

Abstract
Approximately 5% of the human genome consists of segmental duplications that can cause genomic mutations and may play a role in gene innovation. Reticulate evolutionary processes, such as unequal crossing-over and gene conversion, are known to occur within specific duplicon families, but the broader contribution of these processes to the evolution of human duplications remains poorly characterized. Here, we use phylogenetic profiling to analyze multiple alignments of 24 human duplicon families that span >8 Mb of DNA. Our results indicate that none of them are evolving independently, with all alignments showing sharp discontinuities in phylogenetic signal consistent with reticulation. To analyze these results in more detail, we have developed a quartet method that estimates the relative contribution of nucleotide substitution and reticulate processes to sequence evolution. Our data indicate that most of the duplications show a highly significant excess of sites consistent with reticulate evolution, compared with the number expected by nucleotide substitution alone, with 15 of 30 alignments showing a >20-fold excess over that expected. Using permutation tests, we also show that at least 5% of the total sequence shares 100% sequence identity because of reticulation, a figure that includes 74 independent tracts of perfect identity >2 kb in length. Furthermore, analysis of a subset of alignments indicates that the density of reticulation events is as high as 1 every 4 kb. These results indicate that phylogenetic relationships within recently duplicated human DNA can be rapidly disrupted by reticulate evolution. This finding has important implications for efforts to finish the human genome sequence, complicates comparative sequence analysis of duplicon families, and could profoundly influence the tempo of gene-family evolution.
Introduction
Sequence evolution can be described as reticulate (or concerted) if it results in a network of relationships between distinct sequences rather than simple ancestor-descendant relationships. Although such relationships can be caused by recombination between two or more genomes, which is common among lentiviruses such as HIV-1 (Rhodes et al. 2005), they are most commonly associated with paralogous sequence families (e.g., see Newman and Trask 2003; Rozen et al. 2003), where both nonallelic homologous recombination (NAHR) and gene conversion between alleles or paralogues can occur within a single genome. Our understanding of these processes in humans has improved dramatically over the last few years. Rates of gene conversion have been estimated within specific loci by use of techniques such as small-pool PCR (Jeffreys et al. 2004) or repeat-specific PCR within well-characterized repeat pairs (Bosch et al. 2004). Some specific NAHR events that lead to clinical phenotypes as a result of the deletions/duplications they promote have also been dissected in detail (Hurles 2001; Saitta et al. 2004).
Despite these advances, the frequency and patterns of reticulation events within the vast majority of human duplicons are difficult to analyze, because of the lack of associated phenotypes and the number and complexity of sequence repeats. Comparative analysis of sequence data is therefore often used to infer these events, and specific NAHR and conversion events are being identified at a growing number of human genes, including Opsin, Complement C4, and HLA genes (Jakobsen et al. 1998; Jaatinen et al. 2002; Verrelli and Tishkoff 2004). With the near-completion of the human genome sequence, there is now an opportunity to initiate more-extensive analyses of reticulate evolution within our genome. This is particularly important within the ∼5% of the finished human genome sequence that consists of recently formed segmental duplications (Bailey et al. 2002; She et al. 2004a). These regions contain >6% of all human RefSeq exons (Bailey et al. 2002) and harbor examples of both novel chimeric transcripts and genes of unknown function that have undergone rapid positive selection (Johnson et al. 2001; Crosier et al. 2002; Ruault et al. 2003; She et al. 2004a; Ciccarelli et al. 2005). These sequences are also of clinical importance: a large number of duplication/deletion syndromes—such as Charcot-Marie-Tooth, velocardiofacial (VCFS), DiGeorge, William, and Prader-Willi syndromes—are due to recombination between nonallelic copies of specific duplicon familes (reviewed by Shaw and Lupski [2004]), whereas structural variation associated with duplicons (Iafrate et al. 2004; Sebat et al. 2004) can predispose to pathological rearrangements in specific cases (Gimelli et al. 2003). Knowledge of the patterns of reticulation is, therefore, central to our understanding both of genomic mutations that cause disease and of gene evolution. Furthermore, many of the remaining gaps in the human sequence lie within duplicated DNA, and a significant amount of duplicated sequence remains to be integrated into the overall reference sequence (International Human Genome Sequencing Consortium [IHGSC] 2004). If reticulation is common within this sequence, it may pose a further obstacle to the completion of the human sequence map. It is clear, therefore, that patterns of reticulate evolution within human DNA must be assessed.
Many methods routinely used to identify patterns of reticulate evolution require multiple sequence alignments, rather than pairwise alignments, and are complicated by the fact that the products of unequal crossing-over and gene conversion are often indistinguishable at the sequence level (reviewed by Drouin et al. [1999]). Most use sliding-window analyses and/or deviations from trees of sequence relationships to define discontinuities in phylogenetic signal (e.g., Sawyer 1989; Guttman and Dykhuizen 1994; Maynard Smith and Smith 1998; Proutski and Holmes 1998; Weiller 1998; McGuire and Wright 2000; Hurles 2001; Husmeier and McGuire 2003). The fact that these analyses vary extensively in their power, sensitivity, and computational cost has led to the suggestion that multiple methods should be employed in a coordinated fashion (Wiuf et al. 2001). Here, we use a combination of phylogenetic profiling (Weiller 1998), permutation tests (Sawyer 1989; Hurles 2001), and a novel quartet method to assess the relative contribution of reticulation and nucleotide substitution to the evolution of >8 Mb of recently duplicated human DNA. Our results show that reticulation is endemic within these closely related human duplications and, in many instances, has led to a 20-fold excess of sites consistent with reticulation relative to the expected value. This indicates that completion of the human sequence map will be even more difficult than is currently realized and that the contribution of reticulation events to the evolution of gene families may be greater than is currently appreciated.
Material and Methods
Generation of Multiple Alignments
Sequences of individual BAC clones that map to regions rich in segmental duplications (see the “Results” section) were analyzed using NIX (Williams et al. 1998). Paralogues were identified through inspection of BLAST (Altschul et al. 1990) output, and individual clones that maximized the overlap of distinct paralogues were identified using Pairwise FLAG (see Web Resources). Sequences were aligned using the Clustal V implementation within the Lasergene suite of software (DNAstar) by use of a gap-creation penalty of 10 and a gap-extension penalty of 3, to favor gap extension in regions with high mutation rates, such as di- and tetranucleotide repeats. Terminal regions of nonalignment were subsequently trimmed, and each alignment was manually edited to ensure that regions of poor/arbitrary alignment were optimized through the introduction of gap characters. All alignments were then stripped of all positions containing one or more gaps, and PAUP version 4.0 β10 (Swofford 2003) was used to construct neighbor-joining (NJ) trees under a Felsenstein 84 (F84) model of evolution with 1,000 bootstrap replicates. To remove ambiguity in tree topology, if any tree contained a node with <90% bootstrap support, then one sequence at that node was removed from the alignment and the tree was reconstructed. If necessary, this process was repeated until all nodes had >90% bootstrap support. Two alignments were retained within the data set despite the presence of one weak node (Morph-1 and SMA-1), because removal of further sequences did not improve the bootstrap values of the reduced trees. Trees were also generated from these alignments by use of maximum parsimony, and, in all cases, the topology was identical to the NJ trees.
A total of 30 alignments generated in this way were used for subsequent analysis. The criteria for inclusion of an alignment in the data set were the presence of a minimum of four distinct sequences (to allow the use of quartet methods) and sequence identity across >15 kb. These alignments therefore only represent duplicon families with ⩾4 copies that have been sequenced as part of the human genome project. When necessary because of uneven clone overlaps or duplications extending over multiple clones, a single duplicon was represented by more than one nonoverlapping alignment. In addition, two alignments (chAB4 and Morpheus) were each split into two separate alignments because of the presence of two diverged clades, which effectively removed one long internal branch (see generation of alignment data set in the “Results” section).
These alignments and their NJ trees represent the primary data (referred to as “CpG-positive data”). A parallel set of data consisting of all alignments in which CpG dinucleotides had been removed was also generated, and NJ trees were developed as described above (referred to as “CpG-negative data”). The CpG-negative alignments are only 1.4%–3.8% shorter than the CpG-positive alignments but contain 18%–47% fewer variable positions (not shown), which illustrates the high frequency of mutations within CpG dinucleotides. In several cases, the bootstrap values within the CpG-negative trees fell below 90%. However, in only one case (Morph-1) was the topology of the CpG-negative tree different from that of the CpG-positive tree.
In addition to the test data, 13 multiple alignments were generated using primate sequences from nine genes within Target 1 of the National Institutes of Health Intramural Sequencing Center (NISC) comparative vertebrate sequencing initiative (see NISC Comparative Vertebrate Sequencing Web site). This encompasses ∼1.5 Mb of DNA surrounding the CFTR locus in 7q31.2 and includes the WNT2, CAV1, MET, and ST7 genes. These alignments, which exceed 270 kb in total length and include >1.8 Mb of sequence from up to seven primate species, are control alignments because the sequence is single copy and levels of reticulation are assumed to be low. However, it is noteworthy that the mean pairwise distances within these trees are larger than those of the test data set (0.042 vs. 0.01) because of the evolutionary relationships between the species used.
Generation of Simulated Data
One hundred simulated alignments for each CpGpositive and CpG-negative alignment were artificially evolved with Seq-Gen (Rambaut and Grassly 1997) by use of the appropriate alignment length, tree topology, and branch lengths from each NJ tree. In all cases, a F84 model of evolution was used, and both the nucleotide frequencies and the transition/transversion rates were taken from the observed data.
Phylogenetic Profiling and Network Analysis
Phylogenetic profiling was performed using PhylPro (Weiller 1998). This method correlates local pairwise distance measures between sequence windows on both sides of a single position within a multiple alignment. By use of sliding windows of fixed size, putative recombination sites can be identified as sharp changes in correlation between distance measures in the two windows. In all cases, a range of window sizes from 15 to 80 parsimony-informative sites and 40–120 variable sites were analyzed because the optimal window size is influenced by the level of divergence between sequences in the alignment and by the nature of reticulation within the data. In each case, a simulated data set (see the “Generation of Simulated Data” section) was used to generate a control profile and was analyzed using the same parameters. All network analyses were performed using SplitsTree 4 (D. H. Huson and D. Bryant, unpublished data; see Algorithms in Bioinformatics: SplitsTree4 Web site), with parsimony splits and equal angle display.
Quartet Analysis
A method to identify evidence of reticulation was developed that examines the patterns of character states at positions within a multiple alignment that do not support the best tree generated from that alignment. Unlike other methods that adopt this approach, such as the homoplasy test used to assess horizontal gene transfer in bacteria (Maynard Smith and Smith 1999), this method does not require the estimation of alternative trees between taxa and only analyzes specific subsets of quartets within the data. Consider the alignment of four sequences s1, s2, s3, and s4, with sij designating the jth position of the ith sequence and with the phylogenetic relationships between the four sequences described by a tree with the topology 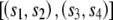 (shown graphically in fig. 1A). Let us consider the quartet of bases {s1j,s2j,s3j,s4j} in the four sequences at position j of the alignment. In quartets where either s1j≠s2j, s1j=s3j, and s2j=s4j or s1j≠s2j, s2j=s3j, and s1j=s4j (e.g., fig. 1B), a minimum of two substitution events needs to be invoked, one within each clade of the tree, in which both mutations are to the same nucleotide. However, these quartets can also be accounted for by one substitution followed by a reticulation event (conversion/recombination). We designate these quartets here as reticulate. Since the probability of multiple substitution events can be derived from branch lengths, substitutions rates, and the composition of the ancestral sequence, we can compare the observed number of reticulate quartets with that expected under a model of substitution alone. An excess of reticulate quartets can be interpreted as evidence of reticulation.
(shown graphically in fig. 1A). Let us consider the quartet of bases {s1j,s2j,s3j,s4j} in the four sequences at position j of the alignment. In quartets where either s1j≠s2j, s1j=s3j, and s2j=s4j or s1j≠s2j, s2j=s3j, and s1j=s4j (e.g., fig. 1B), a minimum of two substitution events needs to be invoked, one within each clade of the tree, in which both mutations are to the same nucleotide. However, these quartets can also be accounted for by one substitution followed by a reticulation event (conversion/recombination). We designate these quartets here as reticulate. Since the probability of multiple substitution events can be derived from branch lengths, substitutions rates, and the composition of the ancestral sequence, we can compare the observed number of reticulate quartets with that expected under a model of substitution alone. An excess of reticulate quartets can be interpreted as evidence of reticulation.
Figure 1.

