

Abstract
We introduce a new method for detection of recombination hotspots from population genetic data. This method is based on (a) defining an (approximate) penalized likelihood for how recombination rate varies with physical position and (b) maximizing this penalized likelihood over possible sets of recombination hotspots. Simulation results suggest that this is a more powerful method for detection of hotspots than are existing methods. We apply the method to data from 89 genes sequenced in African American and European American populations. We find many genes with multiple hotspots, and some hotspots show evidence of being population-specific. Our results suggest that hotspots are randomly positioned within genes and could be as frequent as one per 30 kb.
Introduction
Recombination is a fundamental evolutionary process that shapes patterns of sequence variation by breaking up allelic associations. Recombination is intertwined with the other fundamental molecular evolutionary processes of mutation and selection, since recombination itself may be mutagenic and because recombination aids selection by reducing Hill-Robertson inference. In addition, understanding recombination rates is crucial for practical applications in human genetics, most notably in the use of association mapping of complex traits. However, recombination rates per generation are small, which means that pedigree studies can reveal recombination rate variation only at the megabase scale. The recent development of sperm typing allows the consideration of very large numbers of male meioses, to reveal fine-scale variation in recombination rates, with resolution limited only by the density of polymorphic markers. Sperm typing has revealed the existence of narrow recombination-rate hotspots of ∼1–2 kb in size. But sperm typing is laborious and is not applicable to genomewide studies, so increasing attention has been paid to the inference of recombination-rate hotspots through use of population-genetics models, to infer recombination rates from population data. Such population-genetics approaches yield recombination-rate estimates that differ from those of sperm typing and pedigree studies in a number of important respects. Population-genetics methods estimate a population-scaled compound parameter rather than the raw recombination rates given by the pedigree and sperm-typing methods. The time scale over which recombination rates are inferred with population-genetics methods is also different from the other methods, since those methods consider the history of polymorphism in the sample rather than simply measure present-day recombination rates (sperm typing) or recombination rates in recent generations (pedigree studies). Finally, population-genetics methods estimate a sex-averaged recombination rate (at least for autosomal data), in contrast to sperm typing, which measures only male recombination rates, and pedigree studies, which can measure both male and female recombination rates.
Recent evidence from both sperm data (Jeffreys et al. 2001, 2005) and population data (Crawford et al. 2004; McVean et al. 2004) show that there is large local variation in the recombination rate across the human genome. A simple qualitative description of this is that there are relatively large regions (on the order of 10–100 kb) of the genome that have a small “background” recombination rate, and these regions are separated by recombination “hotspots.” Recombination hotspots are generally 1–2 kb in width (Jeffreys et al. 2001, 2005) and have a recombination rate that is ⩾1 orders of magnitude larger than the background rate.
Currently, there is little understanding about the biological factors that produce hotspots. Jeffreys and Neumann (2002) show a hotspot that is controlled by the nucleotide at a single polymorphic site. Comparisons of hotspots in humans and chimps (Ptak et al. 2005; Winckler et al. 2005) show that recombination hotspots are not conserved between humans and chimps. Thus, hotspots appear to evolve over time scales substantially shorter than the 10–12 million years of evolution that separate humans and chimps. Whether the rate of recombination-rate evolution is sufficiently quick to cause different human populations to have different hotspots is an open question, although large recombination-rate differences among individuals have been reported.
Detection of hotspots is important both for the design and analysis of association studies aimed at finding genetic factors of diseases and for the correct interpretation of patterns of diversity in population data. Furthermore, detection of a large number of recombination hotspots from the extensive human population data that is currently being generated will produce substantial data that can be used to address questions relating to the biology and evolution of these hotspots.
Currently, there are three methods of detection of hotspots from population data (Li and Stephens 2003; Fearnhead et al. 2004; McVean et al. 2004). A recent comparison of these three methods on a 206-kb region of chromosome 1 for which sperm typing had been separately used to detect hotspots (Jeffreys et al. 2005) suggests that the method of Fearnhead et al. (2004) is the most powerful. Of eight hotspots found by sperm typing, the method of Fearnhead et al. (2004) found seven, with one false-positive result. By comparison, the method of McVean et al. (2004) found four hotspots, with zero false-positive results, and that of Li and Stephens (2003) found five, with three false-positive results. Here, we present an extension of the method of Fearnhead et al. (2004) that, in simulation studies, produces an increase in power (for comparable false-positive rates, the new method found 65% of hotspots, as compared with just 53% for the old method [see the “Results” section]). We have applied our new method to polymorphism data for African American (AA) and European American (EA) samples from 89 genes, and we have found strong evidence of multiple recombination hotspots in single genes and some indication of potential differences in hotspots between the two populations.
Material and Methods
Data
We used human sequence data generated by the SeattleSNPs Program for Genomic Applications. The SeattleSNPs database provides polymorphism data that are based on resequencing of a large number of candidate genes thought likely to be involved in genetic disease and so in no way represents a random sampling of the genome. Sequencing spanned the transcribed regions. We chose 89 genes (AGTRAP, ALOX12, ALOX15, ALOX5AP, APOH, C3, CAT, CD36, CD9, CHUK, CKM, CRF, CSF2, CSF3R, CYP4A11, DCN, F10, F12, F13A1, F3, F5, F9, GP1BA, HABP2, ICAM1, IFNAR1, IFNGR1, IFNGR2, IGF2AS, IL10RB, IL11, IL11RA, IL12RB2, IL15RA, IL16, IL17, IL1R1, IL1R2, IL1RN, IL20, IL21R, IL26, IL2RA, IL2RB, IL4R, IL5RA, IL6, IL7R, IL9R, IRAK4, ITGA2, ITGA8, JAK3, MAP3K8, MMP3, NFKBIA, NOS3, PLAUR, PLG, PLTP, PON1, PON2, PPARA, PPARG, PROC, PTGS1, RIPK1, SELE, SELL, SELP, SERPINA5, SFTPA1, SFTPA2, SFTPB, STAT3, STAT6, TF, TFPI, THBS4, TIRAP, TNFAIP2, TNFRSF1A, TNFRSF1B, TRAF2, TRAF6, TRPV6, TTRAP, TYK2, and VCAM1 [see table 1 and the SeattleSNPs Web site for more information]) from an early version of the data set, discarding only those genes that appeared to have too few SNPs or to be too short (in physical distance) to provide sufficient power to infer hotspots.
Table 1.
