

Abstract
Age-related macular degeneration (ARMD) with abnormal deposit formation under the retinal pigment epithelium (RPE) is the major cause of blindness in the Western world. basal laminar deposits are found in early ARMD and are composed of excess basement membrane material produced by the RPE. Here, we demonstrate that mice lacking the basement membrane component collagen XVIII/endostatin have massive accumulation of sub-RPE deposits with striking similarities to basal laminar deposits, abnormal RPE, and age-dependent loss of vision. The progressive attenuation of visual function results from decreased retinal rhodopsin content as a consequence of abnormal vitamin A metabolism in the RPE. In addition, aged mutant mice show photoreceptor abnormalities and increased expression of glial fibrillary acidic protein in the neural retina. Our data demonstrate that collagen XVIII/endostatin is essential for RPE function, and suggest an important role of this collagen in Bruch's membrane. Consistent with such a role, the ultrastructural organization of collagen XVIII/endostatin in basement membranes, including Bruch's membrane, shows that it is part of basement membrane molecular networks.
Keywords: age-related macular degeneration, collagen XVIII, endostatin, retinal pigment epithelium
Introduction
The retinal pigment epithelium (RPE) is essential for vision, supplying 11-cis retinal to photoreceptors, and performing the daily phagocytosis of the shed distal tips of the photoreceptor outer segments. Abnormalities of the RPE are found in human age-related macular degeneration (ARMD), the major cause of blindness in the Western world, and are associated with morphological changes at the Bruch's membrane/RPE interface with pathological sub-RPE deposit formation, termed basal laminar deposits (Curcio and Millican, 1999). They precede the atrophic as well as the exudative type of ARMD (Sarks, 1976). Early-type basal laminar deposits contain a predominantly amorphous electron-dense material, composed of excess basement membrane (BM) material produced by the RPE (van der Schaft et al, 1994). The pathogenetic mechanisms involved in basal laminar deposits formation in early ARMD remain unknown, in part because of the lack of a convincing mouse model.
To study the role of Bruch's membrane abnormalities for RPE function and deposit formation, we investigated the eyes of mice lacking collagen XVIII/endostatin. This collagen is a component of almost all vascular and epithelial BMs (Muragaki et al, 1995), and is also a component of Bruch's membrane (Fukai et al, 2002). Collagen XVIII molecules contain 10 triple-helical (COL) domains that are separated by non-triple-helical (NC) regions (Oh et al, 1994a). A potent inhibitor of angiogenesis, endostatin (O'Reilly et al, 1997) is a proteolytic fragment of the C-terminal NC1 domain of collagen XVIII. Little is known about the physiological role of collagen XVIII and endostatin. Col18a1−/− mice are viable and fertile, but show developmental defects in hyaloid vessel regression (Fukai et al, 2002). Inactivating mutations in the human gene for collagen XVIII, COL18A1, have been identified in patients with Knobloch syndrome (Sertie et al, 2000), who have progressive retinal degeneration, high myopia, and occipital encephalocele. The underlying histopathological changes in the eyes of Knobloch syndrome patients are unknown, and the disease mechanism is not understood. Eye abnormalities in Knobloch syndrome patients suggest an important role of collagen XVIII and/or endostatin in ocular structures, and imply that there are functional alterations in ocular BMs of these patients and of Col18a1−/− mice.
In this study, we demonstrate that collagen XVIII/endostatin is essential for the function of the RPE. Aged Col18a1−/− mice have reduced visual function with pathological electroretinograms (ERGs). Histologically, we find massive age-dependent accumulation of electron-dense deposits between the RPE and Bruch's membrane. The deposits contain excess BM material and are similar to basal laminar deposits. These changes are associated with an abnormal vitamin A metabolism in the RPE. Rhodopsin content in the retina is reduced, explaining the progressive loss of vision. Glial fibrillary acidic protein (GFAP) expression is increased in the neural retina in association with RPE and photoreceptor abnormalities, as seen in ARMD (Guidry et al, 2002). The RPE abnormalities suggest an important role of this collagen for BM function. To gain insights into the structural basis for this role, we used immuno-electron microscopy (immuno-EM) to determine the ultrastructural organization of collagen XVIII/endostatin in Bruch's membrane and in other BMs, and show that this collagen is anchored in a polarized fashion in perlecan-containing BM scaffolds.
Results
Lack of collagen XVIII/endostatin results in an age-dependent attenuation of visual function
We performed electroretinography experiments with dark-adapted Col18a1−/− mice and wild-type littermates at 2 and 16 months of age. All Col18a1−/− mice had abnormal visual function, with ERGs showing significantly reduced a- and b-wave amplitudes, and prolonged implicit times (Figures 1A and B). Attenuation of visual function increased with age, 16-month-old Col18a1−/− mice having only about 48% of normal b-wave amplitudes (Figure 1B).
Figure 1.