Examples of reticulate and bimutational quartets. See description of quartet classification in the “Material and Methods” section.
For a control, we examine the second class of quartets that also require a minimum of two substitutions, one within each clade (e.g., fig. 1C). However, in these quartets, the inferred mutations are to different nucleotides, and postulating a reticulation event does not reduce the number of substitutions that must be inferred. We designate these quartets as bimutational. They satisfy the criteria s1j≠s2j, s3j≠s4j, and either s1j=s3j and s2j≠s4j or s2j=s3j and s1j≠s4j. The expected number can again be predicted from branch lengths, substitution rates, and ancestral sequence composition. We can, therefore, look for evidence of reticulation and control for the consistency of our data sets by calculating the frequency of reticulate and bimutational quartets over all alignment positions—and for all possible sequence quartets—within a given alignment. To do this, we assume that the relationships between the sequences are described by the NJ trees obtained from the multiple alignment, with confidence intervals for the quartet frequencies computed by bootstrapping over all positions in the alignment 1,000 times. To take into account the potential influence of multiple substitutions on quartet frequencies, the observed values are then compared with the frequencies obtained from 100 simulated data sets generated as described in the previous section.
Quartet Analysis of Tract Length
We can use quartet analysis to establish the length of tracts that are consistent with reticulation between two sequences. We confine our analysis to tracts that contain at least one reticulate quartet. We designate a tract beginning at position lb and ending at le as consistent with reticulation between the sequences s1 and s2 if s1l=s2l for lb<l<le, s1lb≠s2lb, and s1le≠s2le and if there is at least one position lc, lb<lc<le, for which there is a pair of sequences si and sj that fulfill the phylogeny 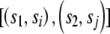 and where silc=sjlc and s1lc≠silc. The length of such a tract is L=le-lb-1.
and where silc=sjlc and s1lc≠silc. The length of such a tract is L=le-lb-1.
The average tract length overall for sequence pairs of a data set was determined and was compared with the distribution derived from 1,000 control data sets generated by random permutation of the alignment positions. An average tract length longer than expected is consistent with reticulation. Tracts longer than the 99th percentile tract in the control data sets were retained for further analysis. To generate a minimal set of tracts, overlapping pairwise tracts with one sequence in common were assumed to be due to the same event, and the shorter one was discarded.
Partition Analysis of Reticulation Event Number
In a subset of alignments, the expected frequency of reticulate quartets is negligible (see the “Results” section), which makes it possible to assume that all of the reticulate quartets within the observed data are due to reticulation events. In these cases, it is possible to examine the distribution of reticulate quartets and to estimate the minimum number of reticulation events required to produce the observed data. This analysis was performed on the CpG-negative data by analyzing the output from the Partimatrix program (Jakobsen et al. 1997). This displays the consistency of all partitions within the data set. Each parsimony-informative site with two character states supports a single partition within the data, consisting of two groups of taxa, each with a single character at that position. The tree that describes the phylogenetic relationship between the sequences in the alignment will be supported by one partition for each internal branch. Partitions that do not support the tree are interpreted as being due to homoplasious substitutions (i.e., independent substitutions to the same character state) and/or reticulation events. When the expected frequency of reticulate quartets is negligible under a model of evolution by nucleotide substitution, it means that few or no homoplasious changes are expected to be observed, given the tree topology, branch length, and sequence length. Under these exceptional circumstances, it follows that observed partitions that do not support the tree are due to reticulation events.
As an example of this method, the cladogram for one alignment (c9orf36) is shown in figure 2A. All the partitions supported by parsimony-informative sites within the alignment are represented in the partition matrix shown in figure 2B. The three partitions compatible with the tree are to the left of the vertical dotted line, those incompatible with the tree are to the right. The minimum number of events can be defined by counting the number of independent runs of sites that are incompatible with the tree. For the example shown here, there are a minimum of four reticulation events, calculated as follows. Position 1 (352 bp) is incompatible with the tree and so is defined as a reticulation event. The six sites between 1648 bp and 1861 bp (inclusive) represent a second event because they are incompatible with the tree but are all compatible with each other (i.e., partition 1A and 1B can be present on the same tree). However, the adjacent site (position 1935) is incompatible both with the tree and with partition 1B and so is indicative of a third reticulation event. The fourth and final reticulation is defined by positions 5904 bp and 7427 bp, which are incompatible with the tree. Although they are separated by two positions in partition 1D (6496 bp and 6801 bp), all three partitions (1D, 5, and 1A) are compatible and so could be the product of a single event encompassing positions 5904–7427 bp. By traversing all positions within the alignment in this manner, one can define a minimal number of reticulation events. This can be divided by the total length of all sequences in the alignment to give a minimum estimate of the density of reticulation events. These estimates are only valid for alignments for which the expected frequency of reticulate quartets is negligible, and they are minimal estimates only (see the “Discussion” section).
Figure 2.

Estimate of reticulation-event density. A, Cladogram of c9orf36 alignment. Sequences are defined by their RPCI11 BAC clone names. The three partitions that support the tree (18, 19, and 1D) are indicated. B, Partition matrix of proximal 8.2 kb of c9orf36 alignment. Sites support 16 different partitions; the two sequence groups that define each partition are indicated by black and white circles above the matrix, and the partitions that support the tree are to the left of the vertical dashed line. Each informative position is represented by a separate row of squares (numbered on the right). The specific partition defined by each informative site is indicated by a white square containing a black dot. All partitions compatible with this partition are shown as white squares, and all partitions incompatible with it are shown in black. Positions that support alternative partitions are assumed to be the result of reticulation. The four reticulation events inferred from the data are numbered 1–4, and the maximal extents of the sequences affected are indicated by dashed horizontal lines.
Results
Generation of Alignment Data Set
To investigate patterns of reticulate evolution within recently duplicated human DNA, we have generated a data set consisting of 30 multiple alignments encompassing >8 Mb of DNA (see the “Material and Methods” section) from regions of the genome rich in recent segmental duplications (table 1). Eleven alignments are from the pericentromeric region of human chromosome 9 (Humphray et al. 2004), and 13 come from six previously characterized duplicon clusters, including the Morpheus genes on chromosome 16 (Johnson et al. 2001), the creatine transporter–related genes in 16p11 (Eichler et al. 1996), the DiGeorge/VCFS region in 22q11 (Saitta et al. 2004), the site of an ancestral centromere in 15q25 (Ventura et al. 2003), the spinal muscular atrophy (SMA) region in 5q13.2 (Courseaux et al. 2003), and the chAB4 sequence family (Cserpan et al. 2002). A further six consist predominantly of sequences from subtelomeric regions of the genome and include RPL23A-, COB-W–, and PGM5-related sequences (Fan et al. 2002). These alignments were generated from individual clones to avoid the incorporation of potential assembly errors into the data set (Bailey et al. 2002) and to include chAB4 sequences that have not been integrated into the current genome assembly. The resulting alignments vary in length from 14.1 to 108.3 kb, with the length constrained by clone overlap and the extent of contiguous sequence identity (see the “Material and Methods” section). All sequences within the alignments show very high sequence identities, with mean percentage identity within alignments that varies from 96% (chAB4-1) to 99.6% (15q25).
Table 1.
Details of Multiple Alignments[Note]
| Alignment Groupand Alignment | GCContent | No. ofSequences | Length ofUngappedAlignment | No. ofVariablePositions inAlignmenta | LowestBootstrapValue inNJ Tree | Mean F84DistancebetweenSequences | AccessionNumberof ReferenceSequence | CytogeneticLocationof ReferenceSequence | Position onSpecifiedChromosome(Mb) | StartNucleotideb | StopNucleotideb |
| Telomeric: | |||||||||||
| 19pter | 44.9 | 10 | 16,761 | 693 | 92 | .012 | AC010507 | 19p13.3 | .15 | 18371 | 35303 |
| 22qter (RPLA) | 39.2 | 10 | 14,183 | 560 | 100 | .017 | AC002055 | 22q13.33 | 49.5 | 23507 | 40664 |
| 7pter (GTFII-I) | 44.8 | 7 | 25,733 | 841 | 99 | .012 | AC139136 | 7p22.3 | .1 | 101307 | 123977 |
| 11pter | 44.3 | 7 | 22,303 | 899 | 100 | .015 | AC069287 | 11p15.5 | .1 | 38627 | 114838 |
| 2qfus (COB-W) | 36.6 | 6 | 65,887 | 1,672 | 100 | .011 | AC016745 | 2q13 | 113.9 | 81331 | 148627 |
| 9pter (PGM5) | 41.1 | 7 | 51,672 | 1,621 | 100 | .013 | AL449043 | 9p24.3 | .5 | 17976 | 71624 |
| Duplicons 9: | |||||||||||
| fk506 | 40.5 | 6 | 68,980 | 1,449 | 100 | .01 | AL591867 | 9p12 | 42.9 | 10230 | 82500 |
| c9orf36 | 41.8 | 6 | 61,129 | 714 | 100 | .005 | AL590491 | 9p12 | 41.4 | 63624 | 125701 |
| Alphoid | 40.7 | 4 | 55,146 | 281 | 100 | .003 | AL512605 | 9q13 | 67.9 | 16171 | 73537 |
| CASPR3 | 38.4 | 5 | 99,373 | 2,413 | 100 | .013 | AL162501 | 9p12 | 39.2 | 1 | 101926 |
| PCC-SR | 40.7 | 6 | 40,151 | 909 | 100 | .012 | AL592525 | 9p13.1 | 40.1 | 92656 | 134714 |
| VSPA | 41.2 | 5 | 51,345 | 939 | 100 | .01 | AL354718 | 9p11.2 | 44.4 | 88333 | 147110 |
| KGF | 40.4 | 6 | 46,259 | 1,836 | 100 | .019 | BX088717 | 9p13.1 | 43.5 | 1 | 46577 |
| SRP19 | 45.3 | 5 | 65,495 | 633 | 100 | .005 | AL953889 | 9q12 | 65.3 | 1 | 73032 |
| SAT5 | 36.5 | 6 | 47,464 | 1,727 | 100 | .017 | AL669942 | 9p11.2 | 45.9 | 1 | 71767 |
| ZNF91 | 38.7 | 5 | 57,388 | 3,839 | 100 | .033 | AL353626 | 9q12 | 64.4 | 8194 | 69485 |
| ANKRD20A | 37.7 | 4 | 85,750 | 844 | 100 | .005 | AL355000 | 9p11.2 | 45.6 | 1 | 85626 |
| Other duplicon: | |||||||||||
| Morph-1 | 46.6 | 5 | 17,802 | 177 | 88 | .005 | AC138932 | 16p13.11 | 14.9 | 84377 | 102113 |
| Morph-2 | 46.5 | 5 | 16,184 | 310 | 98 | .009 | AC138904 | 16p11.2 | 28.2 | 158027 | 176221 |
| 16p11-1 | 40.8 | 6 | 108,314 | 635 | 100 | .003 | AC136613 | 16p11.2 | 32.1 | 26678 | 160871 |
| 22q11-1 (POM21) | 47.2 | 6 | 60,487 | 482 | 100 | .004 | AC008132 | 22q11.21 | 17.1 | 84949 | 145598 |
| 22q11-2 | 42.4 | 9 | 14,130 | 385 | 92 | .01 | AC008079 | 22q11.21 | 16.9 | 117088 | 131322 |
| 22q11-3 (GGT1) | 57.4 | 7 | 13,277 | 138 | 97 | .004 | AC023491 | 22q11.21 | 19.9 | 80248 | 93536 |
| 15q24 (Golgin) | 48.4 | 10 | 46,068 | 474 | 100 | .004 | AC044860 | 15q25.3 | 83.5 | 104032 | 154535 |
| chAB4-1 | 39.1 | 6 | 77,693 | 4,188 | 99 | .021 | AC138771 | NA | NA | 78797 | 157423 |
| chAB4-2 | 39.2 | 4 | 76,302 | 5,506 | 100 | .04 | AL355793 | NA | NA | 9687 | 94151 |
| 16p11-2 (CT1) | 47.5 | 5 | 73,684 | 2,037 | 100 | .012 | AC133561 | 16p11.2 | 33.7 | 86042 | 162485 |
| SMA-1 | 35.2 | 7 | 84,843 | 491 | 86 | .003 | AC140139 | 5q13.2 | 70.6 | 1 | 84843 |
| SMA-2 | 36.3 | 5 | 72,614 | 372 | 100 | .003 | AC145138 | 5q13.2 | 70.4 | 126437 | 199372 |
| SMA-3 | 39.5 | 4 | 58,769 | 260 | 100 | .003 | AC138827 | 5q13.2 | 68.9 | 85001 | 143650 |
| Controls: | |||||||||||
| ASZ1 | 36.0 | 7 | 20,011 | 1,987 | 100 | .043 | AC002465 | 7q31.2 | 116.6 | 118627 | 143479 |
| CAPZA2 | 36.8 | 7 | 30,266 | 3,063 | 100 | .044 | AC002543 | 7q31.2 | 116.1 | 78003 | 119125 |
| CAV1 | 40.3 | 7 | 36,625 | 3,705 | 100 | .042 | AC006159 | 7q31.2 | 115.7 | 5310 | 44554 |
| CAV2 | 36.8 | 7 | 13,334 | 1,477 | 100 | .048 | AC002066 | 7q31.2 | 115.7 | 76577 | 90958 |
| CFTR | 33.2 | 7 | 11,319 | 1,079 | 100 | .042 | AC000111 | 7q31.2 | 116.8 | 44520 | 75032 |
| MET | 38.0 | 7 | 30,039 | 2,624 | 100 | .037 | AC002080 | 7q31.2 | 115.9 | 92482 | 123416 |
| TESS | 37.8 | 7 | 14,557 | 1,472 | 100 | .044 | AC073130 | 7q31.2 | 115.4 | 143191 | 159417 |
| WNT2 | 41.6 | 7 | 23,124 | 1,933 | 100 | .035 | AC002465 | 7q31.2 | 116.5 | 16471 | 44579 |
| ST7-1 | 38.6 | 6 | 19,870 | 1,846 | 100 | .046 | AC002542 | 7q31.2 | 116.2 | 13440 | 34364 |
| ST7-2 | 37.7 | 6 | 18,194 | 1,628 | 100 | .044 | AC002542 | 7q31.2 | 116.3 | 34693 | 54088 |
| ST7-3 | 39.5 | 6 | 15,619 | 1,397 | 98 | .043 | AC002542 | 7q31.2 | 116.3 | 54129 | 70947 |
| ST7-4 | 39.2 | 6 | 14,961 | 1,298 | 100 | .042 | AC002542 | 7q31.2 | 116.3 | 71291 | 86572 |
| ST7-5 | 36.8 | 6 | 22,765 | 1,671 | 100 | .037 | AC002542 | 7q31.2 | 116.4 | 86735 | 110087 |
Note.— Since we have made no assumptions concerning the relationships between sequences, some alignments may contain allelic copies of duplicons. NA = not available.