Inferred Hotspots: Single Analysis of Each Population
|
Hotspot Position(s)c by Population |
||||
| Gene | Sizea(kb) | Sb | AA | EA |
| AGTAP | 18.543 | 63 | 2.406–3.406 | |
| ALOX5AP | 32.08 | 134 | 2.409–3.909 and 8.409–10.659 | 8.909–10.409 |
| ALOX12 | 16.822 | 57 | ||
| ALOX15 | 13.205 | 67 | 1.189–3.439 | 1.439–3.939 |
| APOH | 20.067 | 82 | 17.435–19.185 | |
| C3 | 45.277 | 193 | 3.212–4.212, 13.462–16.212, and 36.212–39.212 | 3.212–4.212, 27.962–28.962, and 36.462–39.212 |
| CAT | 35.889 | 99 | 1.979–3.729 | |
| CD9 | 40.587 | 96 | 5.643–6.643 | |
| CD36 | 29.120 | 88 | 19.520–22.520 | |
| CHUK | 5.364 | 22 | ||
| CKM | 19.934 | 42 | 7.774–10.524 | |
| CRF | 9.929 | 22 | ||
| CSF2 | 5.700 | 15 | ||
| CSF3R | 18.436 | 65 | ||
| CYP4A11 | 13.598 | 69 | ||
| DCN | 39.164 | 107 | 19.849–20.849 | 20.052–20.052 |
| F3 | 16.372 | 33 | ||
| F5 | 74.814 | 216 | 36.130–39.130 and 42.630–45.630 | 32.130–34.630 and 41.630–43.130 |
| F9 | 34.921 | 74 | 3.072–6.072 | |
| F10 | 28.545 | 55 | 23.401–24.651 | |
| F12 | 10.148 | 26 | ||
| F13A1 | 178.078 | 121 | 2.252–3.752 | 2.252–3.752, 7.002–8.002, 74.002–77.002, and 147.752–150.752 |
| GP1BA | 6.093 | 19 | ||
| HABP2 | 39.238 | 194 | 13.351–14.351 and 15.351–18.351 | 15.071–18.071 and 28.821–29.821 |
| ICAM1 | 16.739 | 30 | ||
| IFNAR1 | 33.935 | 94 | ||
| IFNGR1 | 32.499 | 46 | ||
| IFNGR2 | 35.946 | 62 | 10.451–13.451 | 28.451–29.451 |
| IGF2AS | 18.994 | 50 | ||
| IL1R1 | 27.650 | 78 | 7.530–9.530 and 10.780–12.780 | |
| IL1R2 | 22.455 | 106 | 17.082–18.082 | 17.332–18.332 |
| IL1RN | 19.178 | 101 | 15.149–16.649 | |
| IL2RA | 53.821 | 190 | 26.486–27.736 | 26.236–27.736 and 50.236–51.486 |
| IL2RB | 24.764 | 101 | 7.175–8.675 | 2.675–3.675 |
| IL4R | 26.231 | 119 | ||
| IL5RA | 42.815 | 152 | .320–2.570, 18.820–20.320, and 23.820–26.820 | |
| IL6 | 7.498 | 28 | ||
| IL7R | 22.893 | 103 | ||
| IL9R | 15.045 | 57 | 1.961–2.961 | |
| IL10RB | 35.634 | 143 | 15.090–15.340 and 29.340–31.340 | 2.356–3.856 and 14.356–15.356 |
| IL11 | 9.324 | 27 | ||
| IL11RA | 9.732 | 16 | ||
| IL12RB2 | 91.363 | 136 | .634–3.634, 8.634–11.634, 21.634–24.634, 55.884–56.884, and 87.634–90.134 | 1.384–2.384 |
| IL15RA | 35.153 | 131 | .122–1.122 and 26.122–27.122 | |
| IL16 | 20.028 | 62 | 15.172–16.172 | |
| IL17 | 6.158 | 22 | ||
| IL20 | 6.030 | 13 | ||
| IL21R | 25.504 | 58 | 5.310–6.310, 7.560–10.060, and 13.560–14.560 | 3.560–4.810 |
| IL26 | 33.365 | 98 | 28.203–31.203 | |
| IRAK4 | 32.369 | 78 | 27.439–28.939 | |
| ITGA2 | 104.312 | 236 | 8.860–11.860, 25.860–27.360, and 100.110–101.110 | 8.598–9.598 |
| ITGA8 | 206.133 | 145 | .107–2.607, 112.357–114.107 | .357–3.107 |
| JAK3 | 20.967 | 73 | ||
| MAP3K8 | 30.917 | 89 | ||
| MMP3 | 12.058 | 37 | 2.606–3.856 | |
| NFKBIA | 44.703 | 92 | 19.000–20.750 | |
| NOS3 | 24.244 | 49 | ||
| PLAUR | 23.943 | 108 | 6.561–7.811, 13.061–14.311, 17.311–18.311, and 19.311–22.061 | 6.782–9.282 |
| PLG | 54.569 | 117 | 2.356–5.356 | 1.155–4.155 |
| PLTP | 17.934 | 31 | 11.493–12.743 | 11.743–12.743 |
| PON1 | 28.959 | 104 | 5.062–6.562 | 5.312–6.312 |
| PON2 | 30.882 | 85 | ||
| PPARA | 82.267 | 96 | 23.836–26.836 | |
| PPARG | 148.541 | 121 | 142.553–145.553 | 145.800–146.809 |
| PROC | 11.867 | 36 | 3.861–4.861 | |
| PTGS1 | 28.043 | 92 | 5.425–7.425 | |
| RIPK1 | 39.985 | 87 | 12.337–13.837 and 33.337–34.337 | |
| SELE | 13.825 | 62 | ||
| SELL | 24.384 | 98 | 12.833–15.083 | 14.117–15.617 |
| SELP | 43.717 | 122 | 20.427–23.427 and 28.427–31.427 | 21.677–22.927 |
| SERPINA5 | 7.575 | 49 | .106–1.606 | .356–1.606 |
| SFTPA1 | 23.895 | 114 | 11.052–12.552 | |
| SFTPA2 | 17.274 | 105 | 12.706–15.206 | 12.706–14.956 |
| SFTPB | 9.741 | 27 | 1.163–4.163 | 1.663–2.663 |
| STAT3 | 76.609 | 97 | 9.050–10.050, 46.050–47.050, and 62.050–65.050 | |
| STAT6 | 18.638 | 32 | ||
| TF | 36.754 | 116 | ||
| TFPI | 38.066 | 93 | ||
| THBS4 | 50.899 | 111 | 40.295–43.295 | |
| TIRAP | 20.200 | 56 | ||
| TNFAIP2 | 15.510 | 45 | ||
| TNFRSF1A | 15.115 | 34 | ||
| TNFRSF1B | 44.877 | 114 | 40.895–42.645 | 14.145–17.145 and 41.145–42.645 |
| TRAF2 | 43.187 | 102 | 38.887–41.387 | 11.387–12.387 and 39.637–41.137 |
| TRAF6 | 26.142 | 52 | ||
| TRPV6 | 26.679 | 102 | 8.777–11.527 | |
| TTRAP | 20.089 | 79 | 16.899–19.149 | |
| TYK2 | 31.650 | 97 | ||
| VCAM1 | 22.293 | 54 | 12.375–15.375 | 12.125–15.125 |
Size is the amount sequenced between the first and last SNP (inclusive).
S denotes the number of SNPs across both populations.
Hotspot positions are given (in kb) from the start of the region sequenced.
The SeattleSNPs data set contains information from two populations: 23 unrelated EAs from CEPH pedigrees and 24 AAs from the African-American Human Variation Panel. On average, genes in the SeattleSNPs data set are 20–25 kb long and contain 115 segregating sites, of which 100 are segregating in AAs, and 65 in EAs. For the analyses of recombination-rate variation, we required haplotypes that are not directly ascertained in the SeattleSNPs data set but that are instead inferred using PHASE v2.1 software (Stephens et al. 2001; Stephens and Donnelly 2003). Note that only sites with a minor-allele frequency >5% were used for haplotype reconstruction and, hence, for hotspot detection. Simulations indicate that the estimation of haplotypes by PHASE causes very little bias in recombination-rate estimation (Smith and Fearnhead 2005).
Approximate Likelihood Methods
Our approach to detection of hotspots is based on use of the approximate marginal-likelihood (AML) method of Fearnhead and Donnelly (2002) to obtain (approximate) likelihood curves for the recombination rate within short subregions of each data set. Each subregion consists of six (consecutive) SNPs; we calculated likelihood curves for each such subregion within our data set. The likelihood curves were calculated using the program sequenceLDsr, which is available at P.F.'s Web site. This approach uses importance sampling (see Stephens and Donnelly [2000] and Fearnhead and Donnelly [2001] for more details) and is based on (a) simulation of a set of possible genealogical histories for the data, (b) calculation of an (approximate) likelihood curve for the recombination rate for each history, and (c) combination of these curves, by use of a suitable weighted average, to produce the final likelihood curve for the subregion. In practice, we used 100,000 histories in our calculation (step a above) for each subregion, and the computational cost was 10–30 minutes for each subregion.
Note that we attempt to detect recombination hotspots, using recombination models that consider only crossing over; that is, gene conversion is not explicitly modeled. However, previous simulation studies have indicated that variation in gene-conversion rates should be revealed to some extent by methods that consider only crossing over (Smith and Fearnhead 2005).
This AML has been shown to give a very good approximation of the true likelihood curve and is 1–3 orders of magnitude quicker to compute (Fearnhead and Donnelly 2002). Although the likelihood curve is calculated under the assumption of a constant-sized, panmictic population, inferences that are based on it are reasonably robust (in terms of estimation of relative rates of recombination) to a variety of deviations from this model (see Smith and Fearnhead 2005). The likelihood curve is also calculated under the assumption of a constant recombination rate across the subregion. Smith and Fearnhead (2005) show that, if the recombination rate varies across the subregion, then the method estimates an average recombination rate across the subregion.
