

Abstract
Marker-trait association analysis is an important statistical tool for detecting DNA variants responsible for genetic traits. In such analyses, an analysis model of the mean genetic effects of the genotypes is often specified. For instance, the effect of the disease allele on the trait is often specified to be dominant, recessive, additive, or multiplicative. Although this model-based approach is powerful when the analysis model is correctly specified, it has been found to have low power sometimes when the specified model is incorrect. We introduce an approach that does not require the specification of a particular genetic model. This approach is built upon a constrained maximum likelihood in which the mean genetic effect of the heterozygous genotype is required to not exceed those of the two homozygous genotypes. The asymptotic distribution of the likelihood-ratio statistic is derived for two special cases. A simulation study suggests that this new approach has power comparable to that of the model-based method when the analysis model is correctly specified. This approach uses one marker at a time (i.e., it is a single-marker analysis). However, given the latest findings that powerful inferential procedures for haplotype analyses can be constructed from single-marker analyses, we expect this approach to be useful for haplotype analyses.
Introduction
Marker-trait association analysis is an important statistical tool for detecting DNA variants responsible for genetic traits. It can provide higher mapping resolution than do methods based on closely related meiosis events. In such analyses, it is common to specify an analysis model of the average genetic effects of the genotypes. For instance, the effect of the disease allele on the trait is often specified to be dominant, recessive, additive, or multiplicative. When the specified model is close to the underlying trait model, this model-based approach provides a powerful means of detecting association. However, when the specified model is different from the underlying model, its power may be low (Slager and Schaid 2001; Freidlin et al. 2002; Schaid et al. 2005). In real-data analyses, the underlying genetic model is often unknown. For instance, one promising approach to investigation of the gene-regulation mechanism is to map the expression levels of genes by treating them as quantitative traits (Brem et al. 2002; Schadt et al. 2003; Yvert et al. 2003; Morley et al. 2004). Currently, gene-expression arrays contain thousands of DNA probes, and each probe provides a quantitative measurement of its expression level. Given this large number of traits, the model-based method may miss many true-positive signals. It is desirable to develop methods that have power for a wide range of genetic models.
Analytical methods that have power for a wide range of genetic models are desired not only for single-marker analysis but also for haplotype analysis in which multiple markers are involved. Some recent developments indicate that single-marker tests can be used to construct powerful inference procedures for haplotype analysis. Chapman et al. (2003) found that regression analysis based on a linear combination of tagSNPs is more powerful than the traditional haplotype analysis that is based on haplotype frequencies. Roeder et al. (2005) further found that inferential procedures based on single-marker tests, after correction for multiple testing by permutation or curve fitting, is at least as powerful as the regression method proposed by Chapman et al. (2003). In a more recent study, Schaid et al. (2005) described a testing procedure for haplotype analysis that requires specification of a “kernel.” One promising way of specifying a kernel is to derive it from single-marker tests (Schaid et al. 2005).
There have been some studies, mostly on dichotomous traits, that have investigated methods that do not rely on a particular analysis model. Freidlin et al. (2002) proposed a maximin efficiency robust test and a test (named “MAX”) based on the maximum of test statistics under several analysis models. They found that the MAX test is generally more powerful than the other one. Freidlin et al. (2002) assumed an a priori ordering of the mean genetic effects for the three genotypes that are induced from the allele to be tested, by assuming that the marker allele associated with the disease allele is known. Such an ordering can be difficult to make—for instance, in the expression-level mapping example mentioned above. To remove this restriction, Zheng (2003) proposed a “max and min scores” approach. Another method that does not require the specification of an analysis model is to simply compare mean genotypic effects by use of standard statistical methods such as analysis of variance. But such an approach may have low power due to increased degrees of freedom.
Instead of deriving tests from several model-based statistics or simply comparing the mean genotypic effects, we adopt a constrained-likelihood analysis under a so-called no-overdominance constraint. This constraint requires that the mean genetic effect of the heterozygous genotype not exceed those of the two homozygous genotypes—that is, it is neither larger than the larger of the mean genetic effects of the two homozygous genotypes nor smaller than the smaller of the two. We note that this constraint is satisfied by the commonly assumed analysis models—dominance, recessive, additive, and multiplicative.
In the following section, we introduce our approach in terms of a generalized linear model. We then apply this approach to quantitative traits and dichotomous traits. The asymptotic distribution of the constrained likelihood-ratio statistic for each kind of trait is introduced. Simulation studies were conducted to assess the performance of our method, under different generating models, in comparison with some popular methods. Technical details are given in appendixes A and B.
Methods
Let A denote the allele being tested for association with a trait. The three genotypes induced from allele A are indexed by j (j=0,1,2), where j is the number of copies of allele A. Denote the population frequency of genotype j by pj. In some situations, the values of pj are known. For instance, for the F2 population, p0=p2=0.25 and p1=0.5. Suppose that there are nj individuals with genotype j. Given the total number of individuals n:=n0+n1+n2, the triplet (n0,n1,n2) follows a trinomial distribution with a parameter vector (p0,p1,p2). The trait value of the ith individual of genotype j is denoted by yji. For dichotomous traits, it is defined that yji=1 for cases and yji=0 for controls. The sample mean for genotype j is denoted by 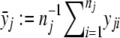 .
.