Because of variation in alignment length and the number of sequences aligned, the number of variable positions ranges from as few as 138 (22q11-3) to 5506 (chAB4-2).
Nucleotide positions of reference sequence in alignment.
Application of Phylogenetic Profiling to Human Duplicon Alignments
As a preliminary analysis of reticulation, we used phylogenetic profiling (Weiller 1998) to investigate the consistency of the phylogenetic signal within all 30 multiple alignments. Examples of the resulting profiles are shown in figures 3 and 5. All profiles show sharp changes in correlation, with most of the alignments producing a small number (1–7) of clearly defined changes consistent with NAHR events. For example, the phylogenetic profile of the alignment from the ancestral centromere region in 15q25 (fig. 3A) has a dip in correlation, with a minima at −0.3 that lies within sequence related to the chondroitin sulphate gene. The simulated control profile shows no dips in correlation <0.7. The source of this change in correlation is clear from the phylogenetic trees of the two regions it defines (fig. 3B), which differ in the position of a single sequence: ac044860. The movement of this sequence between two distinct clades is consistent with it being a recombination product between two sequences, one related to the sequences AC135735, AC127482A, and AC127482B, the other to AC005630 and AC010725.
Figure 3.

Identification of reticulation events by use of phylogenetic profiling. A, Control and observed profiles of 21-kb section of 15q25 alignment created using a window size of 30 parsimony-informative sites. The extent of gene-related sequences is indicated. The X-axis shows position within alignment (in kb); the Y-axis shows correlation. B, NJ trees generated using subalignments from regions 1 and 2. The clades indicated with an asterisk (*) are supported by bootstrap values of 99%–100%. The scale (F84 distance) is the same for both trees. All sequences are indicated by the last three digits of their accession numbers. Sequences included are AC044860, AC127482, AC135735, AC135995, AC005630, and AC010725. AC127482 contains two copies of the duplication, A and B. C, Schematic structure of both SMA alleles (Var1 and Var 2) adapted from Schmutz et al. (2004). The positions of the SMN1 and SMN2 genes are indicated. The extent of duplicated sequence is shown in gray, with the position of the most abundant duplicated segments (V1.1–V2.3) indicated. The gap in the sequences is represented by a pair of dashed lines. The scale is in megabases. D, Control and observed profiles spanning the ∼85-kb SMA-1 alignment, created using a window of 20 parsimony-informative sites. The X-axes show informative sites; the Y-axes show correlation. E, Parsimony networks of all six repeats within allele 1 (left) and all nine repeats within both alleles (right). Scale is in nucleotide differences. Sequences aligned (in order from V1.1 to V2.3) are AC138957, AC131392, AC138866, AC138959, AC138911, AC140139, AC139500, AC108108, and AC138930. Examples of alignments of informative sites used to generate the profiles are provided in figure 4.
Figure 4.


Examples of sequence alignments used to generate profiles. Partial sequence alignments stripped of all invariant and uninformative sites are shown, to highlight changes in phylogenetic signal within the profiles presented in figures 3 and 5. Each alignment is shaded with respect to a reference sequence shown in gray, with all identities to the sequence shown in black. A, 15q24. B, SMA-1. C, 22qter. D, chAB4-2 minima 1. E, chAB4-2 minima 2. F, 22q11.1.
Figure 5.

Reticulations identified by phylogenetic profiling. For ease of presentation, only parsimony-informative positions are plotted, with a window size of 80, 50, and 40 parsimony-informative positions in panels A, B, and C, respectively. The number of positions identical to a reference sequence within the alignment (used to calculate the correlation) is shown for both windows flanking the numbered minima. Thus, in panel A, AP006327 is identical to the reference at 66 of 80 parsimony-informative positions to the left of the minima at position 257, but only 15 of 80 sites to the right of the minima. All numbered minima are >2 times lower than any observed in the control profiles (not shown). The chromosome 22q11-1 alignment (C) has >7 minima that exceed this control threshold.
In contrast, the alignment from the 22q11 and SMA regions gives more complex results in which the profile of each sequence overlaps to produce a more chaotic signal (figs. 3C–3E and 5C). This is most striking in the SMA-1 alignment that spans ∼85 kb of the repeat and includes sequence from two alleles of this genome region (Schmutz et al. 2004). The basic repeat structure of this region is shown in figure 3C, with the repeats in each allele numbered according to physical position. The phylogenetic profile of this alignment (fig. 3D) shows multiple overlapping troughs (>15), with all sequences affected by changes in correlation, which is indicative of a very complex evolutionary history throughout the length of the alignment. The profile also identifies a putative 7-kb recombination hotspot defined by two tandem repeats in which sequence identities rise to 99.96% (see fig. 6). To illustrate the complexity of the sequence relationships further, the phylogenetic networks of all the repeats present in both alleles are also shown (fig. 3E). The network of repeats from allele 1 is relatively simple, with a single major split in the data. The split between V1.2/V1.5 and all other repeats is favored by ∼60 parsimony-informative positions, but ∼15 positions place V1.2 with V1.6/V1.4 and V1.5 with V1.1/V1.3. Four repeats (V1.1, V1.3, V1.4, and V1.6) are independent of the network. However, when the three repeats from allele 2 are included, a further five splits are introduced into the network, which makes it clear that there is no simple relationship between the position of each repeat within alleles 1 and 2 and their phylogeny. Only repeats 1 and 2 of allele 1 (V1.1/V1.3) share a common origin independent of the network.
Figure 6.

Delineation of putative hotspot in SMA-1 region. Output from SimPlot (version 3.2) developed by S. Ray (Lole et al. 1999) shows identity of all sequences within the SMA1 alignment to AC138959 (500-bp window with a 20-bp step size). All nine sequences share ∼99.96% identity within the region of 59–67 kb. B, Detailed view, showing landmarks within the 56–70-kb region. The region of maximal identity between the sequences is defined by an L1PA3 fragment (∼58 kb) and a highly variable AT dinucleotide repeat (68 kb). A further L1PA3 repeat distal to this AT dinucleotide creates a flanking direct repeat (both LIPA3s span positions 5721–6155 of the consensus L1 sequence).
Application of a Quartet Test for Reticulation to Human Duplicon Alignments
The phylogenetic profiling confirms both that reticulate evolution is extremely common within recent duplications—with all 30 alignments showing evidence of such events—and that the patterns of reticulation vary enormously, from well-defined exchange events consistent with small numbers of unequal crossovers to extremely complex patterns likely to involve extensive gene conversion events (fig. 3). To assess the relative contribution of reticulation and nucleotide substitution to the evolution of these sequences, we have developed a method that analyzes the extent and nature of phylogenetic signal that contradicts the trees generated from our multiple alignments (see the “Material and Methods” section and fig. 1 for details). This analyzes the frequency of quartets within an alignment in which either two independent mutations to the same nucleotide have occurred or one mutation and one reticulation event have occurred (reticulate quartets; see fig. 1B). An excess of these quartets relative to the expectation provides evidence of reticulate evolution within the alignment. As a control, we analyze the frequency of quartets in which we can infer that two mutations to different nucleotides have occurred (bimutational quartets; see fig. 1C).
The results of this analysis performed on all 30 CpG-positive alignments and the 13 primate control alignments are shown in figure 7A. The mean percentage of reticulate quartets in the simulated data (shown in red) is <1% for all alignments, with the exception of chAB4-1 and chAB4-2, which are the two most divergent alignments (table 1). In contrast, the observed frequencies of reticulate quartets (shown in black) vary from 0.1% (VSPA) to 47% (ANKRD20A), and it is clear that there is a significant excess of reticulate quartets, relative to the expected numbers, in virtually every alignment, including the single-copy primate control alignments. The control group shows the most modest excess, with observed values 2–4-fold higher than expected (range 1.15%–4.73% of informative quartets). In contrast, all six telomeric alignments show a 7–10-fold excess, with reticulate quartets accounting for between 4.2% and 9.3% of the total. However, the most extreme excess is observed in the alignments from the pericentromeric region of chromosome 9 and other nontelomeric duplicons, with 16 of 24 having an excess of ⩾20 fold. In 15 alignments, reticulate quartets account for >10% of all informative quartets, and, in 11 alignments (Alphoid, ANKRD20A, 22q11-1, 22q11-3, chAB4-1, chAB4-2, both Morpheus alignments, and all three SMA alignments), the frequency is >20%. This represents an enormous excess relative to the expectation.
Figure 7.

Quartet analysis of multiple alignments. A, Reticulate quartets in CpG-positive data expressed as a percentage of all informative quartets. B, Bimutational quartets in CpG-positive data expressed as a percentage of all informative quartets. The insert shows the same data at a higher resolution. C, Reticulate quartets in CpG-negative data expressed as a percentage of all informative quartets. Bars on observed data show 95% bootstrap values, and bars on simulated data show 95% CIs. In the 22q11.1 CpG-negative alignment, reticulate quartets represent >50% of all informative quartets. This is a result of low bootstrap values within the NJ tree.
In sharp contrast to the results for reticulate quartets, there is little discernable difference between the observed and simulated results for bimutational quartets (fig. 7B). Both observed and expected quartet frequencies are consistently low in all alignments, and only the two most diverged alignments (chAB4-1 and chAB4-2) have frequencies exceeding 4%. Only the CASPR3 alignment shows a significant excess relative to the expectation. In the 42 remaining alignments, there is extensive overlap between the bootstrap values obtained from the real data (black bars) and the 95% CIs calculated from the simulations (red bars).
It is noteworthy that, in 16 alignments, the frequency of reticulate quartets (in which both changes are to the same nucleotide) is in >20-fold excess relative to both the simulated data and the observed frequency of bimutational quartets (in which the changes are to two different nucleotides). For instance, the median frequency of reticulate quartets in the SMA-1 simulated alignments is 0.0% (95th percentile 0.82%) compared with an observed value of 28.85% and an observed frequency of bimutational quartets of 1% (fig. 7A and 7B). It is clear from these results that reticulate quartets are in significant excess within the data, often representing >1 in 5 informative quartets but that bimutational quartets are not in excess. This is wholly consistent with the expectation if the sequence evolution of the aligned duplications involves extensive reticulate processes.
