the position of the simulated disease locus at 62.5 cM. In both cases, trait locus genotypes were hidden during calculation of linkage statistics, and the trait locus is in linkage equilibrium with all the genotyped markers.
Figure 4.

Analysis of affected sibship data with one parent genotyped, with and without the modeling of LD. This simulated data set included 500 sibships, each with three affected siblings and one genotyped parent.
Results from 5,000 simulations are summarized in two tables, one corresponding to simulations with no missing data for genotyped individuals (table 3) and the other corresponding to 5% missing data (table 4). The first three columns of each table summarize the average peak LOD score in chromosomes with a simulated disease locus. The next three columns summarize empirical power. It is clear that, although ignoring marker-marker LD produces the largest LOD scores, it also produces the lowest power. LOD scores are randomly inflated by LD between markers and lose the ability to discriminate evidence for linkage. It appears that, although selecting one marker from each cluster is preferable to ignoring marker-marker LD, modeling disequilibrium is the best option, providing greater power in all cases when some parental genotypes are missing and similar power to the other strategies when all parental genotypes are available.
Table 3.
Comparison of Different Analysis Strategies under the Alternative Hypothesis, without Missing Data
|
Average Peak LOD |
Power for α=.05 |
|||||
| Analysis Strategy | Ignore LD | Model LD | Independent SNPs | Ignore LD | Model LD | Independent SNPs |
| No parents genotyped: | ||||||
| 2 sibs per family | 9.774 | .965 | .818 | .094 | .261 | .227 |
| 3 sibs per family | 15.912 | 1.705 | 1.274 | .125 | .568 | .428 |
| 4 sibs per family | 13.498 | 2.932 | 1.997 | .189 | .886 | .718 |
| One parent genotyped: | ||||||
| 2 sibs per family | 3.644 | .882 | .740 | .081 | .199 | .179 |
| 3 sibs per family | 4.630 | 1.557 | 1.234 | .162 | .517 | .423 |
| 4 sibs per family | 4.574 | 2.693 | 2.062 | .466 | .815 | .690 |
| Two parents genotyped: | ||||||
| 2 sibs per family | .864 | .864 | .760 | .170 | .172 | .169 |
| 3 sibs per family | 1.489 | 1.488 | 1.296 | .444 | .444 | .400 |
| 4 sibs per family | 2.494 | 2.494 | 2.140 | .780 | .777 | .724 |
Table 4.
Comparison of Different Analysis Strategies under the Alternative Hypothesis, with 5% Missing Data
|
Average Peak LOD |
Power for α=.05 |
|||||
| Analysis Strategy | Ignore LD | Model LD | Independent SNPs | Ignore LD | Model LD | Independent SNPs |
| No parents genotyped: | ||||||
| 2 sibs per family | 8.280 | .950 | .796 | .094 | .268 | .217 |
| 3 sibs per family | 14.126 | 1.673 | 1.234 | .128 | .555 | .426 |
| 4 sibs per family | 12.857 | 2.885 | 1.923 | .192 | .880 | .700 |
| One parent genotyped: | ||||||
| 2 sibs per family | 3.234 | .869 | .723 | .084 | .192 | .168 |
| 3 sibs per family | 4.501 | 1.535 | 1.196 | .162 | .511 | .394 |
| 4 sibs per family | 4.618 | 2.661 | 1.977 | .454 | .804 | .685 |
| Two parents genotyped: | ||||||
| 2 sibs per family | .923 | .852 | .735 | .166 | .170 | .163 |
| 3 sibs per family | 1.562 | 1.476 | 1.252 | .445 | .452 | .387 |
| 4 sibs per family | 2.548 | 2.470 | 2.060 | .765 | .775 | .696 |
In the simulations described above, cluster boundaries were known without error (that is, analyses used the same cluster boundaries used to generate the data). We also repeated analysis by calculating cluster boundaries with the use of pairwise r2 measures estimated from the available data. For each data set, we defined clusters to include any pair of markers for which pairwise r2 exceeded 0.10 or 0.20, together with all intervening markers. In this setting, we typically recovered ∼15 (with r2 threshold of 0.10) or ∼12 (with r2 threshold of 0.20) of the original 20 simulated clusters. Some of the estimated clusters included only two markers, rather than the original three. In this setting, we observed a slight bias in average LOD scores (e.g., when no parents were genotyped, average LOD scores were ∼0.18 when an r2 threshold of 0.20 was used and ∼0.07 when an r2 threshold of 0.10 was used). Although the results illustrate that even low levels of unaccounted-for marker-marker LD can inflate LOD scores, they also illustrate that even imperfect knowledge of cluster boundaries can resolve the majority of the bias resulting from marker-marker LD in multipoint linkage analysis.