Detecting Hotspots: Single Population
Consider inferring hotspots within a single gene with S SNPs (or segregating sites). Let li(ρ) denote the log AML curve for the ith subregion of the gene that extends from the ith to the (i+5)th SNP, inclusive. (Our approach can be used for subregions that include either fewer or more than six SNPs, and the optimal width of subregion may depend on the SNP density; here, we follow the approach of Fearnhead et al. [2004]). We use li(ρ) for i=1,…,S-5 to fit a hotspot model of how the recombination rate varies with the position along the gene. In particular, we consider recombination surfaces ρ(x), which gives the local recombination rate (per kb) at any position of the gene, of the form
 |
where ρb is the background recombination rate and h is the number of hotspots, with the ith hotspot having rate ρi and extending from position si to ei. We assume that hotspots do not overlap or touch. See figure 1 for an example.
Figure 1.

An example recombination surface of a gene. This surface consists of a background recombination rate of 1 and three hotspots with lengths of 1 kb, 2 kb, and 2 kb and with recombination rates of 11, 9, and 7, respectively. The position of 20 SNPs are marked by crosses, and these SNPs define 15 subregions denoted by rectangles below the X-axis. The penalized likelihood for the recombination surface is calculated from the AMLs for each of these subregions. The AMLs are calculated under an assumption of constant recombination rate across the subregion. The recombination surface restricted to the 1st, 7th, and 13th subregions (blackened rectangles) are shown at the bottom of the figure, together with the average recombination rate defined by the recombination surface (dashed line). For example, for subregion 1, the recombination surface is 4 kb with a recombination rate of 1 and 1 kb with a recombination rate of 11, which produces an average recombination rate of 3 kb. This average rate is the value of the recombination rate at which the AML for this subregion is evaluated (the ρ1 in eq. [2]).
We define a penalized log likelihood for a recombination surface ρ(x) by
 |
where ρi is the average recombination rate across subregion i, as specified by the recombination surface ρ(x) (see fig. 1), and λ is a positive constant included to penalize overfitting with hotspots. (Note that the
 |
term is itself not a true log likelihood, since dependence between the data within different subregions is being ignored.)
We estimate the number and position of the hotspots by maximizing Pl[ρ(x)] with respect to recombination surfaces of the form (eq. [1]). This gives a “maximum penalized-likelihood estimate” of the recombination surface from which the number and positions of the hotspots can be read. This approach is an extension of that of Fearnhead et al. (2004). The approach of Fearnhead et al. (2004) was to consider each subregion individually and to evaluate the evidence of a hotspot within that subregion by use of a likelihood-ratio test. The approach proposed here aims to use the information in all subregions that contain (part of) a hotspot to detect the hotspot.
Maximization of Penalized Likelihood
We maximize the penalized log likelihood (eq. [2]), using a recursive segmentation that iteratively adds hotspots to the recombination surface. Formally, we proceed as follows:
-
A.
Estimate the background rate, ρ(0)b, and set ρ(0)(x) to be equal to ρ(0)b for all x;
-
B.
Given a current recombination surface ρ(i)(x), evaluate the change in penalized log likelihood obtained by adding different hotspots to this surface, subject to the condition that each new considered hotspot does not overlap or touch a hotspot in ρ(i)(x);
-
C.
Add hotspots to ρ(i)(x) that increase the penalized log likelihood. The order in which hotspots are added is determined by the amount they increase the penalized log likelihood (largest increase first) and are subject to each new hotspot not overlapping or touching any hotspots that have already been added; and
-
D.
If no new hotspots are added in step C, then the estimated recombination surface is ρ(i)(x). Otherwise, re-estimate the background rate, update the current recombination surface ρ(i+1)(x), and return to step B.
To estimate the background recombination rate at each iteration, we use the composite likelihood of Fearnhead and Donnelly (2002), after exclusion of any subregions that contain part of a hotspot. For estimation of the background rate in step A, we first use the method of Fearnhead et al. (2004) to detect subregions that include hotspots, and we omit those in the composite likelihood. When considering new hotspots in step B, we consider all hotspots specified by a grid of possible start positions and lengths for the hotspot. In the analyses here, the grid had start positions every 250 bp, and the hotspots lengths were either 250 bp to 3 kb or 1 kb to 3 kb, in steps of 250 bp. The maximization was performed using the function HotspotEstimate written in R.
This approach does not find the true maximum penalized-likelihood estimate. Sources of error include (a) consideration of only a grid of possible hotspots, (b) misestimation of the background recombination rate, and (c) the recursive procedure fitting a single hotspot to a cluster of hotspots. The errors due to source a are small compared with the uncertainty in the data about the position of hotspots. Any errors that are due to source c can be corrected by reanalysis of hotspots that are detected, to determine whether they are single hotspots or hotspot clusters. Errors due to source b are potentially more problematic; accurately estimating a background rate requires the exclusion of subregions that contain hotspots, but finding such subregions may depend on a reasonable estimate of the background rate. Therefore, if, in step A, we overestimate the recombination rate (e.g., because of too many hotspots within the gene), then that may preclude us from correctly detecting the hotspots that are there.
Specifying the Likelihood Penalty
The accuracy of our penalized-likelihood approach to finding hotspots depends on the choice of the penalty for hotspots λ. To determine a suitable choice for λ, we resorted to simulation studies (see below). We chose λ=16, which gave an approximate false-positive rate of one hotspot per 60 25-kb genes (see the “Results” section for further details).
We also introduced an edge correction to this hotspot penalty. Hotspots at most positions within a gene will occur within at least five different subregions, whereas hotspots at the edge of the gene may occur within fewer subregions. To account for this and for the positive correlation between likelihood curves at nearby subregions, we reduce the hotspot penalty for such hotspots. The penalty incurred for a hotspot ranges from 8 (if it appears in just one subregion) to 16 (if it appears in five or more subregions); these different penalties were calculated from the simulation studies, to maintain the same false-positive rate at the edge of a gene as elsewhere.
Detecting Hotspots: Joint Analysis
We also considered an extension of this approach that jointly infers hotspots that are based on the data from two populations (in our application, EA and AA populations). Our aim is to jointly infer the recombination surfaces for both populations, ρ(1)(x) and ρ(2)(x). Our model for these recombination surfaces depends on γ (the ratio of effective population sizes between the two populations), ρb (the background recombination rate in population 1), and three types of hotspots: those in only population 1, those in only population 2, and those in both populations. We denote by “h(1),” “h(2),” and “h,” respectively, the number of each type of hotspot. Our model for the recombination surface is
 |
We again assume that hotspots do not overlap within each of the two populations. Our model is based on the recombination surfaces of the two populations differing only by a constant of proportionality (the ratio of effective population sizes), except at population-specific hotspots. We include the possibility of population-specific hotspots because of the biological evidence that hotspots evolve over time. For an example of our two-population recombination surface, see figure 2.
Figure 2.

An example two-population recombination surface. The recombination surfaces for population 1 (red) and population 2 (blue) are shown for a hypothetical gene. In this example, the effective population size of population 2 is 1.5 times that of population 1; the two populations share two hotspots, and each has one population-specific hotspot.
We define a penalized log likelihood for a recombination surface ρ(x) by
 |
where l(j)i(·) is the log AML for subregion i in population j, and ρ(j)i is the average recombination rate in subregion i of population j, as defined by ρ(j)(x). We assume the same penalty for each of the three possible hotspots, since each hotspot introduces the same number of parameters (three: size, start, and end of hotspot) regardless of its type.
We maximize equation (4) with respect to recombination surfaces of the form (3). We perform the maximization, using a recursive segmentation procedure similar to the one described above, implemented in R via a function HotspotEstimate2. We estimate the ratio of effective population sizes on the basis of the ratio in the estimates of average recombination rate in the two populations. We again used simulation to choose λ=18, which gave an approximate false-error rate of one hotspot per 20 25-kb genes.