Influence of CpG Distribution on Reticulation Signal
Our method assumes that substitution patterns at different nucleotides within an alignment are independent. However, in eukaryotic genomes, this assumption is violated in CpG dinucleotides in which the high frequency of spontaneous deamination leads to an excess of C→T transitions. Genomewide estimates suggest that this occurs at a frequency ∼1 order of magnitude higher than substitution rates (Fryxell and Moon 2005). Both the high frequency of these events and the specificity of the mutational event could increase the observed frequency of reticulate quartets to above that expected. To establish whether this phenomenon is contributing to the observed excess within our data set, we removed all positions that contained one or more nucleotides within a CpG dinucleotide from our alignments (CpG-negative data) (see the “Material and Methods” section). We then performed simulations and reanalyzed the data, the results of which are shown in figure 7C. Athough the confidence intervals associated with both observed and simulated CpG-negative data were larger than those for the CpG-positive data—as a result of the smaller number of variable sites in each alignment (see the “Material and Methods” section)—the observed frequency of reticulate quartets within the test data was not significantly affected. In fact, the results were strikingly similar to those obtained with the CpG-positive data (fig. 7A), with a 7.3–11.4-fold excess within telomeric alignments and a >20-fold excess in 16 alignments of duplicons from the pericentromeric region of chromosome 9 or nontelomeric duplicons. This confirms that CpG mutability is not responsible for the observed excess of reticulate quartets within our data set. However, the removal of CpG dinucleotides reduced the excess of reticulate quartets observed in the 13 primate control alignments by ∼40%, from a mean of 3.36% in the CpG-positive data to 2.07% in the CpG-negative data. This indicates that a large proportion of the excess observed in the CpG-positive primate controls (fig. 7A) can be accounted for by mutability of CpGs. Despite this, the small differences between the observed and simulated primate control data remain significant in 6 of 13 cases (data not shown).
Correlation of Levels of Reticulation with Alignment Identity
Both gene conversion and NAHR are homology-dependent processes, which are promoted by localized regions of near-perfect identity (Stankiewicz and Lupski 2002). We would therefore predict that levels of reticulation are not independent of sequence identity. To investigate this, we plotted the ratios of observed to expected frequencies of reticulate quartets within the CpG-negative data (fig. 7C) against the average pairwise identity within each alignment (table 1). The results are shown in figure 8, and a highly significant correlation between the excess of reticulate quartets and alignment identity is observed (r2=0.599). This is particularly clear from the fact that 15 of 16 alignments in which the average pairwise identity is ⩾99% (table 1) have an observed:expected ratio of >10, compared with only 2 of 14 alignments in which the average identity is <99% (CASPR3 and KGF).
Figure 8.

Reticulation in relation to sequence identity. Linear regression of log-transformed data is shown as a solid line (r2=0.599), and 95% CIs are shown as dashed lines.
Impact of Reticulation upon Tracts of Sequence Identity
The quartet analysis provides striking evidence that reticulation is common within recently duplicated human DNA. However, it provides no information on the distribution of such events. If reticulation is occurring, we would expect to see clustering of reticulate quartets, because each event may affect multiple nucleotides. To establish whether this expectation is met, we performed permutation tests using two independent methods. We calculated the number of reticulate quartets within each tract of perfect identity between all pairs of sequences within each alignment of the CpG-negative data set, and we compared this to expectations from 1,000 pseudosamples in which alignment position had been randomized (see the “Material and Methods” section). These results, in the form of observed:expected ratios, are presented in figure 9. The 13 primate control alignments yield ratios that vary from 0.92 to 1.11, and in no case are the observed data significantly different from the simulations. This establishes that the small excess of reticulate quartets within these controls shows no significant clustering, which is inconsistent with this excess being due to reticulation. In contrast, 25 of the 30 alignments of human duplicons have an observed mean tract length that exceeds 95% of the 1,000 simulations (P<.05), and 19 of the 30 exceed 99% of the simulations (P<.01). The ratio of observed to expected frequencies within these alignments varied from 1.3 (chAB4-1) to 3.0 (SMA-3). This means that tracts of perfect identity within the SMA-3 alignment are ∼3 times longer than expected given the observed phylogenetic relationships. We also analyzed the CpG-positive data by use of the permutation test method described by Hurles (2001), which corrects directly for the influence of CpG deamination. These analyses, which look at all pairwise alignments (including nearest neighbors), identified a significant excess in the length of tracts of identity in all test alignments, confirming the results obtained with the CpG-negative data (see table 2).
Figure 9.

Tract length increase in duplicons. The ratio of observed to expected tract lengths is shown for control and duplicon alignments.
Table 2.
Examples of Sawyer CpG Permutation Tests
| A. 22q11.2 | |||||||||
| SSUF (above diagonal) and MUF (below diagonal) for Sequencesa |
|||||||||
| ac007325 |
ac007708 |
ap000550 |
ap000552 |
ac007981B |
ac007981A |
ac011718A |
ac008103 |
ac008079 |
|
| Sequences | Observed Valuesb | ||||||||
| ac007325 | 2.76E+06 | 2.93E+06 | 2.67E+06 | 3.34E+06 | 1.02E+07 | 4.86E+06 | 4.14E+06 | 2.94E+06 | |
| ac007708 | 559 | 9.22E+06 | 7.89E+06 | 4.11E+06 | 3.61E+06 | 2.99E+06 | 4.79E+06 | 5.11E+06 | |
| ap000550 | 598 | 1989 | 8.20E+06 | 4.30E+06 | 3.14E+06 | 2.86E+06 | 3.92E+06 | 4.07E+06 | |
| ap000552 | 500 | 1629 | 1160 | 3.98E+06 | 3.24E+06 | 2.84E+06 | 4.16E+06 | 4.73E+06 | |
| ac007981B | 738 | 652 | 821 | 652 | 3.08E+06 | 2.97E+06 | 7.72E+06 | 3.59E+06 | |
| ac007981A | 2262 | 896 | 679 | 679 | 525 | 9.08E+06 | 2.67E+06 | 2.68E+06 | |
| ac011718A | 944 | 653 | 542 | 542 | 569 | 1308 | 3.35E+06 | 8.12E+06 | |
| ac008103 | 1163 | 858 | 761 | 784 | 1009 | 472 | 981 | 3.95E+06 | |
| ac008079 | 622 |
775 |
598 |
1120 |
569 |
716 |
2339 |
981 |
|
| Replicate Valuesc |
|||||||||
| ac007325 | 2.10E+06 | 2.09E+06 | 2.07E+06 | 2.18E+06 | 3.89E+06 | 3.92E+06 | 2.24E+06 | 2.30E+06 | |
| ac007708 | 441.382 | 6.26E+06 | 5.82E+06 | 4.49E+06 | 2.14E+06 | 2.21E+06 | 4.03E+06 | 3.99E+06 | |
| ap000550 | 437.204 | 1058.71 | 8.09E+06 | 3.81E+06 | 2.07E+06 | 2.16E+06 | 3.40E+06 | 3.46E+06 | |
| ap000552 | 434.442 | 1010.36 | 1310.24 | 3.91E+06 | 2.13E+06 | 2.26E+06 | 3.59E+06 | 3.83E+06 | |
| ac007981B | 452.949 | 819.1 | 709.144 | 727.638 | 2.11E+06 | 2.33E+06 | 5.25E+06 | 3.30E+06 | |
| ac007981A | 729.553 | 443.177 | 432.326 | 443.705 | 442.061 | 5.21E+06 | 1.81E+06 | 2.06E+06 | |
| ac011718A | 737.733 | 464.614 | 453.975 | 475.009 | 479.727 | 916.778 | 2.03E+06 | 2.69E+06 | |
| ac008103 | 463.104 | 746.998 | 651.127 | 685.725 | 924.422 | 393.87 | 431.065 | 3.03E+06 | |
| ac008079 | 475.254 |
744.433 |
656.231 |
720.788 |
632.355 |
433.431 |
534.728 |
598.686 |
|
| Associated P Valuesd |
|||||||||
| ac007325 | .001 | 0 | .001 | 0 | 0 | .02 | 0 | 0 | |
| ac007708 | .092 | .003 | .012 | .794 | 0 | .001 | .054 | .019 | |
| ap000550 | .054 | .004 | .392 | .104 | 0 | .001 | .066 | .039 | |
| Ap000552 | .206 | .025 | .62 | .373 | 0 | .003 | .052 | .029 | |
| Ac007981B | .016 | .826 | .196 | .623 | 0 | .005 | .003 | .154 | |
| Ac007981A | 0 | 0 | .012 | .014 | .156 | .001 | 0 | .001 | |
| Ac011718A | .102 | .041 | .154 | .218 | .163 | .052 | 0 | 0 | |
| Ac008103 | 0 | .212 | .191 | .208 | .271 | .148 | 0 | .008 | |
| Ac008079 | .076 | .346 | .611 | .026 | .619 | .008 | 0 | .018 | |
| B. 16p11.1 | ||||||
| SSUF (above diagonal) and MUF (below diagonal) for Sequencesa |
||||||
| ac138927 |
ac138815 |
ac136425 |
ac133485 |
ac136613 |
ac140862 |
|
| Sequences | Observed Valuesb | |||||
| ac138927 | 1.02E+08 | 1.02E+08 | 4.44E+08 | 1.85E+08 | 7.53E+07 | |
| ac138815 | 4102 | 6.86E+08 | 9.25E+07 | 8.20E+07 | 4.70E+08 | |
| ac136425 | 3152 | 13657 | 1.00E+08 | 8.52E+07 | 6.25E+08 | |
| ac133485 | 10405 | 2694 | 2694 | 1.36E+08 | 7.87E+07 | |
| ac136613 | 6839 | 2565 | 2565 | 3437 | 6.36E+07 | |
| ac140862 | 3152 |
10314 |
10692 |
2550 |
1634 |
|
| Replicate Valuesc |
||||||
| ac138927 | 6.85E+07 | 6.94E+07 | 2.72E+08 | 1.01E+08 | 5.69E+07 | |
| ac138815 | 1803.29 | 5.52E+08 | 6.86E+07 | 7.35E+07 | 3.96E+08 | |
| ac136425 | 1826.31 | 10011.5 | 6.95E+07 | 7.43E+07 | 5.07E+08 | |
| ac133485 | 5656.85 | 1800.6 | 1837.98 | 9.74E+07 | 5.71E+07 | |
| ac136613 | 2487.9 | 1915.79 | 1933.22 | 2407.7 | 6.21E+07 | |
| ac140862 | 1757.38 |
8686.63 |
10509.1 |
1786.85 |
1911.86 |
|
| Associated P Valuesd |
||||||
| ac138927 | 0 | 0 | 0 | 0 | 0 | |
| ac138815 | 0 | .049 | 0 | .016 | .079 | |