Consider the following generalized linear model for phenotype y with link function g(·): E(y)=μ and g(μ)=α+δ1x1+(δ1+δ2)x2, where xj, j=1,2, is an indicator of genotype j satisfying xj=1 if the individual is of genotype j and xj=0 otherwise. Depending on the random component of the generalized linear model, there may be another nuisance parameter β (possibly a vector). For instance, for normal data, β is the variance of the random component. In the constrained-likelihood approach introduced here, we require a “no-overdominance” constraint on the three mean genotypic effects. That is, the three genotypic means satisfy either α⩽α+δ1⩽α+δ1+δ2 or α⩾α+δ1⩾α+δ1+δ2. This constraint implies that δ1 and δ2 cannot be of different signs, and it can be equivalently written δ1δ2⩾0, which corresponds to the first and third quadrants on the δ1-δ2 plane. When there is no association for allele A, δ1=δ2=0. Let Θ0={(δ1,δ2,α,β):δ1=δ2=0} and Θ1={(δ1,δ2,α,β):δ1δ2⩾0}. The hypotheses of interest are
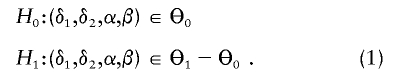 |
The other parameters p0, p1, p2, α, and β are nuisance parameters. The requirement δ1δ2⩾0 contains many commonly used genetic models as special cases. For instance, when the effect of allele A is dominant, we have δ2=0, and there is no restriction on δ1. When the effect of allele A is recessive, we have δ1=0, and there is no restriction on δ2.
The likelihood function of the data {yji} is
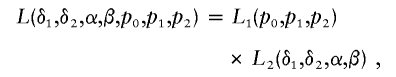 |
where L1(p0,p1,p2)=Pr(n1,n2,n3|p0,p1,p2,n) is the trinomial probability of (n1,n2,n3), given n, and where L2(δ1,δ2,α,β)=j=02i=1njPr(yji|δ1,δ2,α,β) is the conditional probability of the trait values {yji}, given (n1,n2,n3).
The hypotheses in (1) can be tested using the likelihood-ratio statistic. Since L1(p0,p1,p2) and L2(δ1,δ2,α,β) involve two nonoverlapping sets of parameters and since only L2(δ1,δ2,α,β) contains the parameters of interest, the likelihood-ratio statistic equals
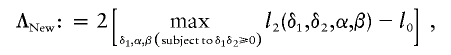 |
where l2(δ1,δ2,α,β)=log[L2(δ1,δ2,α,β)] and 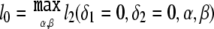 . The nuisance parameters p0, p1, and p2 do not appear in the calculation of ΛNew. However, we show below that the asymptotic distribution of ΛNew can depend on p0, p1, and p2.
. The nuisance parameters p0, p1, and p2 do not appear in the calculation of ΛNew. However, we show below that the asymptotic distribution of ΛNew can depend on p0, p1, and p2.
To compute ΛNew, it is essential to compute the constrained maximum of l2(δ1,δ2,α,β). For this purpose, define
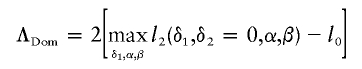 |
to be the likelihood-ratio statistic for the dominance model and
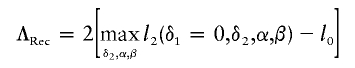 |
to be the likelihood-ratio statistic for the recessive model. Further define ΛLarger=max{ΛDom,ΛRec}. In principle, the constrained maximum of l2(δ1,δ2,α,β) is straightforward to compute. According to standard optimization theory, there are two possible situations regarding the optimal values of δ1 and δ2. They are either in the interior of the region satisfying δ1δ2>0 or on the border of this region. The constrained maximum of l2(δ1,δ2,α,β) equals its unconstrained maximum in the former case and equals ΛLarger in the latter case.
Specifically, the constrained maximum of l2(δ1,δ2,α,β) can be obtained as follows. Do the unconstrained maximization of l2(δ1,δ2,α,β), and denote the values of δ1, δ2, α, and β for which l2(δ1,δ2,α,β) is maximized by 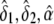 , and
, and 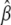 , respectively. The constrained maximum of l2(δ1,δ2,α,β) equals
, respectively. The constrained maximum of l2(δ1,δ2,α,β) equals 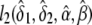 if
if 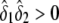 and equals ΛLarger otherwise.
and equals ΛLarger otherwise.
Next, we discuss two particular applications of this constrained-likelihood approach, one to quantitative traits and the other to dichotomous traits. For each application, the asymptotic distribution of the likelihood-ratio statistic ΛNew is derived. There should be many other possible applications, depending on the specification of the random component and the link function of the generalized linear model.
Quantitative Traits
Quantitative traits are usually modeled through a normal distribution, and the “canonical” link function is the identity function g(μ)=μ. Assume that the variance of the normal distribution is the same, regardless of the genotype, and denote this common variance by σ2. The parameter σ2 corresponds to the nuisance parameter β in our generalized-linear-model setup. Now, the function l2 becomes
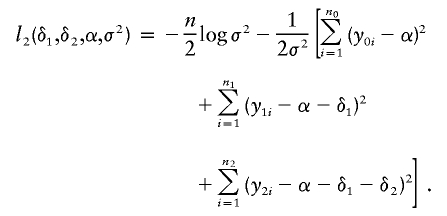 |
The unconstrained maximum-likelihood estimates of α, δ1, δ2, and σ2 are 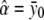 ,
, 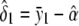 ,
, 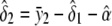 , and
, and
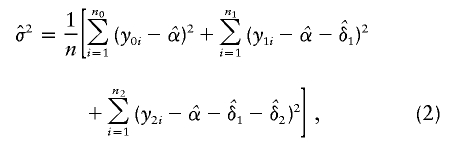 |
respectively. For the recessive model (δ1=0), the maximum-likelihood estimates of α and δ2 are 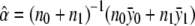 and
and 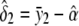 , respectively. For the dominance model (δ2=0), the maximum-likelihood estimates of α and δ1 are
, respectively. For the dominance model (δ2=0), the maximum-likelihood estimates of α and δ1 are 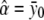 and
and 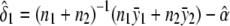 , respectively. The variance σ2 is estimated by fixing
, respectively. The variance σ2 is estimated by fixing 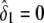 (for the recessive model) or
(for the recessive model) or 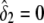 (for the dominance model) in equation (2). Under the null hypothesis, we have δ1=δ2=0, and the maximum-likelihood estimate of α is
(for the dominance model) in equation (2). Under the null hypothesis, we have δ1=δ2=0, and the maximum-likelihood estimate of α is 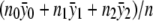 . Given these results, it is straightforward to compute the statistic ΛNew.