Coalescent Simulations
We simulated polymorphism data, using coalescent models for two different purposes: simulations without recombination hotspots were used to choose the likelihood penalty parameter for the penalized-likelihood method, and simulations with recombination hotspots were used to test the power of recombination-hotspot detection methods. For both classes of simulations, three sets of 100 data sets each were generated corresponding to three alternative demographic histories: a null history of constant population size and a panmictic population, a demographic history thought to match that of AAs, and a demographic history thought to match that of EAs. All simulations were for 50 samples of 25-kb sequences, chosen to match the SeattleSNPs data set. For simulation sets 2, 3, 5, and 6, we simulated genotype data by randomly combining the 50 haplotypes to produce 25 genotypes, and we used PHASE to infer haplotypes. The inferred haplotypes were then used in our analysis.
Simulation set 1: constant recombination rate and null demographic model
These sequence data were simulated in two stages. First, the ms program (Hudson 2002) was used to simulate a tree file (consisting of a set of genealogies and branch lengths for different portions of the sequence) under the standard neutral coalescent, with a constant rate of crossing-over across the sequence. The recombination rate ρ=4Ner was determined for each simulation by simulating r from the empirical distribution of autosomal crossing-over rate in the deCODE pedigree study (Kong et al. 2002) and by applying the standard assumption of Ne=10,000. The average autosomal crossing-over rate in the deCODE pedigree study (excluding centromeres) is 1.2 cM/Mb, which means that the average ρ is 0.48 per kb. DNA sequence data was then simulated, according to a two-allele finite-sites mutation model, on the basis of the tree file, using the seq-gen program (Rambaut and Grassly 1997). The population-scaled mutation parameter θ was set to 0.9 per kb, similar to average nucleotide diversity in the pooled AA and EA SeattleSNPs data set, and mutation rates were modeled as constant among sites.
Simulation sets 2 and 3: constant recombination rates and AA and EA demographic histories
Coalescent simulations invoking complex human demographic scenarios were simulated using the Cosi program of Schaffner et al. (in press), available at the Cosi Web site. The Cosi program is designed to simplify simulation of complex demographic scenarios, including asymmetric migration rates between subpopulations, admixture, populations splitting into subpopulations, and various population-size changes, including exponential growth and bottlenecks. Cosi allows for both crossing-over and gene conversion, as well as variation in rates of crossing-over (determined by a piecewise constant recombination surface), and assumes the infinite-sites model of mutation (with mutation positions converted into discrete base-pair positions). We used the detailed model of human demography, which was calibrated by Schaffner et al. (in press), using several large data sets of human sequence variation. The model of Schnaffner et al. (in press) simulates four populations that include an AA population and a European population. Unlike the African population in America, the European population in America has experienced very little recent admixture with other populations, so a European population is equivalent to an EA population. The mutation rate of 1.3×10-8 per base pair per generation, with no variation among sites, was chosen to give the correct average numbers of segregating sites in the AA and EA populations. The crossing-over rate, constant across the sequence for each simulation, was simulated from the empirical deCODE distribution, as described above. The same low level of gene conversion, 4.5×10-9 per bp per generation, was applied to each simulation (with a gene conversion tract length of 500 bp at all gene-conversion events). As a check of these model parameters, we compared our simulated data with the real SeattleSNPs data, with respect to two important summary statistics affected by demography, Tajima’s D and the FST measure of genetic differentiation. For a SeattleSNPs collection of 201 genes, mean D was −0.53 for the AA population and +0.20 for the EA population. For simulation sets of 1,000 sequences, the mean D was −0.67 for the AA population and +0.03 for the EA population. For the SeattleSNPs genes, mean FST between the two populations was 9.9%, and we obtained a mean FST of 8.8% in our simulations (we used equation 3 in the work of Hudson et al. [1992] to estimate FST, with negative FST estimates adjusted to zero).
Simulation set 4: recombination hotspots and the null demographic model
These sequence data were simulated as for simulation set 1, except for the addition of recombination hotspots. The background crossing-over rate was chosen using the deCODE distribution, as for simulation set 1, but, in addition, a single crossing-over hotspot was simulated. The hotspot rate was chosen with ρ distributed uniformly between 20 and 30 per kb (so, on average, 50 times higher than background). Hotspot width was distributed uniformly between 1 kb and 2 kb, and hotspot position in the sequence was chosen at random (more precisely, the start of the hotspot was distributed uniformly between 0 kb and 24 kb in the sequence). We simulated data, using the approach described in appendix C of Li and Stephens (2003), to convert the output of Hudson’s ms program.
Since the deCODE distribution represents the average crossing-over rate for both background and hotspot regions, we could have chosen a different distribution, concentrated on smaller values, for the background rate. Precisely how to do this is unclear, and the approach we take should be considered conservative (in terms of estimating power), since we allow for larger background rates. Furthermore, our results (see below) suggest that the hotspot intensity (ratio of hotspot rate to background rate) had, at most, a small effect on the power of our approach.
Simulation sets 5 and 6: recombination hotspots and AA and EA demographic histories
These sequence data were simulated as for simulation sets 2 and 3, except for the addition of recombination hotspots. The details of the hotspots were the same as for simulation set 4, except that the Cosi program requires recombination rates in terms of r, the recombination probability per meioses, and we chose r to be in the range of 5×10-4 to 7.5×10-4 per kb.
Polymorphism Frequencies
The SeattleSNPs data were examined for a signature of biased gene conversion (BGC) (Marais 2003) on polymorphism frequencies—that is, the higher frequency of AT→GC mutations compared with GC→AT mutations. The direction of mutation of SNPs (GC→AT or AT→GC) was inferred by parsimony with use of the chimpanzee outgroup information that is incorporated in the SeattleSNPs data sets. Coding SNPs were ignored to avoid the effects of selection, as were those SNPs possibly generated by CpG mutations, for which parsimony may be unreliable (for more details, see Webster and Smith [2004]).
Results
Method for Detecting Hotspots
We first performed a detailed simulation study, both to choose the value of the penalty in our penalized likelihood and to evaluate the power of our new method for detecting hotspots and its robustness to deviations from the simple model under which the AMLs are calculated. We compared our new approach with that of Fearnhead et al. (2004), an existing method that performed better than did LDhot (McVean et al. 2004) and Hotspotter (Li and Stephens 2003) at detecting hotspots in a 206-kb region of chromosome 1 (Jeffreys et al. 2005).
The method of Fearnhead et al. (2004) requires a threshold for a likelihood-ratio test to be set. We chose both this threshold and our likelihood penalty from analyses of ∼300 simulated 25-kb data sets, none of which contained recombination hotspots. For each method, we chose two values for the respective constants, one that gave a false-error rate of approximately one hotspot per 100 genes and one that gave a false-error rate of approximately five hotspots per 100 genes. We then analyzed ∼300 data simulated data sets, each of which contained a hotspot under both methods with each choice of constant. See the “Materials and Methods” section for full details of the simulations and table 2 for the results of the power and false-positive rates of these two methods. For a fair comparison with the likelihood-ratio method, we allowed hotspots of lengths between 250 bp and 3 kb in the penalized-likelihood approach (rather than use the knowledge we have about hotspot lengths to impose a minimum hotspot length of 1 kb, information that cannot be incorporated into the likelihood-ratio test). The power results in table 2 count a hotspot to be found if an estimated hotspot overlaps with the true hotspot, and an estimated hotspot is a false-positive result if it does not. The new penalized-likelihood approach is consistently more powerful, for similar false-positive rates, than the existing likelihood-ratio approach. The performance of the penalized-likelihood method is only slightly worse for the EA and AA data sets than for the null data sets.
Table 2.
Power and False-Positive Rates for New and Existing Approaches for Hotspot Detection[Note]
|
Power (False-Positive) Ratesb for |
||||
| Penalized-Likelihood Approach When |
Likelihood-Ratio Approach When |
|||
| Data Simulation and DemographicScenarioa | λ=13 | λ=16 | c=10 | c=12 |
| Simulations without recombination hotspots: | ||||
| Null | … (3) | … (0) | … (1) | … (0) |
| EA | … (3) | … (1) | … (3) | … (1) |
| AA | … (5) | … (1) | … (5) | … (2) |
| Simulations with recombination hotspots: | ||||
| Null | 78 (3) | 75 (2) | 65 (2) | 60 (2) |
| EA | 72 (3) | 63 (1) | 71 (4) | 56 (1) |
| AA | 70 (7) | 67 (4) | 56 (5) | 44 (2) |
| Average | 73 (4) | 65 (1.5) | 62 (3.3) | 53 (1.3) |
Note.— For each method, we chose two values of a user-specified parameter: the penalty, λ, for the penalized-likelihood approach, and the threshold, c, for the likelihood-ratio approach. We allowed hotspot lengths between 250 bp and 3 kb for the penalized-likelihood approach. Each row relates to results from ∼100 data sets.