Finally, we simulated microsatellite mapping panels composed of markers with four equally frequent alleles (75% heterozygosity) distributed ∼5 cM or ∼10 cM apart. Table 5 summarizes observed information content and power for the microsatellite panels. It is clear that clusters of three SNPs in LD spaced ∼5 cM apart provide higher information content, higher expected LOD scores, and higher empirical power than microsatellite markers spaced either 5 cM or 10 cM apart. Within each cluster, SNPs were selected at random among HapMap SNPs with frequency >5% and average minor-allele frequency of ∼24%. Also, note that SNP scans have higher power despite the fact that empirical significance thresholds were slightly lower for microsatellite scans than for scans with clustered SNPs (table 5, footnote).
Table 5.
Comparison of SNP and Microsatellite (Short Tandem Repeat Polymorphism [STRP]) Maps
|
Average Peak LOD |
Information Content |
Power for α=.05a |
|||||||
| Analysis Strategy | Clustered SNPs | 5-cMSTRPs | 10-cMSTRPs | Clustered SNPs | 5-cMSTRPs | 10-cMSTRPs | Clustered SNPs | 5-cMSTRPs | 10-cMSTRPs |
| No parents genotyped: | |||||||||
| 2 sibs per family | .965 | .835 | .691 | .394 | .304 | .197 | .261 | .222 | .204 |
| 3 sibs per family | 1.705 | 1.448 | 1.029 | .537 | .431 | .288 | .568 | .533 | .386 |
| 4 sibs per family | 2.932 | 2.312 | 1.452 | .641 | .537 | .371 | .886 | .795 | .553 |
| One parent genotyped: | |||||||||
| 2 sibs per family | .882 | .778 | .626 | .705 | .534 | .349 | .199 | .205 | .159 |
| 3 sibs per family | 1.557 | 1.371 | 1.021 | .785 | .652 | .449 | .517 | .478 | .384 |
| 4 sibs per family | 2.693 | 2.342 | 1.646 | .833 | .728 | .522 | .815 | .782 | .654 |
| Two parents genotyped: | |||||||||
| 2 sibs per family | .864 | .790 | .638 | .806 | .710 | .506 | .172 | .160 | .157 |
| 3 sibs per family | 1.488 | 1.425 | 1.146 | .838 | .756 | .557 | .444 | .485 | .393 |
| 4 sibs per family | 2.494 | 2.434 | 1.895 | .859 | .787 | .594 | .777 | .784 | .695 |
Empirical significance thresholds for the STRP and SNP scans were determined by analyzing 5,000 data sets generated under the null. Thresholds for the scan with clustered SNPs are given in table 1 (under the heading “Model LD”). For the 5-cM STRP scan, thresholds for families with 2, 3, and 4 affected siblings were 1.28, 1.15, and 1.16 for those with no parents genotyped, 1.25, 1.24, and 1.24 for those with one parent genotyped, and 1.41, 1.29, and 1.34 for those with two parents genotyped. For the 10-cM STRP scan, thresholds for families with 2, 3, and 4 affected siblings were 1.14, 1.10, and 1.11 for those with no parents genotyped, 1.17, 1.04, and 1.06 for those with one parent genotyped, and 1.21, 1.20, and 1.14 for those with two parents genotyped.