| ac136425 | .006 | .08 | 0 | .006 | .053 | |
| ac133485 | .003 | .016 | .022 | 0 | 0 | |
| ac136613 | 0 | .045 | .054 | .024 | .299 | |
| ac140862 | .003 | .203 | .403 | .037 | .762 | |
| C. 15q25 | ||||||||||
| SSUF (above diagonal) and MUF (below diagonal) for Sequencesa |
||||||||||
| ac005630 |
ac010724 |
ac010725 |
ac012064 |
ac044860 |
ac126605 |
ac127482A |
ac135735 |
ac135995 |
ac127482B |
|
| Sequences | Observed Valuesb | |||||||||
| ac005630 | 2.03E+07 | 4.77E+07 | 5.96E+07 | 5.96E+07 | 2.21E+07 | 2.46E+07 | 2.51E+07 | 1.84E+07 | 2.00E+07 | |
| ac010724 | 1251 | 3.23E+07 | 3.22E+07 | 3.22E+07 | 7.70E+07 | 5.32E+07 | 5.67E+07 | 5.99E+08 | 7.05E+07 | |
| ac010725 | 2790 | 3144 | 3.75E+07 | 3.75E+07 | 2.56E+07 | 2.15E+07 | 2.19E+07 | 2.13E+07 | 2.02E+07 | |
| ac012064 | 5090 | 2222 | 2094 | 1.28E+09 | 3.45E+07 | 3.17E+07 | 3.16E+07 | 2.97E+07 | 3.28E+07 | |
| ac044860 | 5090 | 2222 | 2094 | 26962 | 3.45E+07 | 3.17E+07 | 3.16E+07 | 2.97E+07 | 3.28E+07 | |
| ac126605 | 2076 | 3408 | 2315 | 2186 | 2186 | 4.78E+08 | 4.60E+08 | 6.15E+07 | 7.00E+08 | |
| ac127482A | 2202 | 2415 | 1341 | 2233 | 2233 | 19905 | 5.53E+08 | 4.96E+07 | 4.15E+08 | |
| ac135735 | 2202 | 3.41E+03 | 1341 | 2.23E+03 | 2.23E+03 | 18145 | 18408 | 5.27E+07 | 3.12E+08 | |
| ac135995 | 1099 | 17645 | 1500 | 2222 | 2222 | 3.41E+03 | 2415 | 3408 | 6.04E+07 | |
| ac127482B | 2076 |
3408 |
1301 |
2186 |
2186 |
21368 |
14606 |
12844 |
3408 |
|
| Replicate Valuesc |
||||||||||
| ac005630 | 1.66E+07 | 3.82E+07 | 2.40E+07 | 2.37E+07 | 1.59E+07 | 1.68E+07 | 1.66E+07 | 1.49E+07 | 1.54E+07 | |
| ac010724 | 1070.39 | 1.72E+07 | 2.09E+07 | 2.07E+07 | 7.90E+07 | 4.44E+07 | 4.37E+07 | 4.02E+08 | 6.97E+07 | |
| ac010725 | 2132.64 | 1104.21 | 2.34E+07 | 2.31E+07 | 1.61E+07 | 1.67E+07 | 1.66E+07 | 1.49E+07 | 1.56E+07 | |
| ac012064 | 1444.75 | 1297.21 | 1415.97 | 1.01E+09 | 2.00E+07 | 1.90E+07 | 1.89E+07 | 1.88E+07 | 1.95E+07 | |
| ac044860 | 1444.71 | 1276.36 | 1402.35 | 26009.2 | 1.98E+07 | 1.87E+07 | 1.86E+07 | 1.85E+07 | 1.92E+07 | |
| ac126605 | 1028.07 | 3367.85 | 1044.33 | 1252.88 | 1232.08 | 7.08E+07 | 6.82E+07 | 6.03E+07 | 2.69E+08 | |
| ac127482A | 1087.79 | 2415.92 | 1072.94 | 1193.06 | 1163.52 | 3490.57 | 3.96E+08 | 4.31E+07 | 9.06E+07 | |
| ac135735 | 1066.64 | 2371.26 | 1071.77 | 1190.58 | 1176.55 | 3393.34 | 13169.2 | 4.32E+07 | 8.19E+07 | |
| ac135995 | 1033.86 | 12976.1 | 1033.37 | 1248.82 | 1223.41 | 2971.57 | 2479.57 | 2497.68 | 5.87E+07 | |
| ac127482B | 1009.95 |
3050.66 |
1015.19 |
1214.84 |
1198.33 |
8883.66 |
4216.35 |
3894.4 |
2914.25 |
|
| Associated P Valuesd |
||||||||||
| ac005630 | .004 | .015 | 0 | 0 | 0 | 0 | 0 | .002 | 0 | |
| ac010724 | .163 | 0 | 0 | 0 | .531 | .042 | .003 | .042 | .396 | |
| ac010725 | .086 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ac012064 | 0 | .011 | .029 | .139 | 0 | 0 | 0 | 0 | 0 | |
| ac044860 | 0 | .005 | .027 | .391 | 0 | 0 | 0 | 0 | 0 | |
| ac126605 | 0 | .386 | .001 | .007 | .004 | 0 | 0 | .377 | 0 | |
| ac127482A | .001 | .412 | .1 | .003 | .001 | 0 | .066 | .083 | 0 | |
| ac135735 | 0 | .047 | .102 | 0 | .003 | 0 | .095 | .038 | 0 | |
| ac135995 | .308 | .099 | .03 | .003 | .004 | .227 | .469 | .071 | .326 | |
| ac127482B | .002 | .252 | .089 | .003 | .002 | .001 | 0 | 0 | .195 | |
SSUF is the sum of the squares of all tracts of perfect identity between two sequences (uncondensed fragments), and MUF is the longest tract of perfect identity (maximal uncondensed fragment).
The values for both the SSUF and the MUF statistics (Sawyer 1989) from the observed data.
The equivalent results for the simulated data from 1,000 replicates.
Associated P values for both SSUF and the MUF statistics. Pairwise comparisons for which the observed identities are significantly longer than the simulated data (P > .05) are in bold italics. The majority of pairwise alignments give significant results with both statistics, which is consistent with widespread reticulate evolution.
To identify the tracts most likely to be due to reticulation, we established the position of all independent regions of perfect identity within our alignments that are significantly longer than expected (longer than the 99th percentile for regions of perfect identity within the randomized simulations). Despite this stringent cutoff, the alignments contain 187 such tracts, encompassing ∼450 kb or ∼5% of the total aligned sequence (see table 3). The size of the tracts in each alignment varies enormously, since the expected distribution is a function of branch lengths within each tree. However, 74 of the tracts are >2 kb in length, with the largest spanning >18 kb. This confirms that a significant fraction of the very high sequence identity that exists between paralogues within the human genome is a result of gene conversion and unequal crossing-over between existing duplicated sequences rather than recent de novo duplication.
Table 3.
Tracts of Perfect Identity Created by Reticulation Events[Note]
| Tract Number | Alignment | Length(bp) | StartNucleotidea | StopNucleotidea | Sequences(Accession Numbers or Clone Names) |
| 1 | 15q25 | 18,993 | 6569 | 25562 | ac126605, ac127482 |
| 2 | SMA-3 | 13,861 | 44695 | 58556 | ac138827, ac142525 |
| 3 | SMA-2 | 11,356 | 28177 | 39533 | ac118459, ac140140 |
| 4 | 15q25 | 10,962 | 25983 | 36945 | ac127482, ac126605 |
| 5 | SMA-2 | 10,286 | 41211 | 51497 | ac118459, ac140140 |
| 6 | SMA-1 | 10,066 | 66715 | 76781 | ac131392, ac138930 |
| 7 | SMA-1 | 8,337 | 58724 | 67061 | ac131392, ac138957 |
| 8 | SMA-2 | 8,084 | 45731 | 53815 | ac138866, ac145138 |
| 9 | SMA-2 | 7,660 | 8351 | 16011 | ac138866, ac139510 |
| 10 | SMA-2 | 7,042 | 20485 | 27527 | ac138866, ac139510 |
| 11 | fk506 | 6,879 | 26760 | 33639 | 286O1, 50I9, 242D12 |
| 12 | 16p11 | 6,830 | 49357 | 56187 | ac138927, ac136613 |
| 13 | fk506 | 6,803 | 34173 | 40976 | 286O1, 50I9, 242D12 |
| 14 | SMA-3 | 6,369 | 51300 | 57669 | ac145138, ac147599 |
| 15 | 15q25 | 6,333 | 1 | 6334 | ac126605, ac135735 |
| 16 | SMA-2 | 6,324 | 64653 | 70977 | ac138866, ac139510 |
| 17 | SMA-1 | 6,259 | 45384 | 51643 | ac138957, ac139500 |
| 18 | SMA-2 | 6,222 | 32116 | 38338 | ac138866, ac139510 |
| 19 | SMA-1 | 6,114 | 76783 | 82897 | ac131392, ac138930 |
| 20 | c9orf36 | 5,979 | 31785 | 37764 | 144A16, 101E5, 95K23 |
| 21 | 22q11-1 | 5,904 | 7892 | 13796 | ac007708, ac009288 |
| 22 | Morph-Z1 | 5,863 | 9278 | 15141 | AC138896B, AC138896A |
| 23 | SMA-1 | 5,714 | 20225 | 25939 | ac138866, ac139500 |
| 24 | 22q11-1 | 5,663 | 51286 | 56949 | ac007708, ac007981 |
| 25 | 15q25 | 5,487 | 31951 | 37438 | ac005630, ac012064 |
| 26 | 16p11 | 5,285 | 57855 | 63140 | ac138927, ac136613 |
| 27 | 22q11-1 | 5,135 | 33594 | 38729 | ac007708, ac012330 |
| 28 | SMA-1 | 5,035 | 17684 | 22719 | ac131392, ac138957 |
| 29 | fk506 | 4,911 | 3728 | 8639 | 286O1, 50I9, 242D12 |
| 30 | fk506 | 4,624 | 52270 | 56894 | 286O1, 50I9 |
| 31 | SMA-1 | 4,372 | 4493 | 8865 | ac138957, ac138930 |
| 32 | 16p11 | 4,273 | 12274 | 16547 | ac138927, ac136425 |
| 33 | SMA-3 | 4,103 | 5544 | 9647 | ac138827, ac142525 |
| 34 | 9qter | 3,878 | 11807 | 15685 | AC133919, AC139136 |
| 35 | fk506 | 3,837 | 56896 | 60733 | 286O1, 50I9 |
| 36 | 22q11-1 | 3,644 | 19169 | 22813 | ac008132, ac012330 |
| 37 | KGF | 3,465 | 42284 | 45749 | 211N8, 104G3 |
| 38 | c9orf36 | 3,436 | 53464 | 56900 | 144A16, 347J14 |
| 39 | KGF | 3,421 | 39150 | 42571 | 624e13, 262H14 |
| 40 | ANKRD20A | 3,397 | 73304 | 76701 | 78N10, bA793G16 |
| 41 | 22q11-1 | 3,330 | 1 | 3331 | ac008132, ac012330 |
| 42 | 22q11-1 | 3,279 | 12946 | 16225 | ac007708, ac023491 |
| 43 | SMA-3 | 3,267 | 9924 | 13191 | ac142525, ac147599 |
| 44 | fk506 | 3,233 | 48171 | 51404 | 286O1, 50I9 |
| 45 | ANKRD20A | 3,123 | 76703 | 79826 | 78N10, bA793G16 |
| 46 | 22q11-1 | 3,020 | 16147 | 19167 | ac008132, ac009288 |
| 47 | c9orf36 | 3,015 | 24210 | 27225 | 144A16, 101E5, 95K23 |
| 48 | 22q11-1 | 2,993 | 18697 | 21690 | ac009288, ac023491 |
| 49 | 16p11 | 2,881 | 61109 | 63990 | ac133485, ac136425 |
| 50 | Alphoid | 2,861 | 36381 | 39242 | 381O7, 460E7 |
| 51 | SRP19 | 2,686 | 11262 | 13948 | 12A20, 292F9, 374M1 |
| 52 | 16p11 | 2,685 | 76193 | 78878 | ac133485, ac136613 |
| 53 | SRP19 | 2,641 | 47076 | 49717 | 7g23, 292F9, 157L3 |
| 54 | c9orf36 | 2,593 | 56902 | 59495 | 144A16, 347J14 |
| 55 | 16p11 | 2,583 | 66607 | 69190 | ac133485, ac136613 |
| 56 | SMA-3 | 2,534 | 14070 | 16604 | ac138827, ac142525 |
| 57 | 16p11 | 2,466 | 28584 | 31050 | ac136425, ac138927 |
| 58 | ANKRD20A | 2,420 | 70882 | 73302 | 78N10, bA793G16 |
| 59 | 16p11 | 2,419 | 51291 | 53710 | ac136425, ac133485 |
| 60 | fk506 | 2,326 | 738 | 3064 | 286O1, 50I9 |
| 61 | Morph-Z1 | 2,315 | 13887 | 16202 | AC145285B, AC145285A |
| 62 | Alphoid | 2,311 | 42621 | 44932 | 381O7, 460E7 |
| 63 | 22q11-2 | 2,308 | 4713 | 7021 | ac011718A, ac008079 |
| 64 | 16p11 | 2,292 | 63142 | 65434 | ac138927, ac136613 |
| 65 | CT-1 | 2,292 | 12495 | 14787 | ac133561, ac136440 |
| 66 | Alphoid | 2,280 | 34959 | 37239 | 15J10, 460N11 |
| 67 | Morph-Z1 | 2,279 | 5877 | 8156 | AC145285B, AC145285A |
| 68 | 22q11-2 | 2,258 | 9665 | 11923 | ac007325, ac007981A |
| 69 | KGF | 2,172 | 36996 | 39168 | 211N8, 262H14 |
| 70 | c9orf36 | 2,170 | 29613 | 31783 | 144A16, 101E5 |
| 71 | Morph-Z2 | 2,136 | 9984 | 12120 | AC138932, AC126755A |
| 72 | Morph-Z1 | 2,080 | 4182 | 6262 | AC138904, AC145285A |
| 73 | c9orf36 | 2,053 | 22155 | 24208 | 144A16, 95K23 |