We considered three demographic scenarios: the null model assumes a panmictic constant population and is the model under which the likelihoods are calculated; EA and AA refer to data simulated jointly under demographic models that roughly match the patterns of diversity seen in EA and AA populations.
Power is given as a percentage, and the false-positive rate as number of hotspots per 100 genes.
We can further examine the accuracy of methods for estimation of the actual position of the hotspot. The median absolute error for estimating the edge of a hotspot is 400 bps for the EA data sets and 320 bps for the AA data sets, with 80% and 86%, respectively, of inferred hotspots containing the middle of the true hotspot. The increased accuracy for the AA data sets is due to the higher density of segregating sites in those data sets.
The AA and EA data sets simulated to produce the results in table 2 were each simulated jointly in pairs; this models the generation of data from a single gene that is sampled in two diverse populations. We tested our method for detecting hotspots, given data from two populations on these pairs of data sets. Again, we used the results of the analysis of simulated data sets that did not contain a hotspot to choose the penalty in the penalized likelihood, and we chose λ=18, which gave a false-positive rate of approximately five hotspots per 100 genes. Since it is the approach we take for the real data, we fixed a minimum hotspot length of 1 kb in our analysis—although imposing this minimum has only a small effect on the results. Table 3 gives the results of the joint analysis, in terms of power and false-positive rates for each population individually and in terms of hotspots that were inferred jointly in both populations.
Table 3.
Power and False-Positive Rates for the New Penalized-Likelihood Approach[Note]
|
HotspotsInferred in Population(s) |
|||
| Rate | EA | AA | Both |
| Power (%) | 86 | 85 | 81 |
| False-Positivea | 5 | 7 | 3 |
Note.— A penalty λ=18 was used, and we allowed hotspots to have lengths of 1–3 kb.
Number of hotspots per 100 genes.
The joint analysis gives improved power for detecting hotspots, as compared with the analyses of single populations with similar false-positive rates. In particular, the method performs well for hotspots inferred in both populations. The joint analysis also gives slightly more accurate inference of the hotspot boundaries, with median absolute error of 320 bps and 95% of inferred hotspots containing the middle of the true hotspot. Although there is limited data, a simple analysis of the features of the data sets for which hotspots were detected suggests that the most important feature is the number of SNPs inside or near (within 2 kb) the hotspot (logistic regression P<.01); by comparison, the width or amount of recombination within the hotspot or background recombination rate had little effect (across the range of values that these varied for our simulation study).
Hotspots in SeattleSNPs Genes
We applied our method for inferring hotspots to 3 Mb of sequence data from 89 genes (see the “Materials and Methods” section). We allowed for hotspots of 1–3 kb in size, since sperm-typing results suggest that hotspots tend to be within this range. Imposition of a suitable minimum length for the hotspot should improve later inferences about the features of detected hotspots.
A summary of the results is given in table 4, and details of the position of the inferred hotspots are given in tables 1 and 5. The results differ across the two analyses, with the joint analysis inferring more hotspots, although this is consistent with the higher power and slightly higher false-positive rate observed in the simulation study. There is also a difference in the number of hotspots inferred in the AA sample compared with the EA sample. One possible explanation of the increased power in the AA population is the increased SNP density in that population—although we did not observe any difference in power in our simulation study, where we had the same difference in SNP density across the two populations.
Table 4.
Inferred Hotspots in 89 Genes
|
No. of Inferred Hotspots,by Population,as Determined by |
|||||
| Separate Analysisa |
Joint Analysisb |
||||
| No. ofHotspotsin Gene(kb) | EA | AA | EA | AA | Both |
| 0 | 51 | 42 | 33 | 26 | 36 |
| 1 | 26 | 32 | 36 | 39 | 37 |
| 2 | 8 | 9 | 10 | 14 | 10 |
| 3 | 3 | 4 | 3 | 4 | 4 |
| 4 | 1 | 1 | 6 | 5 | 2 |
| 5 | 0 |
1 |
1 |
1 |
0 |
| Totalc | 55 | 71 | 94 | 104 | 77 |
The separate analyses used a likelihood penalty of 16.
The joint analysis used a likelihood penalty of 18.
Total number of hotspots inferred across the 89 genes.
Table 5.
Inferred Hotspots: Joint Analysis[Note]
|
Hotspot position(s)a by Population |
|||
| Gene | Both | AA | EA |
| AGTRAP | 1.156–4.156 | ||
| ALOX5AP | 2.159–5.159 and 8.659–10.409 | ||
| ALOX15 | .939–3.689 | ||
| APOH | 17.435–19.185 | ||
| C3 | 3.212–4.212, 13.462–15.462, and 36.212–39.212 | 27.962–28.962 | |
| CAT | 1.979–3.729 | 7.229–9.729 | |
| CD9 | 5.643–6.893 | ||
| CKM | 7.524–10.524 | ||
| CSF3R | 7.093–10.093 | ||
| CYP4A11 | 1.458–2.708 | ||
| DCN | 19.599–21.099 | ||
| F5 | 34.630–37.630 and 42.380–45.380 | ||
| F9 | 3.072–6.072 | ||
| F12 | 4.115–5.365 | ||
| F13A1 | 1.752–4.002 and 6.752–9.752 | 74.252–75.752 and 147.502–150.502 | |
| HABP2 | 6.601–7.601, 11.351–12.351, and 15.101–18.101 | 13.351–14.351 | 28.351–29.851 |
| I11RA | 2.027–3.277 | ||
| I12RB2 | .634–3.634, 19.884–22.384, and 55.884–56.884 | 33.634–36.134 | |
| ICAM1 | 10.948–13.948 | ||
| IFNAR1 | 20.324–21.574 | ||
| IFNGR1 | 26.986–29.986 | 1.986–3.236 | |
| IFNGR2 | 11.451–14.451 | 27.451–30.451 | |
| IL1R1 | 7.530–9.530 | 10.780–12.780 | 15.780–18.780 |
| IL1R2 | 17.082–18.082 | ||
| IL1RN | 15.149–16.649 | ||
| IL2RA | 25.986–27.736 and 48.486–51.236 | ||
| IL2RB | 7.175–8.675 | ||
| IL5RA | .320–2.570 and 24.320–27.320 | 27.820–29.070 | 18.820–20.820 |
| IL10RB | 14.340–15.590 and 29.340–31.340 | 2.090–3.090 | |
| IL15RA | 25.334–27.334 | .084–1.084 | |
| IL16 | 15.172–16.172 | ||
| IL21R | 3.560–5.060, 5.310–8.310, 8.560–9.810, and 12.810–15.310 | 2.310–3.310 | |
| IL26 | 28.641–31.391 | ||
| IRAK4 | 7.882–9.632 and 27.382–29.132 | ||
| ITGA2 | 8.348–11.348 and 54.098–56.598 | 26.098–27.098 and 100.098–101.098 | 1.598–3.348 and 59.598–62.598 |
| ITGA8 | .857–3.857, 75.357–77.107, 112.107–115.107, and 189.607–190.607 | 49.107–50.607 | |
| JAK3 | 6.539–9.539 | ||
| MAP3K8 | 10.006–12.756 | 7.006–8.006 | |
| MMP3 | 2.856–3.856 | ||
| NFKB1A | 19.500–20.750 | ||
| PLAUR | 6.311–8.811, 13.061–14.311, and 19.561–20.811 | 17.311–18.311 | |
| PLG | 2.356–5.356 | ||
| PLTP | 11.493–12.993 | ||
| PON1 | 5.062–6.562 | ||
| PON2 | 8.679–10.179 and 27.929–29.429 | ||
| PPARA | 73.086–76.086 | 23.836–26.836, 34.586–35.586, and 48.086–49.836 | |
| PPARG | 139.053–141.803 and 146.553–147.803 | ||
| PROC | 3.861–4.861 | ||
| PTGS1 | 5.425–7.675 | ||
| RIPK1 | 12.337–13.587 | 33.337–34.337 | |
| SELL | 13.083–15.333 | ||
| SELP | 20.677–23.677 | 28.427–31.427 | |
| SERPINA5 | .106–1.606 | ||
| SFTPA1 | 11.052–12.552 | ||
| SFTPA2 | 12.456–15.206 | ||
| SFTPB | 1.163–4.163 | ||
| STAT3 | 9.050–10.050, 45.800–47.550, and 62.050–65.050 | ||
| TF | 10.857–13.857 | ||
| THBS4 | 40.545–43.545 | ||
| TNFRSF1B | 14.645–17.395 and 40.895–42.645 | ||
| TRAF2 | 38.887–41.387 | ||
| TRAF6 | .609–3.609 | ||
| TRPV6 | 8.777–11.527 | ||
| TTRAP | 16.899–19.149 | ||
| TYK2 | 1.687–4.687 | ||
| VCAM1 | 12.375–15.375 | ||
Note.— Hotspots are classified on the basis of whether they appear in both or in just the AA or just the EA populations. Only genes with at least one hotspot are shown.