. Given these results, it is straightforward to compute the statistic ΛNew.
Let γ=(p0p2)1/2[(1-p0)(1-p2)]-1/2 and κ=(2π)-1arccos(γ). In appendix A, it is shown that, as n→∞,
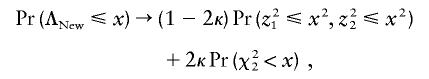 |
where (z1,z2)t follows a standard bivariate normal distribution with correlation coefficient γ and where χ22 follows a χ2 distribution with 2 df. It is also shown in appendix A that, as n→∞,
The statistic ΛLarger was proposed as a robust test statistic (Freidlin et al. 2002), but its asymptotic distribution was not given. The correlation coefficient γ depends on the genotype frequencies p0 and p2. When their true values are unknown, the genotype frequencies p0 and p2 can be consistently estimated by their respective sample frequencies.
Dichotomous Traits
Dichotomous traits are usually modeled through binomial distribution, and the “canonical” link function is the logit function g(μ)=log[μ/(1-μ)]. In this situation, the log-likelihood function l2 becomes, up to an additive constant,
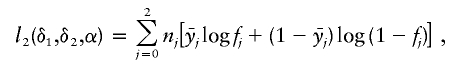 |
where f0:=exp(α)/[1+exp(α)], f1:=exp(α+δ1)/[1+exp(α+δ1)], and f2:=exp(α+δ1+δ2)/[1+exp(α+δ1+δ2)] are the penetrances of the trait for the three genotypes that have 0, 1, and 2 copies of allele A, respectively. We note that the Armitage trend test typically assumes δ1=δ2 (Sasieni 1997).
The likelihood function l2(δ1,δ2,α) depends on δ1, δ2, and α through f0, f1, and f2. The constraint δ1δ2⩾0 holds if and only if f0⩾f1⩾f2 or f0⩽f1⩽f2 holds. It is easy to obtain that the unconstrained maximum-likelihood estimate of fj is 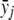 , j=0,1,2. For the dominance model where f1=f2 (equivalent to δ2=0), the maximum-likelihood estimate of f0 is
, j=0,1,2. For the dominance model where f1=f2 (equivalent to δ2=0), the maximum-likelihood estimate of f0 is 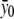 , and the maximum-likelihood estimate of f1=f2 is
, and the maximum-likelihood estimate of f1=f2 is 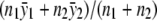 . For the recessive model where f0=f1 (equivalent to δ1=0), the maximum-likelihood estimate of f0=f1 is
. For the recessive model where f0=f1 (equivalent to δ1=0), the maximum-likelihood estimate of f0=f1 is 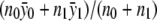 , and the maximum-likelihood estimate of f2 is
, and the maximum-likelihood estimate of f2 is 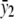 . Under the null hypothesis, we have f0=f1=f2, and the maximum-likelihood estimate is
. Under the null hypothesis, we have f0=f1=f2, and the maximum-likelihood estimate is 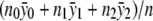 . Given these results, it is again straightforward to compute the statistic ΛNew.
. Given these results, it is again straightforward to compute the statistic ΛNew.
The generalized linear model is appropriate for population samples. For selected samples such as cases and controls, it may not be appropriate. However, it has been shown that simply applying logit regression to case-control data affects only the intercept term α and not the slopes δ1 and δ2 (Prentice and Pyke 1979). Following the arguments of Prentice and Pyke (1979), it can be shown that the intercept term depends on δ1, δ2, and α and that the constraint δ1δ2⩾0 has no impact on the value the intercept term can take (details omitted). So, for a case-control study, one can treat the data as if they were population samples and apply the proposed test.
In appendix B, it is shown that the asymptotic distribution of ΛNew in this situation is identical to that of the likelihood-ratio statistic ΛNew for continuous traits. When true values are unknown, the genotype frequencies p0 and p2 can be estimated by their respective sample frequencies in the combined sample of cases and controls.
Let p denote the frequency of allele A. Under the assumption of Hardy-Weinberg equilibrium (HWE), some quantiles of the asymptotic distribution for ΛNew are tabulated in table 1 for p=0.01, 0.1, 0.3, and 0.5. We note that, for p=0.5, the genotype frequencies are p0=p2=0.25 and p1=0.5, which are the expected genotype frequencies for the F2 population.
Table 1.