| 74 | SMA-3 | 1,992 | 1 | 1993 | ac138827, ac145138 |
| 75 | 22q11-2 | 1,987 | 8678 | 10665 | ac007708, ap000550 |
| 76 | ANKRD20A | 1,972 | 67501 | 69473 | 78N10, bA793G16 |
| 77 | c9orf36 | 1,971 | 37703 | 39674 | 95H8, 101E5, 95K23 |
| 78 | fk506 | 1,967 | 47425 | 49392 | 133N6, 286O1 |
| 79 | ANKRD20A | 1,958 | 54105 | 56063 | ANKRD20A, bA793G16 |
| 80 | 9pter | 1,947 | 1 | 1948 | 403A15, 561O23, 115J7 |
| 81 | Morph-Z2 | 1,940 | 6659 | 8599 | AC138932, AC126755A |
| 82 | c9orf36 | 1,931 | 19862 | 21793 | 144A16, 101E5 |
| 83 | 9qter | 1,910 | 19500 | 21410 | AC133919, AC139136 |
| 84 | ANKRD20A | 1,901 | 14904 | 16805 | ANKRD20A, 195B21 |
| 85 | 9qter | 1,846 | 8436 | 10282 | AC133919, AC139136 |
| 86 | c9orf36 | 1,795 | 20358 | 22153 | 144A16, 95K23 |
| 87 | fk506 | 1,782 | 13484 | 15266 | 286O1, 50I9 |
| 88 | c9orf36 | 1,750 | 1488 | 3238 | 95K23, 347J14 95H8 |
| 89 | 22q11-2 | 1,701 | 11763 | 13464 | ac007708, ac008103 |
| 90 | 22q11-2 | 1,673 | 648 | 2321 | ac007325, ac008103 |
| 91 | c9orf36 | 1,613 | 40232 | 41845 | 393E19, 347J14, 95H8 |
| 92 | SRP19 | 1,607 | 31415 | 33022 | 7g23, 374m1 |
| 93 | 22q11-2 | 1,599 | 6345 | 7944 | ap000552, ac008079 |
| 94 | CT-1 | 1,594 | 10086 | 11680 | ac133561, ac136440 |
| 95 | Sat5 linke | 1,585 | 34170 | 35755 | 399F4, P704L1, 459O16 |
| 96 | CASPR3 | 1,576 | 76849 | 78425 | 133G22, 204I2 |
| 97 | SRP19 | 1,561 | 19231 | 20792 | 7g23, 374m1 |
| 98 | CT-1 | 1,535 | 12024 | 13559 | ac010539, ac133536 |
| 99 | 16qter | 1,528 | 1 | 1529 | AC097645, AL627309 |
| 100 | 16qter | 1,416 | 8804 | 10220 | AC130290, AC137818 |
| 101 | 22q11-2 | 1,355 | 5331 | 6686 | ac007325, ac007981A |
| 102 | 22q11-2 | 1,288 | 4102 | 5390 | ac007325, ac007981B |
| 103 | CASPR3 | 1,275 | 2668 | 3943 | 292B8, 74l19, 133G22 |
| 104 | 22q11-2 | 1,251 | 12747 | 13998 | ac007708, ac008079 |
| 105 | KGF | 1,233 | 27589 | 28822 | 475I24, 211N8 |
| 106 | CT-1 | 1,229 | 393 | 1622 | ac133536, ac140899 |
| 107 | 9pter | 1,213 | 1519 | 2732 | 115J7, 391M20 |
| 108 | 2qfus | 1,204 | 13348 | 14552 | 39F9, 460E7 |
| 109 | 22q11-2 | 1,175 | 282 | 1457 | ap000550, ac007981B |
| 110 | 22qter | 1,157 | 11687 | 12844 | AL954635, AP006313 |
| 111 | KGF | 1,156 | 43903 | 45059 | 475I24, 262H14 |
| 112 | 9pter | 1,148 | 13506 | 14654 | 391M20, 561O23 |
| 113 | CT-1 | 1,139 | 10562 | 11701 | ac010539, ac133536 |
| 114 | CT-1 | 1,135 | 767 | 1902 | ac010539, ac133561 |
| 115 | PCC | 1,122 | 34647 | 35769 | 111G23, 290lL7, 246P17 |
| 116 | CT-1 | 1,108 | 67146 | 68254 | ac133536, ac140899 |
| 117 | 9pter | 1,108 | 23906 | 25014 | 143M1, 395L14 |
| 118 | 22q11-2 | 1,077 | 10351 | 11428 | ap000552, ac008079 |
| 119 | PCC | 1,008 | 34588 | 35596 | 475B17, 211P14 |
| 120 | VSPA | 979 | 49344 | 50323 | 187C18, 416B15 |
| 121 | CT-1 | 961 | 26050 | 27011 | ac010539, ac133536 |
| 122 | chAB4-1 | 943 | 35095 | 36038 | AC145212, BX004987 |
| 123 | KGF | 932 | 45320 | 46252 | 624e13, 262H14 |
| 124 | Sat5 linke | 908 | 39528 | 40436 | 399F4, P704L1 |
| 125 | VSPA | 885 | 50453 | 51338 | 187C18, 416B15 |
| 126 | CT-1 | 870 | 25961 | 26831 | ac133561, ac140899 |
| 127 | KGF | 824 | 40758 | 41582 | 211N8, 104G3 |
| 128 | 11pter | 807 | 14076 | 14883 | ac097645, ac136309 |
| 129 | VSPA | 767 | 46036 | 46803 | 187C18, 416B15 |
| 130 | KGF | 738 | 39063 | 39801 | 475I24, 262H14 |
| 131 | chAB4-1 | 658 | 18530 | 19188 | AL354822, BX004987 |
| 132 | chAB4-1 | 656 | 25609 | 26265 | AL354822, AL355973 |
| 133 | chAB4-1 | 636 | 36509 | 37145 | AC145212 , BX004987 |
| 134 | KGF | 631 | 39730 | 40361 | 211N8, 104G3 |
| 135 | PCC | 609 | 1 | 610 | 246P17, 211P14 |
| 136 | chAB4-1 | 607 | 53293 | 53900 | AC138771, AC145212 |
| 137 | VSPA | 600 | 46254 | 46854 | 12P21, 216M21 |
| 138 | KGF | 598 | 8053 | 8651 | 624e13, 213O5 |
| 139 | chAB4-1 | 588 | 48724 | 49312 | AC138771, AC145212 |
| 140 | KGF | 585 | 41585 | 42170 | 211N8, 104G3 |
| 141 | KGF | 582 | 43319 | 43901 | 624e13, 262H14 |
| 142 | chAB4-1 | 565 | 64513 | 65078 | AC145212, BX004987 |
| 143 | chAB4-1 | 557 | 33669 | 34226 | AC138771, AC145212 |
| 144 | chAB4-1 | 545 | 32859 | 33404 | AL355973, BX004987 |
| 145 | chAB4-2 | 506 | 21120 | 21626 | AL355793, BX572084 |
| 146 | KGF | 501 | 45751 | 46252 | 211N8, 104G3 |
| 147 | PCC | 497 | 52 | 549 | 475B17, 290lL7 |
| 148 | chAB4-2 | 438 | 7373 | 7811 | AL355793, BX572084 |
| 149 | ANK-ZNF | 417 | 11735 | 12152 | ctd2642, hs21c002 |
| 150 | ANK-ZNF | 401 | 17411 | 17812 | hs21c002, 512E23 |
| 151 | chAB4-2 | 389 | 4084 | 4473 | AL355793, BX572084 |
| 152 | ANK-ZNF | 362 | 16647 | 17009 | ctd2642, hs21c002 |
| 153 | chAB4-2 | 334 | 13008 | 13342 | AL355793, BX572084 |
| 154 | ANK-ZNF | 332 | 19565 | 19897 | hs21c002, 318K12 |
| 155 | ANK-ZNF | 326 | 37198 | 37524 | 629P16, 512E23 |
| 156 | chAB4-2 | 321 | 5533 | 5854 | BX004987, BX640538 |
| 157 | ANK-ZNF | 321 | 22478 | 22799 | hs21c002, 318K12 |
| 158 | chAB4-2 | 320 | 12341 | 12661 | AL355793, BX572084 |
| 159 | chAB4-2 | 309 | 13487 | 13796 | AL355793, BX572084 |
| 160 | ANK-ZNF | 304 | 2845 | 3149 | 629P16, 512E23 |
| 161 | chAB4-2 | 297 | 15980 | 16277 | AL355793, BX572084 |
| 162 | chAB4-2 | 297 | 16726 | 17023 | AL355793, BX572084 |
| 163 | chAB4-2 | 295 | 19834 | 20129 | AL355793, BX572084 |
| 164 | chAB4-2 | 289 | 21912 | 22201 | AL355793, BX572084 |
| 165 | ANK-ZNF | 289 | 9673 | 9962 | 629P16, 512E23 |
| 166 | ANK-ZNF | 289 | 9673 | 9962 | 629P16, 318K12 |
| 167 | chAB4-2 | 287 | 6144 | 6431 | AL355793, BX572084 |
| 168 | ANK-ZNF | 282 | 9962 | 10244 | ctd2642, hs21c002 |
| 169 | chAB4-2 | 278 | 24575 | 24853 | BX004987, BX640538 |
| 170 | ANK-ZNF | 277 | 9225 | 9502 | 629P16, 512E23 |
| 171 | ANK-ZNF | 276 | 8444 | 8720 | ctd2642, hs21c002 |
| 172 | chAB4-2 | 274 | 7097 | 7371 | AL355793, BX572084 |
| 173 | chAB4-2 | 274 | 24683 | 24957 | AL355793, BX572084 |
| 174 | ANK-ZNF | 274 | 10662 | 10936 | ctd2642, hs21c002 |
| 175 | chAB4-2 | 273 | 22265 | 22538 | AL355793, BX572084 |
| 176 | chAB4-2 | 272 | 640 | 912 | AL355793, BX572084 |
| 177 | chAB4-2 | 272 | 15141 | 15413 | AL355793, BX572084 |
| 178 | chAB4-2 | 261 | 71866 | 72127 | BX004987, BX572084 |
| 179 | ANK-ZNF | 259 | 13547 | 13806 | 629P16, 318K12 |
| 180 | chAB4-2 | 258 | 5461 | 5719 | AL355793, BX572084 |
| 181 | chAB4-2 | 252 | 20843 | 21095 | BX004987, BX640538 |
| 182 | chAB4-2 | 251 | 35812 | 36063 | BX004987, BX640538 |
| 183 | chAB4-2 | 249 | 11212 | 11461 | AL355793, BX572084 |
| 184 | chAB4-2 | 249 | 18385 | 18634 | AL355793, BX572084 |
| 185 | chAB4-2 | 237 | 24444 | 24681 | AL355793, BX572084 |
| 186 | chAB4-2 | 235 | 17237 | 17472 | BX004987, BX640538 |
| 187 | chAB4-2 | 233 | 2078 | 2311 | AL355793, BX572084 |
Note.— Tracts of identity between any two sequences that are longer than the 99th percentile tract within the corresponding simulated alignments are listed in order of size. The first nucleotide (start position) and last nucleotide (stop position) that define the tracts within each CpG negative alignment are given, together with the sequences involved. The listing of more than two sequences for a particular tract number indicates the presence of more than one overlapping tract of perfect identity larger than the 99th percentile. These tracts are likely to be due to the same reticulation event and so are merged. In these cases, only the longer tract, defined by the first two sequences listed, is indicated.
Nucleotide positions of reference sequence in alignment.
Reticulation Density
Although phylogenetic profiling identifies the products of major NAHR events within most alignments, it is clear that it cannot resolve events when the signal is complex (e.g., fig. 3D). To precisely define events of this nature, or to estimate the frequency of events, is difficult in such data sets because it is unknown whether all sequences within the genome are present in each alignment. Even in cases in which the copy number is known, it is not possible to sample from sequences that have been deleted from ancestral genomes. However, in 11 of our alignments, the expected frequency of reticulate quartets is effectively zero. This implies that virtually all the observed reticulate quartets are the result of reticulation events, as opposed to multiple independent mutational events to the same nucleotide. Under these exceptional circumstances, it is possible to use the distribution of all informative sites within the alignments to define the minimal number of events that is required to generate the observed data (see the “Material and Methods” section and fig. 2). The results of this analysis are summarized in figure 10. A minimum of 189 events are inferred within 11 alignments that span ∼3.54 Mb of DNA. This is much higher than suggested by the phylogenetic profile analyses and gives a minimal density of 1 reticulation event every 18.7 kb. However, it is clear that there is significant variation in the event frequency between alignments. Most give densities between 0.01 and 0.1 events per kb, but the 22q11-3 alignment has a density of 0.262 events per kb, or ∼1 reticulation event every 4 kb. It is noteworthy that VCFS, caused by deletions of 22q11 sequences, is associated with the highest mutation rate of any duplicon-induced pathogenic rearrangement (Shaffer and Lupski 2000), which suggests that the frequency of reticulation events and duplicon-induced mutation may be directly correlated.