Hotspot positions are given (in kb) from the start of the region sequenced.
We are able to compare our results with those of Crawford et al. (2004), who analyzed data from 74 genes, 39 of which are also in our study. Their analysis was based on a Bayesian approach that compared the hypotheses of no hotspot within a gene with that of one hotspot within a gene, and they analyzed each population separately. If we compare their results with those of our separate analyses, we have a slightly higher proportion (69% as compared with 62%) of genes for which a hotspot was inferred in at least one population. Of the 39 genes in common, the two analyses agree on the presence of hotspots in at least one population, or absence from both, in 28 cases. Of the remaining 11, we inferred hotspots not found by Crawford et al. (2004) in 7 genes. It is encouraging that, for all the 20 genes for which both approaches inferred a hotspot, the position of the hotspot was consistent for the two methods. (We define results as consistent if one of the hotspots inferred in the gene by our approach overlapped with the position of the hotspot inferred by Crawford et al. 2004).
Our results suggest hotspots occur more frequently than did the results of Crawford et al. (2004), whose estimate of a hotspot frequency of one every 63 kb was biased by their assumption that genes had, at most, a single hotspot. A simple estimate of the frequency of hotspots from our analysis of the AA population suggests hotspots either every ∼30 kb (on the basis of the results from the joint analysis) or every ∼40 kb (on the basis of results from the separate analysis). Our higher-density estimates are roughly consistent with the sperm-typing results from the major histocompatibility complex (MHC) gene (Jeffreys et al. 2001), in which six hotspots are found in ∼200 kb, and from a 206-kb region of chromosome 1 that contains eight hotspots (Jeffreys et al. 2005).
Our results provide information on the clustering of hotspots within genes, which is unavailable from the analysis of Crawford et al. (2004). There is no evidence from our results against a model in which hotspots are placed uniformly at random along the genome (see fig. 3). The most extreme examples of clustering of hotspots were for HABP2, PLAUR, and C3, in which 5, 4, and 4 hotspots were inferred (from the joint analysis) in 39-kb, 24-kb, and 45-kb sequences, respectively.
Figure 3.

Quantile-quantile plot for residuals of a Poisson model for the number of hotspots per gene. Dashed lines show 90% symmetric CIs. The model assumes the number of hotspots per gene has a Poisson distribution whose mean is proportional to the length of the gene, which is the same as the assumption that hotspots occur randomly within genes.
Hotspot Position within Genes
Although our data suggest that the position of hotspots within the genome (or at least our biased sample of the genome) is random, there remains the issue of what factors might determine where hotspots are located within the genes in our data set. To investigate factors affecting hotspot position within genes, we used our largest set of hotspots, the AA hotspots identified in the joint analysis.
We first considered the position of hotspots within the genes. We calculated the position of the center of each hotspot as a proportion of the entire gene sequence, and we found the mean position to be close to the center of the gene (mean proportional position 0.45). We also tested whether the proportion of hotspot sequence in exons differed from the proportion of background sequence in exons (considering only those genes in which hotspots were found). We found a slightly higher proportion of hotspot sequence to be exonic (5.4%) compared with background sequence (4.2%), although simulations indicated that the observed proportion of exonic sequence in hotspots was not significantly greater than for randomly positioned hotspots distributed across genes, according to the Poisson distribution with mean equal to the observed density of joint AA hotspots (P=.34).
We also searched for primary sequence motif differences between hotspot sequences and background sequences. We first considered testing for motifs that were more common overall in hotspots than in background sequences, similar to the approach of Crawford et al. 2004, but we found this test identified motifs that were found to be highly abundant in just a few hotspots—in particular, motifs repeated in rare large microsatellites. Instead, we looked for motifs found in a high proportion of hotspots, testing for significance by generating random sets of hotspots (uniform density within and among genes). We first tested the complete set of all 48=65,536 octamers for their presence in the 104 hotspots. Four octamers—AAAAAAAA, CAGCCTGG, GGAGGCTG, and TCCCAGCA—were found in the highest proportion (45 of 104) of hotspots, so no single octamer is a powerful predictor of hotspots. To find octamers found more commonly in hotspots than would be expected by chance, we assessed significance for the 185 octamers that were present in >30% of hotspots. We used this relatively low cutoff point because the proportion of hotspots containing a motif is a function of both enrichment in hotspots and also overall frequency in the genome. One of the octamers, ACAGAGCA, had a P value of <1 in 100,000; a further octamer, TCCCAGCA, had a P value <1 in 10,000. However, no octamers had significant P values after a Bonferroni correction.
Comparison of Populations
The results from the joint analysis of the data in the two populations (see table 4) show that a substantial number of hotspots are inferred only in one of the two populations. This is in marked contrast to the results from the simulation study (see table 3). There are various explanations for this difference; one possibility is that the recombination landscape is actually different for the two distinct populations and that there are hotspots that exist solely in one of the two populations. There is evidence to show that recombination hotspots evolve over time (see the Introduction), although there is currently little idea as to whether this evolution has resulted in different hotspots in different populations and, if so, to what extent there are population-specific hotspots.
To quantify the evidence of population-specific hotspots within the 89 genes that we analyzed, we used a simple likelihood-ratio test (similar to that used by Crawford et al. [2004]). For each population-specific hotspot that was inferred by our joint analysis, we first chose a single region that overlapped with the hotspot. We centered each region around the center of the inferred hotspot and chose the width of that region to be the smallest of 1 kb, 2 kb, 3 kb, 4 kb, or 5 kb, such that there were at least six SNPs within the region for each of the two populations. For some hotspots, even the 5-kb regions did not contain six SNPs in each population; in these cases, we decided to use 5-kb regions. Note that these regions are not necessarily any of the subregions used when we originally inferred the hotspots. We chose regions in this way with the aim of getting small regions, so that the majority (ideally all) of each region would be contained within the area of the true (although potentially population-specific) hotspot, while ensuring enough SNPs within each population to have reasonable power for inferring the recombination rates within each hotspot.
For each hotspot, we then calculated the log-likelihood-ratio statistic for different recombination rates (relative to the different effective population sizes) in the two populations:
where l1(·) and l2(·) are the log AMLs for the AA and EA populations, γ is the estimate of the ratio of the average recombination rates in the two populations, 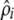 is the maximum-likelihood estimate (MLE) for the recombination rate in population i, and
is the maximum-likelihood estimate (MLE) for the recombination rate in population i, and 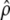 is the MLE for the recombination rate (defined for the AA population), with the assumption that the rate differs only by a factor of γ across the two populations.
is the MLE for the recombination rate (defined for the AA population), with the assumption that the rate differs only by a factor of γ across the two populations.
We converted the likelihood-ratio statistic values into approximate P values, using a χ21 assumption for the likelihood-ratio statistic. Simulations suggests that the likelihood-ratio statistic does at least roughly have a χ21 distribution and that the resulting P values are likely to be conservative (because of the positive dependence of the likelihood curves in the two populations).