Some Quantiles for Test Statistics ΛNew and ΛLarger[Note]
|
Statistic atNominal Significance Level |
||||
| Frequencyof Allele Aand Statistic | .1 | .05 | .01 | .001 |
| p=.01: | ||||
| ΛNew | 4.197 | 5.507 | 8.597 | 13.084 |
| ΛLarger | 3.796 | 5.000 | 7.874 | 12.115 |
| p=.1: | ||||
| ΛNew | 4.152 | 5.457 | 8.536 | 13.008 |
| ΛLarger | 3.778 | 4.985 | 7.867 | 12.113 |
| p=.3: | ||||
| ΛNew | 4.111 | 5.413 | 8.485 | 12.949 |
| ΛLarger | 3.752 | 4.964 | 7.855 | 12.109 |
| p=.5: | ||||
| ΛNew | 4.100 | 5.401 | 8.472 | 12.934 |
| ΛLarger | 3.745 | 4.957 | 7.852 | 12.107 |
Note.— HWE is assumed, so that p0=(1-p)2, p1=2p(1-p), and p2=p2.
Simulation
Simulation studies were done for both quantitative traits and dichotomous traits. The type I error rate and the power were computed on the basis of 10,000 replications.
Quantitative Traits
Consider the following data-generating model:
where G is the genotypic value and ε is an independent environmental factor. Let G=-a for genotype 0, G=d for genotype 1, and G=a for genotype 2. The distribution of ε is taken to be the standard normal distribution whose mean is 0 and whose variance is 1. The heritability for this model is h2:=VG/(VG+1), where VG is the variance of the genotypic effect. Under the assumption of HWE, p0, p1, and p2 can be written as p0=q2, p1=2pq, and p2=p2, where p is the frequency of allele A and q=1-p. According to Falconer and Mackay (1996), the genotypic variance VG=2pq[a+d(q-p)]2+(2pqd)2. Four generating models are considered. They are the dominance model (d=a), the recessive model (d=-a), the additive model (d=0), and an overdominance model (d=2a). Given heritability h2, the value of VG can be obtained from VG=h2/(1-h2). For any given allele frequency p, the value of a for each model can be determined as follows: a=[VG/4pq2(1+q)]1/2 for the dominance model, a=[VG/4p2q(1+p)]1/2 for the recessive model, a=(VG/2pq)1/2 for the additive model, and a=[VG/(2pq(4q-1)2+16p2q2)]1/2 for the overdominance model.
To analyze data from these generating models, seven statistics are computed. They are the proposed statistic ΛNew, the likelihood-ratio statistic ΛDom for the dominant model (δ2=0), the likelihood-ratio statistic ΛRec for the recessive model (δ1=0), the likelihood-ratio statistic ΛAdd for the additive model (δ2=2δ1), the statistic ΛLarger=max{ΛDom,ΛRec}, the statistic ΛLargest:=max{ΛDom,ΛRec,ΛAdd}, and the unconstrained likelihood-ratio statistic ΛUnc that tests whether the three genotypic means are the same or not. The asymptotic distributions for ΛNew and ΛLarger are given in this article. The three statistics ΛDom, ΛRec, and ΛAdd follow an asymptotic χ2 distribution with 1 df. The asymptotic distribution for ΛUnc is a χ2 with 2 df. The statistic ΛLargest was introduced by Freidlin et al. (2002). Since its null distribution is unknown, simulated critical values are used in its power study.
The simulated type I error rates for statistics ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, and ΛUnc are reported in table 2 for allele A frequency p=0.1, 0.3, and 0.5 and for sample size n=100 and 200. These type I error rates are close to their respective nominal significance levels, which suggests that all these tests have valid size. A similar phenomenon is also observed in situations where HWE does not hold (data not shown).
Table 2.
Simulated Type I Error Rates for Quantitative Traits
|
Type I Error Rate atNominal Significance Level |
||||
| Frequencyof Allele A,Sample Size,and Statistic | .1 | .05 | .01 | .001 |
| p=.1: | ||||
| n=100: | ||||
| ΛNew | .0774 | .0366 | .0082 | .0011 |
| ΛDom | .1024 | .0517 | .0116 | .0010 |
| ΛRec | .0669 | .0334 | .0068 | .0009 |
| ΛAdd | .1008 | .0516 | .0108 | .0011 |
| ΛLarger | .0862 | .0435 | .0086 | .0009 |
| ΛUnc | .0787 | .0379 | .0091 | .0011 |
| n=200: | ||||
| ΛNew | .0908 | .0435 | .0097 | .0006 |
| ΛDom | .1075 | .0524 | .0122 | .0011 |
| ΛRec | .0848 | .0418 | .0076 | .0010 |
| ΛAdd | .1095 | .0529 | .0116 | .0009 |
| ΛLarger | .0949 | .0481 | .0102 | .0012 |
| ΛUnc | .0957 | .0457 | .0096 | .0007 |
| p=.3: | ||||
| n=100: | ||||
| ΛNew | .1018 | .0506 | .0116 | .0011 |
| ΛDom | .1064 | .0548 | .0121 | .0015 |
| ΛRec | .1057 | .0518 | .0110 | .0011 |
| ΛAdd | .1062 | .0533 | .0109 | .0013 |
| ΛLarger | .1069 | .0544 | .0126 | .0012 |
| ΛUnc | .1111 | .0537 | .0128 | .0011 |
| n=200: | ||||
| ΛNew | .0971 | .0491 | .0126 | .0014 |
| ΛDom | .1028 | .0543 | .0107 | .0019 |
| ΛRec | .0996 | .0511 | .0116 | .0014 |
| ΛAdd | .1038 | .0532 | .0109 | .0010 |
| ΛLarger | .1063 | .0535 | .0113 | .0017 |
| ΛUnc | .1039 | .0529 | .0124 | .0015 |
| p=.5: | ||||
| n=100: | ||||
| ΛNew | .0972 | .0496 | .0108 | .0012 |
| ΛDom | .1057 | .0508 | .0105 | .0012 |