Applied Example with Psoriasis Data: Analysis of a Map Including SNPs and Microsatellites
This data set also includes a mixture of different pedigree structures, including several two-, three-, and four-generation pedigrees, each with up to 30 individuals. There are a total of 2,598 genotyped individuals. In these data, genotypes collected for the original microsatellite genome scan were augmented with markers selected for fine mapping, and, thus, the example illustrates the ability of our method to handle data sets including both SNP and microsatellite markers (with 6–13 alleles). We identified four clusters of markers in LD (defined as multiallelic D′ > 0.3). Two of these clusters included only microsatellite markers (two each), another cluster included only SNPs (three), and the final cluster included three SNPs and one microsatellite marker. Estimating haplotype frequencies for these data took <2 min, whereas completing a full multipoint nonparametric analysis took ∼50 min. The results are summarized in figure 5. Using the NPLALL scoring statistic, we observed a peak Kong and Cox (1997) LOD score of 3.21 (gray line in fig. 5) at ∼126 cM when LD between markers was ignored. A second peak, corresponding to a LOD score of 2.61, was observed at ∼145 cM. These two peaks correspond to the two clusters of candidate SNPs selected for fine mapping. When LD between markers was modeled using our clustering approach (dark line in fig. 5), the peak LOD score decreased to 2.73 at ∼128 cM, and there was no second peak at ∼145 cM. Thus, although there is good evidence for linkage of psoriasis to the chromosome 17q locus (peak LOD = 2.73 in this data set), results show that the signal can be inflated when LD between markers is not modeled, even if the data include only a few markers in LD. Although association of SNPs in this region with psoriasis had been reported previously, Stuart et al. (2005) report that there is no evidence for association between psoriasis and any of the SNPs selected for fine mapping in their data. As a result, we expect that changes in LOD scores reflect marker-marker LD, rather than any effects of trait-marker LD.
Figure 5.

Analysis of exemplary psoriasis data set
Discussion
We have described a practical method for handling LD between markers in pedigree analysis. Our method is general and can be incorporated into parametric and nonparametric multipoint linkage analysis of discrete traits, variance-components and regression-based analyses of quantitative traits (Sham et al. 2002), calculation of IBD or kinship coefficients, case selection for follow-up association studies (Fingerlin et al. 2004), and many other likelihood-based pedigree analyses. In addition, we have described a companion EM algorithm for rapid estimation of allele and haplotype frequencies in pedigrees of modest size. These advances will facilitate the use of high-throughput SNP data in studies of human pedigrees.
The arrival of low-cost, high-throughput SNP genotyping presents human geneticists with exciting possibilities but also generates some novel analytical challenges. We show that when LD between available SNP markers is appropriately modeled, SNP linkage panels can outperform standard microsatellite mapping panels in studies of affected sibships. This is an important advance, but we expect that low-cost, rapid SNP genotyping will not only facilitate traditional analysis of human pedigrees but also enable new gene-mapping approaches. For example, genomewide association scans are now within reach (Abecasis et al. 2005), and promising new gene-mapping approaches that use pedigree data to model LD between genotyped markers and unobserved disease alleles are being developed (Cantor et al. 2005; Li et al. 2005).
Our simulation results and analysis of an exemplary data set emphasize the importance of modeling marker-marker LD in pedigree analysis. For example, we show that ignoring marker-marker LD can lead to severe biases in LOD score calculations for affected sibships and that these biases are resolved when our approach is used. Our method also resolves inflation in multipoint estimates of IBD and kinship coefficients (data not shown). When modeling LD between markers with the methods described here is impractical, we recommend that a set of markers that are approximately in linkage equilibrium should be selected for analysis.
How to organize available markers into clusters is an important practical question. Sometimes, the genotyped SNPs will fall naturally into clusters of tightly linked markers separated by gaps with no SNPs, but this will not always be the case. In general, we recommend that a liberal approach be taken when grouping markers into clusters, so that even modest evidence for LD between markers (say r2>0.10) should lead to markers being placed in the same cluster. We are actively comparing different automated strategies for grouping markers that appear to be in LD with each other (G. R. Abecasis, J. E. Wigginton, M. Boehnke, and R. Pruim, unpublished data).