Figure 10.

Reticulation-event density in duplicons. Analysis of 11 alignments for which the expected frequency of reticulate quartets is negligible. All show a >20-fold excess of reticulate quartets relative to the expectation, with expected frequencies in 100 control alignments <0.5% of the observed value at the 50th percentile and <2.0% of the observed value at the 95th percentile. Analyses were performed on CpG-negative data, and the minimum number of events was estimated as shown in figure 2.
Discussion
To date, detailed analyses of reticulate processes, such as unequal crossing-over and gene conversion within nonrepetitive human DNA, have been confined to analyses of duplicons with a stable copy number of 2, which allow donor and acceptor loci to be defined (e.g., Han et al. 2000; Hurles 2001; Bosch et al. 2004; Hurles et al. 2004); analyses of meiotic events between alleles at known hotspots (e.g., Jeffreys et al. 2004); or analyses that take advantage of a phenotype associated with a specific event, such as duplication/deletion (e.g., Saitta et al. 2004; Shaw and Lupski 2004). The analysis presented here therefore represents the first concerted effort to analyze the phylogenetic consistency of a broad cross section of complex human duplicons from a wide variety of genomic locations and with unknown copy number. All 30 alignments analyzed have sharp changes in phylogenetic profile; ∼90% have a significant excess of reticulate quartets relative to the expectation, and a minimum of 5% (450 kb) of the analyzed sequence consists of tracts of perfect identity created by reticulation events. Because the copy number of these duplicons is unknown (and may have varied over time), accurate rate estimates for these events cannot be derived. Despite this, the pattern of sequence variation within a subset of alignments is consistent with an average of one reticulation event every ∼18.7 kb. All these observations provide striking confirmation that human duplicons are not evolving independently but rather are exchanging information at a very high frequency.
The levels of reticulation we have identified are extreme, often affecting >20% of the informative positions in an alignment. Although this is surprisingly high, consideration of the methodology suggests that the true levels may be significantly higher. First, in common with many other methods (Hein 1993; McGuire and Wright 2000; Husmeier and McGuire 2003), the quartet method outlined here will only detect reticulation events that have led to a change in tree topology. Exchanges between sequences that only affect branch lengths will not be detected. Second, the method assumes that the tree generated from each alignment is an accurate reflection of the phylogenetic relationships between the sequences involved. We have deliberately simplified some trees to minimize violations of this assumption (see the “Material and Methods” section), and the existence of sequences that cannot be positioned on a tree with confidence is likely the result of further reticulation events. Furthermore, in several alignments, the sites within each partition that support the tree are often clustered together (see fig. 11), indicating that the phylogenetic relationships are not adequately described by the tree. The most plausible explanation for such clustering is reticulation involving sequences that have not been sampled because of either incomplete sequence coverage or deletion from the human genome. It is clear from these considerations that current methods underestimate the true levels of reticulation in these sequences.
Figure 11.

Distribution of sites indicating suboptimal trees. Partimatrix output shows parsimony-informative sites from the central region of the 15q25 alignment (11.0–33.12 kb). The three partitions that support the tree are to the left of the red line (7, 3, and 17), and the clustering of sites supporting each partition is indicated. If the tree is an accurate representation of the phylogenetic relationships, then the positions supporting each partition should be randomly distributed. For explanation of output, see figure 2 legend.
Since our alignments include >8 Mb of DNA, representing ∼5% of all known human duplicons (IHGSC 2004), it is tempting to speculate that the results presented here are typical of all human duplicons. However, all duplicon families in our sample contain a minimum of four copies, and, although there is extensive empirical evidence that reticulate processes occur in duplicon families with two and three copies (Hurles 2001; Hurles et al. 2004; Bagnall et al. 2005), there may be some correlation between reticulation frequency and duplicon copy number. In addition, our sample is heavily biased toward duplicons with >99% identity, which also reflects the constraints of the inclusion criteria (see the “Material and Methods” section). Approximately one-third are from the single largest region of duplicons within the human genome (9p12-q12 [Humphray et al. 2004]), whereas others are from duplications already known to be associated with deletion/duplication syndromes (SMA in 5q13.2 and DiGeorge/VCFS in 22q11). It is interesting that these duplicons are largely chromosome specific and have relatively localized distributions. In contrast, the subtelomeric and chAB4 repeats that map to multiple chromosomes show a smaller excess of reticulation quartets in our analyses and share lower sequence identities. This may be related to the fact that rates of NAHR and gene conversion are influenced by both sequence identity and the spacing of interacting molecules (Stankiewicz and Lupski 2002; Schildkraut et al. 2005), with dependence on sequence identity supported by the significant correlation we observe between the levels of reticulation and the mean sequence identity of each alignment (fig. 8). The higher sequence identities shared between human intrachromosomal duplications, compared with interchromosomal duplications (Bailey et al. 2002), may therefore be partly the result of different rates of reticulation events between these two classes of duplicon.
It is noteworthy that our primate control alignments also contain an excess of reticulate quartets relative to the expectation. However, the excess observed in the controls is modest in the CpG-positive data and is significantly reduced in magnitude by exclusion of CpG dinucleotides. It is possible that violations of the model of molecular evolution used to generate the simulated data, such as rate heterogeneity or variation in specific substitution rates, may be responsible. These would have the greatest impact on the primate controls, because the control trees are much larger than those generated from the test data, and such violations could influence the frequency of both reticulate and bimutational quartets. The excess could be due to biased gene conversion within single-copy human genomic DNA, which has been described recently by Webster et al. (2005). Although a convenient explanation, this mechanism is unlikely to be entirely responsible, because the reticulate quartets are not clustered in the control alignments (fig. 9). Another possible explanation may be the speciation events within the primate controls. The stochastic fixation of intraspecific sequence variation that crosses species barriers can, in a proportion of polymorphic sites, give rise to apparent homoplasy. With limited sequence data, this has led to the recovery of gene trees in which the topology is distinct from the primate species tree (e.g., Chen and Li 2001). Among closely related species, such stochastic assortment of polymorphic variation would generate reticulate quartets more frequently than bimutational quartets, and these would be randomly distributed in terms of physical position. Thus, the assortment of intraspecific sequence variation during one or more primate speciation events could produce a specific excess of reticulate quartets without any clustering of the affected sites.
Irrespective of the source of these minor inconsistencies within the primate controls, they do not affect the conclusion, based on our analyses, that some of the human duplicons described here are the most reticulate euchromatic sequences described to date in any species, with only the most recombinant strains of HIV virus showing remotely comparable mosaic histories (Vidal et al. 2000).
The Effect of Reticulation on Map Closure and Comparative Analyses
The extreme levels of reticulation described here have important implications for efforts to close remaining gaps in duplicon-rich regions of the human genome, and for gene-family evolution. All genome-sequencing methodologies use the assumption that sequence identity is indicative of physical overlap to facilitate contig construction. The limitation of applying whole shotgun sequencing methods to complex genomes is clear (She et al. 2004b), and the advantage of using haploid libraries to avoid allelic variation during contig construction has been demonstrated (e.g., Skaletsky et al. 2003). Despite this, most of the current human draft sequence has been constructed using more than one diploid library and represents a combination of two or more alleles (IHGSC 2004). This has prevented assembly of contiguous reference sequence in highly duplicated polymorphic regions (e.g., Taudien et al. 2004). In some duplicationrich regions, a “deep coverage” sequencing strategy has been used to sequence both alleles from a single library, with very high levels of sequence redundancy (Martin et al. 2004; Schmutz et al. 2004). Despite these efforts, gaps still remain in these regions, highlighting the difficulty of obtaining sequence closure within such duplications.
The ability to eliminate the confounding influence of diploidy (by analyzing hydatiform moles, for example) is only one aspect of the problem. Even if a single haploid resource were exclusively used, the fact that a minimum of 5% of the sequence in our alignments share perfect identity because of reticulation indicates that perfect overlap between clones is not a totally secure criterion for map construction in duplicated DNA. Our results, therefore, strongly suggest that the human sequence map may never be truly finished and that, even if all remaining sequence gaps are closed, we will only be able to state that the resulting sequence build represents the most parsimonious map we can create given the limitations of the cloning resources used and the complexity of human duplications. These results also illustrate the importance of creating immortalized cell lines from material used to generate large-insert genomic libraries for reference, to allow discrepancies within sequence maps to be unambiguously resolved using methods such as PFGE and Fiber-FISH, should the need arise.
The high levels of reticulation observed here will also significantly affect the conclusions of comparative analyses of duplicated sequences. High interspecific sequence identities between duplicon copies, despite a relatively ancient evolutionary origin, has been noted by several authors and has been interpreted as indirect evidence of gene conversion (e.g., Orti et al. 1998; DeSilva et al. 1999; Shaikh et al. 2001; Crosier et al. 2002). Our data unequivocally confirm this inference and indicate that divergence times based solely on sequence identity between human duplicons could be extremely misleading. It follows that the recent evolutionary origins for many human duplicon families, inferred from human sequence data, must be reexamined using more-comprehensive comparative approaches (e.g., Stankiewicz et al. 2004).
Reticulation and the Identification of Recombination Hotspots
Duplicons are known to promote rearrangement, and several genomewide analyses have explored the physical association between duplicons and clinically important deletions and have developed archived resources to visualize duplicons within the working draft (Bailey et al. 2002; Cheung et al. 2003). More recenty, empirical analyses of large-scale copy-number changes within the human genome (Iafrate et al. 2004; Sebat et al. 2004) have begun to refine our understanding of this relationship. Our phylogenetic profiling identifies the alignments from the SMA and 22q11 microdeletion regions as those with the most complex phylogenetic histories. This is clear both from the phylogenetic profiles (figs. 3C and 5C) and from the high density of inferred reticulation events (fig. 10). Several alignments within the highly polymorphic 9p12-q12 region are also noteworthy because of the presence of a single unstable duplicon within each sequence family (M.S.J., unpublished data). This establishes that regions of known instability have marked disruption of the phylogenetic signal and that signatures of past recombination/conversion events can be readily detected using existing methods. It follows that systematic analysis of this signal may help to distinguish duplicons that are physically unstable from those whose sequence identity is due purely to descent, as suggested by Hurles et al. [2004], and it may pinpoint hotspots of activity such as the SMA-1 hotspot identified here (fig. 6). Thus, although we have demonstrated unprecedented levels of reticulation within the duplicons we have analyzed, a more comprehensive genomewide scan is desirable, both to fully characterize the levels and distribution of reticulation and to correlate this information with genomic instability. Although straightforward in principle, this will be complex in practice. Many methods, including our quartet method, require a multiple alignment containing a minimum of four sequences. This reduces their utility, because they cannot be applied to duplicons with a low copy number (2–3) without generation of comparative data from other species and because accurate multiple alignment still requires extensive user interaction. In contrast, methods that analyze pairwise alignments, such as permutation tests (Sawyer 1989; Hurles 2001), are simpler to apply but are less informative.
Finally, the results obtained here also have important implications for gene-family evolution. Several of the alignments used here contain active genes or ORFs related to known genes (e.g., ANKRD20A, CT1, COB-W, Freak5, and Morpheus genes). Since reticulation events have the potential to rapidly transfer advantageous sequence variants between the members of a multigene family (Dover 1982), it is clear that the impact of these events on the coding potential within the duplicated portion of the human genome is an important area for future research.
Acknowledgments
This work was funded by Wellcome Trust grant 059369. Some of the sequence data were generated by the National Institutes of Health Intramural Sequencing Center.