| ΛRec | .1064 | .0548 | .0130 | .0014 |
| ΛAdd | .1034 | .0508 | .0109 | .0017 |
| ΛLarger | .1055 | .0515 | .0120 | .0010 |
| ΛUnc | .1041 | .0540 | .0109 | .0011 |
| n=200: | ||||
| ΛNew | .0997 | .0478 | .0100 | .0013 |
| ΛDom | .1024 | .0507 | .0115 | .0018 |
| ΛRec | .1043 | .0514 | .0107 | .0009 |
| ΛAdd | .1072 | .0520 | .0095 | .0008 |
| ΛLarger | .1039 | .0531 | .0112 | .0014 |
| ΛUnc | .1031 | .0511 | .0099 | .0013 |
In the power study, the critical values for statistics ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, and ΛUnc are from the respective asymptotic null distributions of these statistics, and the critical values for statistic ΛLargest at allele A frequency p=0.1, 0.3, and 0.5 are obtained from a simulation with 10,000 replications. At significance level 0.001, the power of these seven statistics at allele frequency p=0.1, 0.3, and 0.5, heritability h2=0.05, 0.1, 0.15, and 0.2, and sample size n=100 and 200 is graphed in figures 1, 2, 3, and 4, in which the generating models are dominant, recessive, additive, and overdominant, respectively. It can be seen from these figures that, as expected, the statistic ΛDom performs best when the generating model is dominant, as do the statistic ΛRec when the model is recessive and the statistic ΛAdd when the model is additive. The statistic ΛDom performs worst when the generating model is recessive, and the statistic ΛRec performs worst when it is dominant. Overall, the four statistics ΛNew, ΛLarger, ΛLargest, and ΛUnc seem to have similar power. When the generating model is dominant or recessive, the statistic ΛLarger shows marginally the best power among these four statistics. When the generating model is additive, the statistic ΛLargest is slightly better than any of the other three. For the overdominance model, the statistic ΛNew has slightly more power than the statistic ΛUnc for p=0.1. For p=0.3 and p=0.5, the statistic ΛUnc shows higher power than the statistic ΛNew.
Figure 1.

Power comparison for the quantitative trait when the generating model is dominant. The significance level is 0.001. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Figure 2.

Power comparison for the quantitative trait when the generating model is recessive. The significance level is 0.001. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Figure 3.

Power comparison for the quantitative trait when the generating model is additive. The significance level is 0.001. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Figure 4.

Power comparison for the quantitative trait when the generating model is overdominant. The significance level is 0.001. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Dichotomous Traits
Let 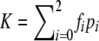 be the prevalence of the trait. The frequency of genotype i would be fipi/K in the cases and (1-fi)pi/(1-K) in the controls. In the absence of association, f0=f1=f2=K, and there is no difference in genotype frequencies between cases and controls. Let γi=fi/f0, i=1,2, be the relative risk of genotype i compared with genotype 0. In the simulation, we consider a dominance model (γ1=γ2), a recessive model (γ1=1), an additive model (γ1=(1+γ2)/2), a multiplicative model (γ1=γ1/22), and an overdominance model (γ1=2γ2). Given population prevalence K and the relative risk γ2, f0 can be determined from f0=K/(p0+γ1p1+γ2p2), from which f1=γ1f0 and f2=γ2f0 can be computed for each model. To simplify the calculation, it is assumed again that HWE holds and that the frequency of allele A is denoted by p.
be the prevalence of the trait. The frequency of genotype i would be fipi/K in the cases and (1-fi)pi/(1-K) in the controls. In the absence of association, f0=f1=f2=K, and there is no difference in genotype frequencies between cases and controls. Let γi=fi/f0, i=1,2, be the relative risk of genotype i compared with genotype 0. In the simulation, we consider a dominance model (γ1=γ2), a recessive model (γ1=1), an additive model (γ1=(1+γ2)/2), a multiplicative model (γ1=γ1/22), and an overdominance model (γ1=2γ2). Given population prevalence K and the relative risk γ2, f0 can be determined from f0=K/(p0+γ1p1+γ2p2), from which f1=γ1f0 and f2=γ2f0 can be computed for each model. To simplify the calculation, it is assumed again that HWE holds and that the frequency of allele A is denoted by p.
The type I error rates for statistics ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, and ΛUnc are reported in table 3 for allele A frequency p=0.1, 0.3, and 0.5 and sample size n=100 and 200. These results suggest that all six statistics have valid type I error rates. A similar finding is also found in simulation studies where HWE fails (data not shown).
Table 3.