We found strongest evidence of a population-specific hotspot in the gene TRPV6 (P=.007), although this P value is far from significant when the multiple testing is accounted for (we tested 44 inferred population-specific hotspots, but these were preferentially chosen from a total of 121 inferred hotspots; thus, we should correct for an equivalent of 121 hypothesis tests). One further hotspot (the putative EA-specific hotspot in HABP2) had a P value <.01.
We studied the putative population-specific hotspot in TRPV6 in more detail. The signal for a population specific hotspot is due to a breakdown of linkage disequilibrium (LD) across the region in the AA population, whereas there is substantial LD across the whole gene in the EA population (four distinct haplotypes from 50 SNPs that segregate in the EA population). However, the four distinct haplotypes found in the EA population consist of three haplotypes that differ only at two SNPs, and the fourth haplotype (which is at frequency 1 in the EA population) appears to be a recent migrant from the AA population. Thus, even the signal here appears to be caused by a combination of a lack of diversity in the EA population and this recent migrant, rather than strong evidence of a population-specific hotspot. (If we remove the migrant haplotype, the remaining haplotypes have almost no information about the amount of recombination in the EA population.)
Other Features of Hotspots
We also looked at features of SNPs and G+C content within the inferred hotspots. Throughout, we used the largest set of hotspots, those detected for the AA population from the joint analysis, and SNPs and G+C content from that population. To see the importance of inferring recombination rates at the kilobase scale—rather than the megabase scale—we also considered the features within the 10% of genes with highest recombination rates, as defined by the deCODE recombination map.
We compared the patterns of G+C content and SNP density for the background region of the AA data (G+C=44.2%; SNP density of one per 310 bp), for the hotspot regions (G+C=46.6%; SNP density of one per 190 bp), and for the nine genes with high deCODE recombination rates (G+C=49.1%; SNP density of one per 260 bp). Both the large-scale and fine-scale high-recombination regions show higher levels of G+C and higher SNP densities. The difference in both G+C content and SNP density is significant (P<.001) for differences between the hotspot and background regions. These results are consistent with known correlations between G+C content and recombination rates and with the possible mutagenic effect of recombination (Hellmann et al. 2003).
One explanation for the correlation between G+C content and recombination rates is BGC: the bias in repair of hetero-mismatches in heteroduplex DNA formed by gene conversion that favors G+C over A+T (see Marais [2003] for a review). To look for evidence of BGC, we calculated the mean polymorphism frequencies of AA GC→AT and AT→GC SNPs within and outside recombination hotspots (see the “Materials and Methods” section).
We see that the signature of BGC, with higher mean frequency of AT→GC polymorphisms relative to GC→AT polymorphisms, is stronger within recombination hotspots than for background regions outside hotspots (see table 6), although the signal is small and not statistically significant. By comparison, a similar analysis of the SeattleSNPs data set found no discernible effect on polymorphism frequencies that were due to large-scale regional recombination-rate variation, as measured by the deCODE pedigree data (Webster and Smith 2004). When we partitioned our AA SNPs by deCODE recombination rates, we found no difference in polymorphism frequencies (see table 6).
Table 6.
Frequencies of Polymorphisms for Different Subsets of AA SNPs
|
Findings by Mutation Direction |
||||||
| GC→AT |
AT→GC |
|||||
| Subset of AA SNPs | Frequencyb(%) | No. of SNPs | Frequencyb(%) | No. of SNPs | Average(%) | Da(%) |
| All SNPs | 18.1 | 3,432 | 20.3 | 2,464 | 19.2 | 2.2 |
| SNPs within hotspots | 20.5 | 360 | 23.5 | 278 | 22.0 | 3.0 |
| Background SNPsc | 17.8 | 3,072 | 19.9 | 2,186 | 18.9 | 2.1 |
| deCODEd | 21.2 | 382 | 21.4 | 226 | 21.3 | .2 |
The difference in mean frequencies.
Mean frequencies of polymorphisms that differ in their effects on GC content.
Without hotspots.
SNPs in genes with high deCODE regional recombination rates.
The proportion of mutations that change GC content and are GC→AT is similar for the complete data and the hotspot regions (58% and 56%, respectively) but significantly smaller than for regions with high deCODE recombination rates (63%; P=.01 for difference in proportion to that of the complete data).
Discussion
We have presented a new method for detecting recombination hotspots from population-genetics data. Our simulation study shows that this method has greater power than what, on the basis of the results in Jeffreys et al. (2005), is currently the best method, that of Fearnhead et al. (2004). Both that approach and our new method are based on use of the AML for subregions of the genomic region of interest. It is intuitive that, for this approach to work well, the subregions must be informative (i.e., contain a sufficient number of SNPs) and be of a size similar to the width of recombination hotspots. (If subregions were much wider than hotspots, the signal from the hotspot would be weaker, since the main part of the subregion analyzed would be outside the hotspot.) This intuition is backed by our simulation results, which suggested that the main factor affecting our method's power for detecting hotspots is the number of SNPs within and near the hotspot. Thus, these approaches are well suited for analyzing the SeattleSNPs data considered in this work as well as the population data described by Jeffreys et al. (2005): in both cases, the density of SNPs is large. It may be that the methods of Li and Stephens (2003) and McVean et al. (2004) will be comparatively more powerful for data for which the SNP density is much smaller.
The main focus of our work has been on detecting recombination hotspots and not on estimating background or hotspot recombination rates. Results of the work of Smith and Fearnhead (2005) suggest that the pairwise likelihood method of McVean et al. (2002) and the approximate likelihood method of Li and Stephens (2003) are more accurate for estimation of recombination rates than the AML that is the basis of our method. As a result, we would suggest that, for estimating recombination rates, one should use the pairwise likelihood approach of McVean et al. (2002), conditional on the hotspot positions found by our method.
The SeattleSNPs data that we analyzed consisted of population data from two distinct populations. The penalized-likelihood approach we propose can be easily extended to deal with data from two populations. This extension is “approximate,” since it ignores the positive dependence that would be expected between the data from the two populations. However, in simulations under models that capture the main features of the data we analyze, we found that this joint analysis performed best. Whether such an approach will work more generally is unclear. The joint analysis is potentially less robust than the separate analysis, since there are extra features of the data, particularly the dependences between the two populations, that will affect the choice of penalization factor and would need to be roughly similar in the simulated and real data.
Our method is dependent on specifying a likelihood penalty λ, an estimate of the background-recombination rate 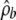 , and, for the joint analysis, an estimate of the ratio of recombination rates between the two populations, γ. An important question is the degree to which results are robust to variations in these. The effect of varying λ is to change the amount of evidence required before a region is determined to be a hotspot. The choice of λ is a simple trade-off between power and false-positive rate; guidelines for choosing λ can be obtained from our simulation study. Although we considered only three possible demographic scenarios (for the separate analysis), the similarity in false-positive rates across these different scenarios gives us some confidence that these results will be roughly correct for a large variety of demographic scenarios.
, and, for the joint analysis, an estimate of the ratio of recombination rates between the two populations, γ. An important question is the degree to which results are robust to variations in these. The effect of varying λ is to change the amount of evidence required before a region is determined to be a hotspot. The choice of λ is a simple trade-off between power and false-positive rate; guidelines for choosing λ can be obtained from our simulation study. Although we considered only three possible demographic scenarios (for the separate analysis), the similarity in false-positive rates across these different scenarios gives us some confidence that these results will be roughly correct for a large variety of demographic scenarios.
The results of the joint analysis appear to be reasonably robust to variations in the choice of γ. For example, in our simulation study, varying γ from 1.5 to 2.3 had only a small effect on the number of both joint and population-specific hotspots inferred (on the order of one or two hotspots for each category). For the real data, an earlier analysis with γ=1.5 (which was based on an estimate of the ratio of effective population size from polymorphism data), rather than γ=3.75, did produce some noticeable differences in the results. In particular, whereas the total number of hotspots in the AA population was almost unaffected, the number of joint hotspots and the total number of hotspots in the EA population were reduced by an order of 10–20.