Web Resources
The URLs for data presented herein are as follows:
- Algorithms in Bioinformatics: SplitsTree4, http://www-ab.informatik.uni-tuebingen.de/software/splits/welcome.html (for D. H. Huson and D. Bryant's work on estimating phylogenetic trees and networks using SplitsTree4)
- BLAST, http://www.ncbi.nlm.nih.gov/BLAST/
- NISC Comparative Vertebrate Sequencing, http://www.nisc.nih.gov/open_page.html?/projects/comp_seq.html (for Target 1)
- Partimatrix, http://www.cecalc.ula.ve/BIOINFO/servicios/herr1/PARTIMATRIX/manual.htm
- Pairwise FLAG, http://bioinformatics.itri.org.tw/prflag/prflag.php
- PhylPro, http://www.rsbs.anu.edu.au/Products&Services/BiotechnologyTransferUnit/phylpro.asp
- Seq-Gen: Sequence-Generator, http://bioweb.pasteur.fr/seqanal/interfaces/seqgen-simple.html
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410 [DOI] [PubMed] [Google Scholar]
- Bagnall RD, Ayres KL, Green PM, Giannelli F (2005) Gene conversion and evolution of Xq28 duplicons involved in recurring inversions causing severe hemophilia A. Genome Res 15:214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE (2002) Recent segmental duplications in the human genome. Science 297:1003–1007 [DOI] [PubMed] [Google Scholar]
- Bosch E, Hurles ME, Navarro A, Jobling MA (2004) Dynamics of a human interparalog gene conversion hotspot. Genome Res 14:835–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FC, Li WH (2001) Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet 68:444–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung J, Estivill X, Khaja R, MacDonald JR, Lau K, Tsui LC, Scherer SW (2003) Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol 4:R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, von Mering C, Suyama M, Harrington ED, Izaurralde E, Bork P (2005) Complex genomic rearrangements lead to novel primate gene function. Genome Res 15:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courseaux A, Richard F, Grosgeorge J, Ortola C, Viale A, Turc-Carel C, Dutrillaux B, Gaudray P, Nahon JL (2003) Segmental duplications in euchromatic regions of human chromosome 5: a source of evolutionary instability and transcriptional innovation. Genome Res 13:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosier M, Viggiano L, Guy J, Misceo D, Stones R, Wei W, Hearn T, Ventura M, Archidiacono N, Rocchi M, Jackson MS (2002) Human paralogs of KIAA0187 were created through independent pericentromeric-directed and chromosome-specific duplication mechanisms. Genome Res 12:67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserpan I, Katona R, Praznovszky T, Novak E, Rozsavolgyi M, Csonka E, Morocz M, Fodor K, Hadlaczky G (2002) The chAB4 and NF1-related long-range multisequence DNA families are contiguous in the centromeric heterochromatin of several human chromosomes. Nucleic Acids Res 30:2899–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSilva U, Massa H, Trask BJ, Green ED (1999) Comparative mapping of the region of human chromosome 7 deleted in Williams syndrome. Genome Res 9:428–436 [PMC free article] [PubMed] [Google Scholar]
- Dover G (1982) Molecular drive: a cohesive mode of species evolution. Nature 299:111–117 [DOI] [PubMed] [Google Scholar]
- Drouin G, Prat F, Ell M, Clarke GD (1999) Detecting and characterizing gene conversions between multigene family members. Mol Biol Evol 16:1369–1390 [DOI] [PubMed] [Google Scholar]
- Eichler EE, Lu F, Shen Y, Antonacci R, Jurecic V, Doggett NA, Moyzis RK, Baldini A, Gibbs RA, Nelson DL (1996) Duplication of a gene-rich cluster between 16p11.1 and Xq28: a novel pericentromeric-directed mechanism for paralogous genome evolution. Hum Mol Genet 5:899–912 [DOI] [PubMed] [Google Scholar]
- Fan Y, Newman T, Linardopoulou E, Trask BJ (2002) Gene content and function of the ancestral chromosome fusion site in human chromosome 2q13-2q14.1 and paralogous regions. Genome Res 12:1663–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryxell KJ, Moon WJ (2005) CpG mutation rates in the human genome are highly dependent on local GC content. Mol Biol Evol 22:650–658 [DOI] [PubMed] [Google Scholar]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, Zuffardi O (2003) Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet 12:849–858 [DOI] [PubMed] [Google Scholar]
- Guttman DS, Dykhuizen DE (1994) Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science 266:1380–1383 [DOI] [PubMed] [Google Scholar]
- Han LL, Keller MP, Navidi W, Chance PF, Arnheim N (2000) Unequal exchange at the Charcot-Marie-Tooth disease type 1A recombination hot-spot is not elevated above the genome average rate. Hum Mol Genet 9:1881–1889 [DOI] [PubMed] [Google Scholar]
- Hein J (1993) A heuristic method to reconstruct the history of sequences subject to recombination. J Mol Evol 36:396–405 [Google Scholar]
- Humphray SJ, Oliver K, Hunt AR, Plumb RW, Loveland JE, Howe KL, Andrews TD, et al (2004) DNA sequence and analysis of human chromosome 9. Nature 429:369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurles ME (2001) Gene conversion homogenizes the CMT1A paralogous repeats. BMC Genomics 2:11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurles ME, Willey D, Matthews L, Hussain SS (2004) Origins of chromosomal rearrangement hotspots in the human genome: evidence from the AZFa deletion hotspots. Genome Biol 5:R55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmeier D, McGuire G (2003) Detecting recombination in 4-taxa DNA sequence alignments with Bayesian hidden Markov models and Markov chain Monte Carlo. Mol Biol Evol 20:315–337 [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large-scale variation in the human genome. Nat Genet 36:949–951 [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium (2004) Finishing the euchromatic sequence of the human genome. Nature 431:931–945 [DOI] [PubMed] [Google Scholar]
- Jaatinen T, Eholuoto M, Laitinen T, Lokki ML (2002) Characterization of a de novo conversion in human complement C4 gene producing a C4B5-like protein. J Immunol 168:5652–5658 [DOI] [PubMed] [Google Scholar]
- Jakobsen IB, Wilson SR, Easteal S (1997) The partition matrix: exploring variable phylogenetic signals along nucleotide sequence alignments. Mol Biol Evol 14:474–484 [DOI] [PubMed] [Google Scholar]
- ——— (1998) Patterns of reticulate evolution for the classical class I and II HLA loci. Immunogenetics 48:312–323 [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Holloway JK, Kauppi L, May CA, Neumann R, Slingsby MT, Webb AJ (2004) Meiotic recombination hot spots and human DNA diversity. Philos Trans R Soc Lond B Biol Sci 359:141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, Rocchi M, Eichler EE (2001) Positive selection of a gene family during the emergence of humans and African apes. Nature 413:514–519 [DOI] [PubMed] [Google Scholar]
- Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC (1999) Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol 73:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J, Han C, Gordon LA, Terry A, Prabhakar S, She X, Xie G, et al (2004) The sequence and analysis of duplication-rich human chromosome 16. Nature 432:988–994 [DOI] [PubMed] [Google Scholar]
- Maynard Smith J, Smith NH (1998) Detecting recombination from gene trees. Mol Biol Evol 15:590–599 [DOI] [PubMed] [Google Scholar]
- McGuire G, Wright F (2000) TOPAL 2.0: improved detection of mosaic sequences within multiple alignment. Bioinformatics 16:130–134 [DOI] [PubMed] [Google Scholar]
- Newman T, Trask BJ (2003) Complex evolution of 7E olfactory receptor genes in segmental duplications. Genome Res 13:781–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orti R, Potier MC, Maunoury C, Prieur M, Creau N, Delabar JM (1998) Conservation of pericentromeric duplications of a 200-kb part of the human 21q22.1 region in primates. Cytogenet Cell Genet 83:262–265 [DOI] [PubMed] [Google Scholar]
- Proutski V, Holmes EC (1998) SWAN: sliding window analysis of nucleotide sequence variability. Bioinformatics 14:467–468 [DOI] [PubMed] [Google Scholar]
- Rambaut A, Grassly NC (1997) Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput Appl Biosci 13:235–238 [DOI] [PubMed] [Google Scholar]
- Rhodes TD, Nikolaitchik O, Chen J, Powell D, Hu WS (2005) Genetic recombination of human immunodeficiency virus type 1 in one round of viral replication: effects of genetic distance, target cells, accessory genes, and lack of high negative interference in crossover events. J Virol 79:1666–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H, Marszalek JD, Minx PJ, Cordum HS, Waterston RH, Wilson RK, Page DC (2003) Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423:873–876 [DOI] [PubMed] [Google Scholar]
- Ruault M, Ventura M, Galtier N, Brun ME, Archidiacono N, Roizes G, De Sario A (2003) BAGE genes generated by juxtacentromeric reshuffling in the hominidae lineage are under selective pressure. Genomics 81:391–399 [DOI] [PubMed] [Google Scholar]
- Saitta SC, Harris SE, Gaeth AP, Driscoll DA, McDonald-McGinn DM, Maisenbacher MK, Yersak JM, Chakraborty PK, Hacker AM, Zackai EH, Ashley T, Emanuel BS (2004) Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum Mol Genet 13:417–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer S (1989) Statistical tests for detecting gene conversion. Mol Biol Evol 6:526–538 [DOI] [PubMed] [Google Scholar]
- Schildkraut E, Miller CA, Nickoloff JA (2005) Gene conversion and deletion frequencies during double-strand break repair in human cells are controlled by the distance between direct repeats. Nucleic Acid Res 33:1574–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J, Martin J, Terry A, Couronne O, Grimwood J, Lowry S, Gordon LA, et al (2004) The DNA sequence and comparative analysis of human chromosome 5. Nature 431:268–274 [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M (2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528 [DOI] [PubMed] [Google Scholar]
- Shaffer LG, Lupski JR (2000) Molecular mechanisms for constitutional chromosomal rearrangements in humans. Annu Rev Genet 34:297–329 [DOI] [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Emanuel BS (2001) Evolutionarily conserved low copy repeats (LCRs) in 22q11 mediate deletions, duplications, translocations, and genomic instability: an update and literature review. Genet Med 3:6–13 [DOI] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR (2004) Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet Spec 1 13:R57–R64 [DOI] [PubMed] [Google Scholar]
- She X, Horvath JE, Jiang Z, Liu G, Furey TS, Christ L, Clark R, Graves T, Gulden CL, Alkan C, Bailey JA, Sahinalp C, Rocchi M, Haussler D, Wilson RK, Miller W, Schwartz S, Eichler EE (2004a) The structure and evolution of centromeric transition regions within the human genome. Nature 430:857–864 [DOI] [PubMed] [Google Scholar]
- She X, Jiang Z, Clark RA, Liu G, Cheng Z, Tuzun E, Church DM, Sutton G, Halpern AL, Eichler EE (2004b) Shotgun sequence assembly and recent segmental duplications within the human genome. Nature 431:927–930 [DOI] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, et al (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR (2002) Molecular-evolutionary mechanisms for genomic disorders. Curr Opin Genet Dev 12:312–319 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR (2004) Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res 14:2209–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL (2003) PAUP*: phylogenetic analysis using parsimony (*and other methods), version 4. Sinauer Associates, Sunderland, MA [Google Scholar]
- Taudien S, Galgoczy P, Huse K, Reichwald K, Schilhabel M, Szafranski K, Shimizu A, Asakawa S, Frankish A, Loncarevic IF, Shimizu N, Siddiqui R, Platzer M (2004) Polymorphic segmental duplications at 8p23.1 challenge the determination of individual defensin gene repertoires and the assembly of a contiguous human reference sequence. BMC Genomics 5:92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, Tishkoff SA (2004) Signatures of selection and gene conversion associated with human color vision variation. Am J Hum Genet 75:363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MT, Smith NG, Hultin-Rosenberg L, Arndt PF, Ellegren H (2005) Male-driven biased gene conversion governs the evolution of base composition in human alu repeats. Mol Biol Evol 22:1468–1474 [DOI] [PubMed] [Google Scholar]
- Weiller GF (1998) Phylogenetic profiles: a graphical method for detecting genetic recombinations in homologous sequences. Mol Biol Evol 15:326–335 [DOI] [PubMed] [Google Scholar]
- Williams GW, Woollard PM, Hingamp P (1998) NIX: a nucleotide identification system at the HGMP-RC. (http://www.hgmp.mrc.ac.uk/NIX/) (accessed June 25, 2005)
- Wiuf C, Christensen T, Hein J (2001) A simulation study of reliability of recombination detection methods. Mol Biol Evol 18:1929–1939 [DOI] [PubMed] [Google Scholar]
- Ventura M, Mudge JM, Palumbo V, Burn S, Blennow E, Pierluigi M, Giorda R, Zuffardi O, Archidiacono N, Jackson MS, Rocchi M (2003) Neocentromeres in 15q24-26 map to duplicons which flanked an ancestral centromere in 15q25. Genome Res 13:2059–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal N, Mulanga-Kabeya C, Nzilambi N, Delaporte E, Peeters M (2000) Identification of a complex env subtype E HIV type 1 virus from the Democratic Republic of Congo, recombinant with A, G, H, J, K, and unknown subtypes. AIDS Res Hum Retroviruses 16:2059–2064 [DOI] [PubMed] [Google Scholar]