Simulated Type I Error Rates for Dichotomous Traits[Note]
|
Type I Error Rate atNominal Significance Level |
||||
| Frequencyof Allele A,Sample Size,and Statistic | .1 | .05 | .01 | .001 |
| p=.1: | ||||
| n=100: | ||||
| ΛNew | .0694 | .0270 | .0047 | .0003 |
| ΛDom | .1094 | .0516 | .0107 | .0007 |
| ΛRec | .1028 | .0164 | .0003 | .0000 |
| ΛAdd | .1041 | .0522 | .0112 | .0008 |
| ΛLarger | .0713 | .0271 | .0060 | .0003 |
| ΛUnc | .0732 | .0329 | .0059 | .0004 |
| n=200: | ||||
| ΛNew | .1158 | .0494 | .0075 | .0006 |
| ΛDom | .1064 | .0520 | .0095 | .0013 |
| ΛRec | .2008 | .0577 | .0039 | .0001 |
| ΛAdd | .1045 | .0531 | .0106 | .0011 |
| ΛLarger | .1057 | .0424 | .0064 | .0008 |
| ΛUnc | .1101 | .0484 | .0078 | .0006 |
| p=.3: | ||||
| n=100: | ||||
| ΛNew | .1068 | .0544 | .0118 | .0009 |
| ΛDom | .0869 | .0570 | .0120 | .0009 |
| ΛRec | .1111 | .0621 | .0145 | .0012 |
| ΛAdd | .1033 | .0536 | .0096 | .0005 |
| ΛLarger | .1134 | .0553 | .0138 | .0011 |
| ΛUnc | .1103 | .0580 | .0119 | .0013 |
| n=200: | ||||
| ΛNew | .0935 | .0445 | .0099 | .0017 |
| ΛDom | .1039 | .0539 | .0081 | .0009 |
| ΛRec | .0978 | .0493 | .0105 | .0020 |
| ΛAdd | .1035 | .0499 | .0082 | .0014 |
| ΛLarger | .0999 | .0499 | .0104 | .0014 |
| ΛUnc | .0971 | .0480 | .0113 | .0016 |
| p=.5: | ||||
| n=100: | ||||
| ΛNew | .0968 | .0487 | .0100 | .0006 |
| ΛDom | .1017 | .0498 | .0101 | .0011 |
| ΛRec | .1012 | .0509 | .0101 | .0006 |
| ΛAdd | .1014 | .0492 | .0106 | .0007 |
| ΛLarger | .1073 | .0546 | .0109 | .0010 |
| ΛUnc | .1039 | .0558 | .0115 | .0007 |
| n=200: | ||||
| ΛNew | .0977 | .0471 | .0084 | .0007 |
| ΛDom | .1008 | .0503 | .0088 | .0011 |
| ΛRec | .1023 | .0539 | .0102 | .0006 |
| ΛAdd | .1053 | .0525 | .0103 | .0009 |
| ΛLarger | .1054 | .0508 | .0088 | .0007 |
| ΛUnc | .1032 | .0488 | .0083 | .0006 |
Note.— Half of the sample is cases, and the other half is controls.
In the power study, the critical values for the seven statistics, including ΛLargest, are obtained in the same manner as in the case of quantitative traits. At significance level 0.001, the power of the seven statistics is simulated for allele A frequency p=0.1, 0.3, and 0.5, prevalence K=0.01, 0.1, and 0.3, and sample size n=100 and 200 under five models; the results for the dominance model, the recessive model, the additive model, the multiplicative model, and the overdominance model are graphed in figures 5, 6, 7, 8, and 9, respectively. In these simulations, the value of γ2 is fixed at γ2=3. Similar simulations were also done for γ2=2, but the patterns were similar and so the results are not reported here. The pattern of the power for these seven statistics is similar to that in the case of quantitative traits.
Figure 5.

Power comparison for the dichotomous trait when the generating model is dominant. The significance level is 0.001. Half of the sample is cases, and the other half is controls. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Figure 6.

Power comparison for the dichotomous trait when the generating model is recessive. The significance level is 0.001. Half of the sample is cases, and the other half is controls. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Figure 7.

Power comparison for the dichotomous trait when the generating model is additive. The significance level is 0.001. Half of the sample is cases, and the other half is controls. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Figure 8.

Power comparison for the dichotomous trait when the generating model is multiplicative. The significance level is 0.001. Half of the sample is cases, and the other half is controls. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
Figure 9.

Power comparison for the dichotomous trait when the generating model is overdominant. The significance level is 0.001. Half of the sample is cases, and the other half is controls. The order of statistics (shaded bars) is (from left to right): ΛNew, ΛDom, ΛRec, ΛAdd, ΛLarger, ΛLargest, and ΛUnc.
For the recessive model, these seven statistics have very similar power when allele A frequency p=0.1. We suspect that this may be caused by the rarity of the genotype homozygous for allele A, since all statistics would be close to the same likelihood-ratio statistic that tests the equality of the genotype effects between the other two genotypes.
Discussion
We have developed a constrained-likelihood approach to marker-trait association analysis. This approach does not require the specification of an analysis model. We investigated two applications of this approach, one for quantitative traits and the other for dichotomous traits. The asymptotic distribution of the constrained likelihood-ratio statistic was derived for both types of traits. Simulation studies suggest that this approach has power to detect association for a range of genetic models. Simulation results also suggest that the power of this approach seems to be close to that of the model-based method when the analysis model is correctly specified. It should be noted that the constrained-likelihood approach has been applied to linkage analyses performed using affected siblings (Holmans 1993) and using extreme discordant sib pairs (Knapp 1998; Freidlin et al. 2003) and to association studies using parent-sibs trios (Zheng et al. 2003) but not to the situation considered in the current article.
This approach provides an alternative to the model-based method and to the statistic ΛUnc that does not have any restriction on the genotypic means. It has power for a wider range of underlying models than does the model-based method, and it is more specific than the statistic ΛUnc. It seems to be an appealing alternative method for cases in which specification of an analysis model is inappropriate—for instance, in studies in which numerous phenotypes are analyzed simultaneously, such as expression QTL mapping (Brem et al. 2002; Schadt et al. 2003; Yvert et al. 2003; Morley et al. 2004).
This approach is related to order-restricted inference methods (Robertson et al. 1988). Order-constrained inference methods are suitable for situations in which the allele being tested for association is expected to increase or decrease the trait value—for instance, gene-mapping association studies of knockout mice. In comparison, the proposed approach does not require any ordering of the mean genotypic effects. What it requires is that the mean effect of the heterozygous genotype not exceed those of the two homozygous genotypes. In other words, if one copy of the allele being tested has an increasing or decreasing effect on the trait, then having an additional copy of this allele will not weaken this trend, regardless of its direction. That is, the allele under investigation shows no overdominance effect.