For computational convenience, our method makes two important assumptions: (1) that there is no recombination within clusters and (2) that there is no LD between clusters. If clusters are relatively small (<0.1 cM in most of our simulations), the assumption of no recombination within clusters causes a small fraction of genotypes to be discarded and produces no noticeable bias in LOD score calculations. When measured in terms of the recombination fraction, clusters are unlikely to grow very large because very low recombination rates are required to maintain substantial levels of LD in human populations. Nevertheless, we repeated our simulations with larger 500-kb windows for each cluster of three markers (corresponding to a recombination rate of ∼0.005 per cluster per generation within simulated pedigrees). Again, this produced no significant change in our conclusions, since (1) our clustering approach still produced LOD scores of ∼0 on average under the null, (2) information content was increased compared with situations in which we focused on a subset of independent markers, and (3) modeling of LD within clusters remained the most powerful analysis strategy.
The effect of LD between clusters is potentially more serious and will depend on the approach used to define the clusters. If there is substantial LD between clusters, we expect that biases in LOD scores for affected sib pair analyses and other statistics will result. Fortunately, in our experience, the patchy nature of LD in the genome (e.g., see Abecasis et al. 2001b; Dawson et al. 2002) means that there are often natural breakpoints in LD that lead to very little disequilibrium between clusters.
We used an EM algorithm to estimate haplotype frequencies within clusters. The method can comfortably handle haplotypes of 10–20 SNPs per cluster in small-sized and medium-sized pedigrees, but it is not practical for handling very large numbers of SNPs within a cluster. In principle, large clusters (>20 SNPs) can be handled with a divide-and-conquer strategy (Abecasis et al. 2001a; Qin et al. 2002), in which haplotype frequencies are first estimated for small stretches with fewer markers.
We have implemented our methods for multipoint analysis with clustered markers and for haplotype frequency estimation in the Merlin package (Abecasis et al. 2002). Our implementation is freely available and, in addition to handling user-defined clusters, includes simple automated algorithms for grouping markers into clusters before analysis. We hope it will enable researchers to more fully realize the benefits of high-throughput SNP genotyping technologies.
Acknowledgments
This work was made possible by research grants from the National Human Genome Research Institute and GlaxoSmithKline. We are indebted to Mike Boehnke, Randy Pruim, and Phil Stuart, for many helpful discussions and comments on early versions of the manuscript. We thank Phil Stuart, J. T. Elder, and Rajan Nair, for making the psoriasis chromosome 17 fine-mapping data available. Finally, we are grateful to the many users of Merlin who have helped us to improve the program by providing helpful feedback and code.
Web Resources
The URL for data presented herein is as follows:
- Merlin, Center for Statistical Genetics, http://www.sph.umich.edu/csg/abecasis/Merlin/
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Cookson WOC (2000) GOLD—graphical overview of linkage disequilibrium. Bioinformatics 16:182–183 10.1093/bioinformatics/16.2.182 [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Ghosh D, Nichols TE (2005) Linkage disequilibrium: ancient history drives the new genetics. Hum Hered 59:118–124 10.1159/000085226 [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Martin R, Lewitzky S (2001a) Estimation of haplotype frequencies from diploid data. Am J Hum Genet 69:S198 [Google Scholar]