The method for obtaining 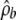 is important, particularly for small genes (or genes with only a small amount of background sequence). Our method—using the likelihood ratio test of Fearnhead et al. (2004) to extract regions that are likely to be hotspots and then using a composite likelihood based on likelihoods from all other subregions to estimate the background rate—is simple and appears to work reasonably well (on the basis of the simulation-study results). Even so, the choice of threshold used for detecting hotspot regions can have a noticeable effect on the results that are obtained; for some genes there can be relatively few subregions that are not classified as hotspots, which can lead to potentially large uncertainty in the estimate of
is important, particularly for small genes (or genes with only a small amount of background sequence). Our method—using the likelihood ratio test of Fearnhead et al. (2004) to extract regions that are likely to be hotspots and then using a composite likelihood based on likelihoods from all other subregions to estimate the background rate—is simple and appears to work reasonably well (on the basis of the simulation-study results). Even so, the choice of threshold used for detecting hotspot regions can have a noticeable effect on the results that are obtained; for some genes there can be relatively few subregions that are not classified as hotspots, which can lead to potentially large uncertainty in the estimate of 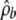 . Better approaches for obtaining
. Better approaches for obtaining 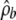 may lead to important improvements in the accuracy of this penalized-likelihood approach.
may lead to important improvements in the accuracy of this penalized-likelihood approach.
One advantage of the joint analysis is that it allows us to directly compare the presence/absence of hotspots in the two populations. For the SeattleSNPs data, we found that a third of the hotspots were inferred to be present in only one of the two populations—much more than is expected on the basis of the simulation study. A simple analysis of these hotspots through use of a likelihood-ratio statistic does not give any evidence in favor of the presence of different recombination hotspots in the two populations. This may be due to lack of power, or there may just be smaller differences in the recombination landscape in the two populations, such as a different ratio of hotspot to background rate, that are causing us to detect a large number of population-specific hotspots.
One important question is whether we would expect to have any power to detect population-specific hotspots, if they exist, given that differences in the recombination rates in the two populations are likely to be recent and that the populations have shared ancestry. One reason to be hopeful about having some power to detect population-specific hotspots is that it is the presence (or absence) of the most-recent recombination events that has most effect on inferences about recombination rates, and it is the recent recombination process that will be most different and most close to independent between the two populations.
Some of the SeattleSNPs data we analyzed has been analyzed by Crawford et al. (2004). An important difference between these two analyses is that our method for detecting hotspots allows us to find multiple hotspots (if they exist) within a gene rather than just answering the question of whether there is at least one hotspot within the gene. Our results show that many genes have multiple hotspots, and, accounting for this, we estimate that the frequency of hotspots is greater (one per 30–40 kb) than that suggested by Crawford et al. (2004) (one per 60 kb). Analysis of the position of the hotspots shows no evidence against the simple hypothesis that hotspots occur randomly.
To what extent the recombination hotspots that have been detected correspond to increased rates of gene conversion and/or increased rates of crossing-over is unclear. Whereas the AML is based on a model that assumes that all recombination events consist of a single recombination break point within the subregion being analyzed (pure crossing-over), the results of Smith and Fearnhead (2005) show that the AML actually infers some (weighted) sum of the crossing-over and gene-conversion rates. Thus, the recombination hotspots indicate a general increase in the rate of gene conversion and/or crossing-over, and our method gives no information about the relative rate of these two processes.
A qualitative look at the population data suggests that some of the hotspots may actually correspond to an increase only in the gene-conversion rate. For example, the hotspot detected at position 19,849–20,849 in DCN in the AA population corresponds to a region in which five neighboring SNPs show little LD (and, e.g., there are nine distinct haplotypes defined by the data at these five SNPs). However, if we consider the 16 SNPs that comprise the 8 SNPs immediately adjacent to either side of this hotspot, then these are in complete LD (as defined by D′) and are consistent with no recombination. In fact, the LD is so strong that these 16 SNPs define only seven distinct haplotypes. This amount of LD is surprising if there is indeed a crossing-over hotspot in the middle of them, and a reasonable conclusion is that this hotspot corresponds solely to an increase in gene conversion (with short tract lengths).
Acknowledgments
This work is dedicated to the memory of Nick Smith. This work was supported by Engineering and Physical Sciences Research Council grant GR/S18786/01. We thank Andrea Jorgensen, for work during her M.Sc. project that motivated this; and two anonymous reviewers, for thoughtful and helpful comments. We particularly thank one of the reviewers for the idea of how to estimate γ.
Web Resources
The URLs for data presented herein are as follows:
- Cosi, http://www.broad.mit.edu/personal/sfs/cosi/
- P.F.'s Web site, http://www.maths.lancs.ac.uk/~fearnhea/
- SeattleSNPs, http://pga.gs.washington.edu/
References
- Crawford DC, Bhangale T, Li N, Hellenthal G, Rieder MJ, Nickerson DA, Stephens M (2004) Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat Genet 36:700–706 10.1038/ng1376 [DOI] [PubMed] [Google Scholar]
- Fearnhead P, Donnelly P (2001) Estimating recombination rates from population genetic data. Genetics 159:1299–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead P, Donnelly P (2002) Approximate likelihood methods for estimating local recombination rates (with discussion) J Roy Stat Soc B 64:657–680 [Google Scholar]
- Fearnhead P, Harding RM, Schneider JA, Myers S, Donnelly P (2004) Application of coalescent methods to reveal fine-scale rate variation and recombination hotspots. Genetics 167:2067–2081 10.1534/genetics.103.021584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann I, Ebersberger I, Ptak SE, Pääbo S, Przeworski M (2003) A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet 72:1527–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338 10.1093/bioinformatics/18.2.337 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA-sequence data. Genetics 132:583–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys AJ, Kauppi L, Neumann R (2001) Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29:217–222 10.1038/ng1001-217 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R (2002) Reciprocal crossover asymmetry and meiotic drive in a human recombination hotspot. Nat Genet 31:267–271 10.1038/ng910 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Neumann R, Panayi M, Myers S, Donnelly P (2005) Human recombination hotspots hidden within regions of strong marker association. Nat Genet 37:601–606 10.1038/ng1565 [DOI] [PubMed] [Google Scholar]
- Kong A, Gubdjartsson DF, Sainz J, Jonsdottir GM, Gudjonson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- Li N, Stephens M (2003) Modelling LD, and identifying recombination hotspots from SNP data. Genetics 165:2213–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G (2003) Biased gene conversion: implications for genome and sex evolution. Trends Genet 19:330–338 10.1016/S0168-9525(03)00116-1 [DOI] [PubMed] [Google Scholar]
- McVean GAT, Awadalla P, Fearnhead P (2002) A coalescent method for detecting recombination from gene sequences. Genetics 160:1231–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GAT, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304:581–584 10.1126/science.1092500 [DOI] [PubMed] [Google Scholar]
- Ptak SE, Hinds DA, Koehler K, Nickel B, Patil N, Ballinger DG, Przeworski M, Frazer KA, Pääbo S (2005) Fine-scale recombination patterns differ between chimpanzees and humans. Nat Genet 37:429–434 10.1038/ng1529 [DOI] [PubMed] [Google Scholar]
- Rambaut A, Grassly NC (1997) Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput Appl Biosci 13:235–238 [DOI] [PubMed] [Google Scholar]
- Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NGC, Fearnhead P (2005) A comparison of three estimators of the population-scaled recombination rate: accuracy and robustness. Genetics (electronically published June 14, 2005; accessed September 7, 2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P (2000) Inference in molecular population genetics (with discussion). J Roy Stat Soc B 62:605–635 10.1111/1467-9868.00254 [DOI] [Google Scholar]
- ——— (2003) A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MT, Smith NGC (2004) Fixation biases affecting human SNPs. Trends Genet 20:122–126 10.1016/j.tig.2004.01.005 [DOI] [PubMed] [Google Scholar]
- Winckler W, Myers SR, Richter DJ, Onofrio RC, McDonald GJ, Bontrop RE, McVean G. AT, Gabriel SB, Reich D, Donnelly P, Altshuler D (2005) Comparison of fine-scale recombination rates in humans and chimpanzees. Science 308:107–111 10.1126/science.1105322 [DOI] [PubMed] [Google Scholar]