This approach provides a single-marker test. There are some recent findings that single-marker tests can be used to construct inferential procedures for haplotype analysis that are more powerful than some commonly used haplotype analysis methods (Roeder et al. 2005; Schaid et al. 2005). So, our approach has some interesting implications for haplotype analysis as well. For instance, Roeder et al. (2005) studied the performance of, among other inferential procedures, a statistic that is the largest single-marker test statistic over a set of markers. By permuting the affection status among cases and controls, it was found that this statistic is more powerful than a test proposed by Chapman et al. (2003). The single-marker test used by Roeder et al. (2005) is based on allele counts and requires HWE for it to be valid (Sasieni 1997). On the other hand, our proposed approach is based on genotypes and does not require HWE for its validity. It will be interesting to assess the performance of the procedures of Roeder et al. (2005) when the single-marker test is substituted with the test proposed here.
The constrained-likelihood approach presented here is conceptually simple. Such simplicity may be useful for its generalization to situations other than those considered here. For instance, one could include covariates in the analysis or consider markers with more than two alleles.
Acknowledgments
We thank Dr. Christopher Bartlett, Dr. Jian Huang, and three anonymous reviewers, for their useful comments. This work is supported in part by National Institutes of Health grant R01-EY-11298 (to V.C.S. and K.W.). V.C.S. is an Investigator of the Howard Hughes Medical Institute.
Appendix A : Asymptotic Distribution of ΛNew: Continuous Traits
It is straightforward to obtain that, under H0, the Fisher information matrix for (δ1,δ2,α,σ2) is
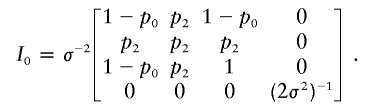 |
According to theorem 16.7 of van der Vaart (1998), when the null hypothesis is true, the likelihood-ratio statistic ΛNew converges to
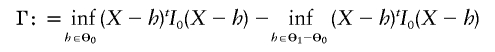 |
as n→∞, where X is normally distributed with mean vector 0 and covariance matrix I-10.
Let 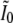 be the partial variance matrix of the first two components of X, given the latter two components. That is,
be the partial variance matrix of the first two components of X, given the latter two components. That is,
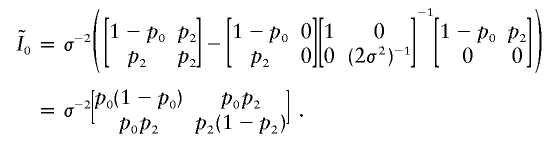 |
After some matrix calculation, it can be shown that Γ can be written as
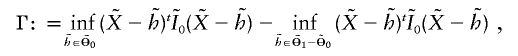 |
where 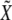 is a bivariate normal random vector with mean vector 0 and variance matrix
is a bivariate normal random vector with mean vector 0 and variance matrix 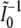 ,
, 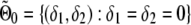 , and
, and 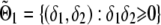 . For Γ to have 2 df, the two components of
. For Γ to have 2 df, the two components of  must be either both strictly positive or both strictly negative. Since the correlation coefficient between these two components is γ=(p0p2)1/2[(1-p0)(1-p2)]-1/2, the probability that the two components are both strictly positive or both strictly negative is 2×(2π)-1arccos(γ)=2κ. When both components of
must be either both strictly positive or both strictly negative. Since the correlation coefficient between these two components is γ=(p0p2)1/2[(1-p0)(1-p2)]-1/2, the probability that the two components are both strictly positive or both strictly negative is 2×(2π)-1arccos(γ)=2κ. When both components of 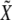 are strictly positive or negative, the constraint δ1δ2⩾0 is not binding and Γ has 2 df.
are strictly positive or negative, the constraint δ1δ2⩾0 is not binding and Γ has 2 df.
The probability that Γ does not have 2 df is 1-2κ. In this case, we have ΛNew=ΛLarger=max{ΛDom,ΛRec}. According to standard asymptotic theory (e.g., that of Cox and Hinkley [1974]), ΛDom is asymptotically equivalent to the score statistic T2Dom, where
and ΛRec is asymptotically equivalent to the score statistic T2Rec, where
The maximum-likelihood estimate of σ2 under the null hypothesis is
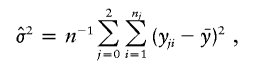 |
where 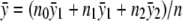 is the grand mean of the trait. The correlation coefficient between TDom and TRec is (n0n2)1/2[(n-n0)(n-n2)]-1/2, which converges to γ=(p0p2)1/2[(1-p0)(1-p2)]-1/2 as n→∞. Asymptotically, TDom and TRec jointly follow bivariate normal distribution (z1,z2)t, whose correlation coefficient is γ and for which both z1 and z2 follow the standard normal distribution. Hence, for any x⩾0, we have
is the grand mean of the trait. The correlation coefficient between TDom and TRec is (n0n2)1/2[(n-n0)(n-n2)]-1/2, which converges to γ=(p0p2)1/2[(1-p0)(1-p2)]-1/2 as n→∞. Asymptotically, TDom and TRec jointly follow bivariate normal distribution (z1,z2)t, whose correlation coefficient is γ and for which both z1 and z2 follow the standard normal distribution. Hence, for any x⩾0, we have
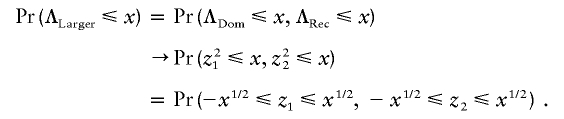 |
Appendix B : Asymptotic Distribution of ΛNew: Dichotomous Traits
The arguments for dichotomous traits are parallel to those for continuous traits given in appendix A. For dichotomous traits, the Fisher information matrix I0 becomes
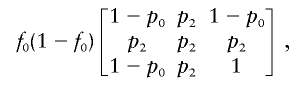 |
and the matrix 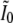 becomes
becomes
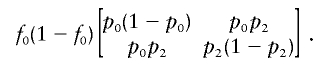 |
Both matrices are proportional to those in appendix A. So, the probability that ΛNew has 2 df remains the same.