- Abecasis GR, Noguchi E, Heinzmann A, Traherne JA, Bhattacharyya S, Leaves NI, Anderson GG, Zhang Y, Lench NJ, Carey A, Cardon LR, Moffatt MF, Cookson WO (2001b) Extent and distribution of linkage disequilibrium in three genomic regions. Am J Hum Genet 68:191–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehnke M (1991) Allele frequency estimation from data on relatives. Am J Hum Genet 48:22–25 [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Feingold E (2004) SNPs made routine. Nat Methods 1:104–105 10.1038/nmeth1104-104 [DOI] [PubMed] [Google Scholar]
- Cannings C, Thompson EA, Skolnick MH (1978) Probability functions on complex pedigrees. Adv Appl Probab 1:26–61 [Google Scholar]
- Cantor RM, Chen GK, Pajukanta P, Lange K (2005) Association testing in a linked region using large pedigrees. Am J Hum Genet 76:538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceppellini R, Siniscalco M, Smith CAB (1955) The estimation of gene frequencies in a random-mating population. Ann Hum Genet 20:97–115 [DOI] [PubMed] [Google Scholar]
- Dawson E, Abecasis GR, Bumpstead S, Chen Y, Hunt S, Beare DM, Pabial J, et al (2002) A linkage disequilibrium map of chromosome 22. Nature 418:544–548 10.1038/nature00864 [DOI] [PubMed] [Google Scholar]
- Dempster A, Laird N, Rubin D (1977) Maximum likelihood from incomplete data via the E-M algorithm. J R Stat Soc Ser B 39:1–38 [Google Scholar]
- Elston RC, Stewart J (1971) A general model for the genetic analysis of pedigree data. Hum Hered 21:523–542 [DOI] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M (1995) Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- Fingerlin TE, Boehnke M, Abecasis GR (2004) Increasing the power and efficiency of disease-marker case-control association studies through use of allele-sharing information. Am J Hum Genet 74:432–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard KA, Wijsman EM (2002) Characteristics of genetic markers and maps for cost-effective genome screens using diallelic markers. Genet Epidemiol 22:205–220 10.1002/gepi.0177 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 10.1038/75514 [DOI] [PubMed] [Google Scholar]
- Hedrick PW (1987) Gametic disequilibrium measures: proceed with caution. Genetics 117:331–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6:95–108 10.1038/nrg1521 [DOI] [PubMed] [Google Scholar]
- Huang Q, Shete S, Amos CI (2004) Ignoring linkage disequilibrium among tightly linked markers induces false-positive evidence of linkage for affected sib pair analysis. Am J Hum Genet 75:1106–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idury RM, Elston RC (1997) A faster and more general hidden Markov model algorithm for multipoint likelihood calculations. Hum Hered 47:197–202 [DOI] [PubMed] [Google Scholar]
- John S, Shephard N, Liu G, Zeggini E, Cao M, Chen W, Vasavda N, Mills T, Barton A, Hinks A, Eyre S, Jones KW, Ollier W, Silman A, Gibson N, Worthington J, Kennedy GC (2004) Whole-genome scan, in a complex disease, using 11,245 single-nucleotide polymorphisms: comparison with microsatellites. Am J Hum Genet 75:54–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy GC, Matsuzaki H, Dong S, Liu WM, Huang J, Liu G, Su X, Cao M, Chen W, Zhang J, Liu W, Yang G, Di X, Ryder T, He Z, Surti U, Phillips MS, Boyce-Jacino MT, Fodor SP, Jones KW (2003) Large-scale genotyping of complex DNA. Nat Biotechnol 21:1233–1237 10.1038/nbt869 [DOI] [PubMed] [Google Scholar]
- Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L (1997) The use of a genetic map of biallelic markers in linkage studies. Nat Genet 17:21–24 10.1038/ng0997-21 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ (1998) Linkage thresholds for two-stage genome scans. Am J Hum Genet 62:994–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Lander ES (1998) Faster multipoint linkage analysis using Fourier transforms. J Comput Biol 5:1–7 [DOI] [PubMed] [Google Scholar]
- Kwok PY (2001) Methods for genotyping single nucleotide polymorphisms. Annu Rev Genomics Hum Genet 2:235–258 10.1146/annurev.genom.2.1.235 [DOI] [PubMed] [Google Scholar]
- Lander ES, Green P (1987) Construction of multilocus genetic linkage maps in humans. Proc Natl Acad Sci USA 84:2363–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange K (1997) Mathematical and statistical methods for genetic analysis. Springer, New York [Google Scholar]
- Lathrop GM, Lalouel J, Julier C, Ott J (1984) Strategies for multilocus linkage in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Boehnke M, Abecasis GR (2005) Joint modeling of linkage and association: identifying SNPs responsible for a linkage signal. Am J Hum Genet 76:934–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise TC, Sachidanandam R, Clark AG, Kruglyak L, Wijsman E, Kakol J, Buyske S, et al (2003) A 3.9-centimorgan-resolution human single-nucleotide polymorphism linkage map and screening set. Am J Hum Genet 73:271–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton FA, Pato MT, Gentile KL, Morley CP, Zhao X, Eisener AF, Brown A, Petryshen TL, Kirby AN, Medeiros H, Carvalho C, Macedo A, Dourado A, Coelho I, Valente J, Soares MJ, Ferreira CP, Lei M, Azevedo MH, Kennedy JL, Daly MJ, Sklar P, Pato CN (2004) Genomewide linkage analysis of bipolar disorder by use of a high-density single-nucleotide-polymorphism (SNP) genotyping assay: a comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am J Hum Genet 74:886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR (2000) Zero-recombinant haplotyping: applications to fine mapping using SNPs. Genet Epidemiol 19:S64–S70 [DOI] [PubMed] [Google Scholar]
- Ott J (1991) Analysis of human genetic linkage. Johns Hopkins University Press, Baltimore [Google Scholar]
- Qin ZS, Niu T, Liu JS (2002) Partition-ligation–expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am J Hum Genet 71:1242–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, et al (2001) A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409:928–933 10.1038/35057149 [DOI] [PubMed] [Google Scholar]
- Schaid DJ, Guenther JC, Christensen GB, Hebbring S, Rosenow C, Hilker CA, McDonnell SK, Cunningham JM, Slager SL, Blute ML, Thibodeau SN (2004) Comparison of microsatellites versus single-nucleotide polymorphisms in a genome linkage screen for prostate cancer-susceptibility loci. Am J Hum Genet 75:948–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid DJ, McDonnell SK, Wang L, Cunningham JM, Thibodeau SN (2002) Caution on pedigree haplotype inference with software that assumes linkage equilibrium. Am J Hum Genet 71:992–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham PC, Purcell S, Cherny SS, Abecasis GR (2002) Powerful regression-based quantitative-trait linkage analysis of general pedigrees. Am J Hum Genet 71:238–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw SS, Oliphant A, Shen R, McBride C, Steeke RJ, Shannon SG, Rubano T, Bahram GK, Fan JB, Chee MS, Hansen MST (2004) A highly informative SNP linkage panel for human genetic studies. Nat Methods 1:113–117 10.1038/nmeth712 [DOI] [PubMed] [Google Scholar]
- Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337 [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart P, Nair RP, Abecasis GR, Nistor I, Hiremagalore R, Chia NV, Qin ZS, Thompson RA, Jenisch S, Weichenthal M, Janiga J, Lim HW, Christophers E, Voorhees JJ, Elder JT (2005) Analysis of RUNX1 binding site and RAPTOR polymorphisms in psoriasis: no evidence for association despite adequate power and evidence for linkage. J Med Genet (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium (2003) The International HapMap Project. Nature 426:789–796 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- Tomfohrde J, Silverman A, Barnes R, Fernandez-Vina MA, Young M, Lory D, Morris L, et al (1994) Gene for familial psoriasis susceptibility mapped to the distal end of human chromosome 17q. Science 264:1141–1145 [DOI] [PubMed] [Google Scholar]
- Weber JL, Broman KW (2001) Genotyping for human whole-genome scans: past, present, and future. Adv Genet 42:77–96 [DOI] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50:118–127 [PubMed] [Google Scholar]
- Yu A, Zhao C, Fan Y, Jang W, Mungall AJ, Deloukas P, Olsen A, Doggett NA, Ghebranious N, Broman KW, Weber JL (2001) Comparison of human genetic and sequence-based physical maps. Nature 409:951–953 10.1038/35057185 [DOI] [PubMed] [Google Scholar]