The calculation of ΛLarger is also parallel. The expressions for TDom and TRec remain the same, but the expression for 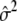 is different. In the situation of dichotomous traits, we need to substitute
is different. In the situation of dichotomous traits, we need to substitute 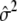 with
with 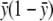 , where
, where 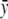 is the proportion of cases among the total number of subjects. This change in
is the proportion of cases among the total number of subjects. This change in 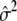 does not affect the asymptotic distribution of ΛLarger or, hence, that of ΛNew.
does not affect the asymptotic distribution of ΛLarger or, hence, that of ΛNew.
Web Resources
The URL for data presented herein is as follows:
- K.W.’s Web site, http://arctica.public-health.uiowa.edu/research.html (for a publicly available R program that implements the method described in this article)
References
- Brem RB, Yvert G, Clinton R, Kruglyak L (2002) Genetic dissection of transcriptional regulation in budding yeast. Science 296:752–755 10.1126/science.1069516 [DOI] [PubMed] [Google Scholar]
- Chapman JM, Cooper JD, Todd JA, Clayton DG (2003) Detecting disease associations due to linkage disequilibrium using haplotype tags: a class of tests and the determinants of statistical power. Hum Hered 56:18–31 10.1159/000073729 [DOI] [PubMed] [Google Scholar]
- Cox DR, Hinkley DV (1974) Theoretical statistics. Chapman and Hall, London [Google Scholar]
- Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics, 4th ed. Prentice Hall, London [Google Scholar]
- Freidlin B, Zheng G, Li Z, Gastwirth J (2002) Trend tests for case-control studies of genetic markers: power, sample size and robustness. Hum Hered 53:146–152 10.1159/000064976 [DOI] [PubMed] [Google Scholar]
- ——— (2003) Efficiency robust tests for mapping quantitative trait loci using extremely discordant sib pairs. Hum Hered 55:117–124 10.1159/000072316 [DOI] [PubMed] [Google Scholar]
- Holmans P (1993) Asymptotic properties of affected–sib-pair linkage analysis. Am J Hum Genet 52:362–374 [PMC free article] [PubMed] [Google Scholar]
- Knapp M (1998) Evaluation of a restricted likelihood ratio test for mapping quantitative trait loci with extreme discordant sib pairs. Ann Hum Genet 62:75–87 10.1017/S0003480098006617 [DOI] [PubMed] [Google Scholar]
- Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman R, Cheung VG (2004) Genetic analysis of genome-wide variation in human gene expression. Nature 430:733–734 10.1038/430733a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice RL, Pyke R (1979) Logistic disease incidence models and case-control studies. Biometrika 66:403–411 [Google Scholar]
- Robertson T, Wright FT, Dykstra RL (1988) Order restricted statistical inference. John Wiley & Sons, New York [Google Scholar]
- Roeder K, Bacanu SA, Zhao X, Devlin B (2005) Analysis of single-locus tests to detect gene/disease associations. Genet Epidemiol 28:207–219 10.1002/gepi.20050 [DOI] [PubMed] [Google Scholar]
- Sasieni PD (1997) From genotypes to genes: doubling the sample size. Biometrics 53:1253–1261 [PubMed] [Google Scholar]
- Schadt EE, Monks SA, Drake TA, Lusis AJ, Che N, Colinayo V, Ruff TG, Milligan SB, Lamb JR, Cavet G, Linsley PS, Mao M, Stoughton RB, Friend SH (2003) Genetics of gene expression surveyed in maize, mouse and man. Nature 422:297–302 10.1038/nature01434 [DOI] [PubMed] [Google Scholar]
- Schaid DJ, McDonnell SK, Hebbring SJ, Cunningham JM, Thibodeau SN (2005) Nonparametric tests of association of multiple genes with human disease. Am J Hum Genet 76:780–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager S, Schaid D (2001) Case-control studies of genetic markers: power and sample size approximations for Armitage’s test for trend. Hum Hered 52:149–153 10.1159/000053370 [DOI] [PubMed] [Google Scholar]
- van der Vaart AW (1998) Asymptotic statistics. Cambridge University Press, Cambridge [Google Scholar]
- Yvert G, Brem RB, Whittle J, Akey JM, Foss E, Smith EN, Mackelprang R, Kruglyak L (2003) Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet 35:57–64 10.1038/ng1222 [DOI] [PubMed] [Google Scholar]
- Zheng G (2003) Use of max and min scores for trend tests for association when the genetic model is unknown. Stat Med 22:2657–2666 10.1002/sim.1474 [DOI] [PubMed] [Google Scholar]
- Zheng G, Chen Z, Li Z (2003) Tests for candidate-gene association using case-parents design. Ann Hum Genet 67:589–597 10.1046/j.1529-8817.2003.00065.x [DOI] [PubMed] [Google Scholar]