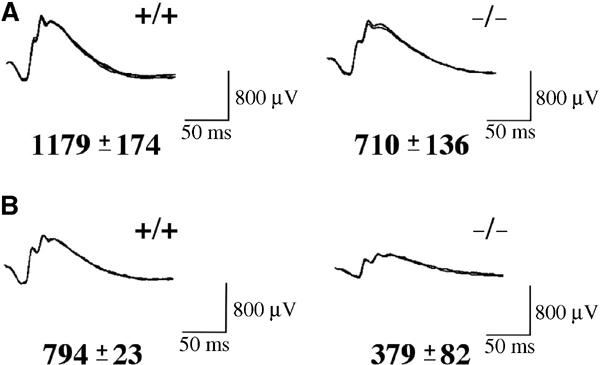
ERGs of Col18a1−/− and wild-type littermates show reduced a- and b-wave amplitudes in mutant mice. (A) Representative ERGs from 2-month-old wild-type and mutant mice. The b-wave amplitude average and standard deviation are indicated. (B) Representative ERGs from 16-month-old wild-type and mutant mice.
Morphological RPE abnormalities in Col18a1−/− mice with age-dependent formation of sub-RPE deposits
Examination of the retina in living Col18a1−/− animals by fluorescence angiography revealed that the retinal vessels were perfused (not shown) and that there was no atrophy of the retina. The eyes of these animals were examined by light and electron microscopy (EM). Bruch's membrane showed no disruption and stained positively for BM components, including laminin and type IV collagen. In wild-type mice, extensive interdigitations were seen between the apical villi of RPE cells and the photoreceptor outer segments (Figure 2A). In contrast, in aged Col18a1−/− mice, morphological abnormalities of the RPE and the outer segments of the photoreceptors could be seen (Figures 2B and C). The outer segments of the photoreceptors appeared disorganized and abnormally bent in mutant mice, and a reduced interdigitation of the apical villi of the RPE with the photoreceptors was apparent when compared to wild-type littermates. Despite the reduced interdigitation, cell polarity of the RPE was maintained in the mutant mice, with apical villi and basal cell membrane infoldings present. In addition, immunohistochemical staining for the polarization marker ezrin showed no difference between mutant and wild-type mice (not shown).
Figure 2.

Col18a1−/− mice show age-dependent morphological abnormalities of the RPE and sub-RPE deposits. (A) Electron micrograph of the RPE region (white double-headed arrow) of a wild-type littermate (16-month-old) with no sub-RPE deposits at the basement membrane (BM) and normal interdigitation (arrow) of the apical villi of the RPE with the photoreceptor outer segments. Scale bar, 1.5 μm. (B) Reduced interdigitation of the apical villi of the RPE with the photoreceptor outer segments (arrow) in a 16-month-old Col18a1−/− mouse. Abnormal deposits are visible at the basal RPE (black double-headed arrow). Scale bar, 1.5 μm. (C) Increased sub-RPE deposits (black double-headed arrow) in a 22-month-old Col18a1 null mouse. The diameter of the remaining RPE cell (white double-headed arrow) is reduced in relation to the sub-RPE deposits. Scale bar, 1.5 μm. (D) The RPE of a 16-month-old wild-type littermate shows no deposits between the basal infoldings (arrow) or membranous debris. Scale bar, 0.5 μm. (E) Higher magnification of electron-dense deposits (arrow) between the basal infoldings of the RPE in mutant mice (16-month-old). Scale bar, 1 μm. (F) Membranous debris (arrowhead) of RPE basal infoldings in aged mutant mice (16-month-old). Amorphous material is indicated by an arrow. Scale bar, 0.3 μm. (G) Electron-dense amorphous sub-RPE deposits (arrow) with membranous debris (arrowhead) in a 16-month-old Col18a1−/−/Hspg2Δ3/Δ3 mutant mouse. Scale bar, 0.5 μm.
In a number of mouse strains, including C57Bl/6 mice examined here, subtle age-dependent accumulation of amorphous electron-dense material between the basal infoldings of the RPE can be seen by EM. However, in aged Col18a1−/− mice the accumulation of such material was dramatically increased when compared to wild-type littermates (Figure 2D), with the material occupying the entire sub-RPE space (Figures 2E and F). This amorphous electron-dense material contained vesicles and membranous debris, and was continuous with the lamina densa of the RPE BM, which had the same electron density when examined by EM. The RPE basal infoldings were wider in mutant mice than in wild-type mice that had no deposits. In 22-month-old mutant mice, the sub-RPE deposits occupied a larger area than the entire RPE diameter (Figure 2C). The deposits were found throughout the entire sub-RPE space of the eye, including the peripheral retinal region. These deposits observed in aged Col18a1−/− mice show striking morphological similarities to basal laminar deposits found in aged human eyes with early ARMD (van der Schaft et al, 1994). Widened RPE basal infoldings and irregular apical villi, as observed in these mutant mice, are also found in human eyes with basal laminar deposits (van der Schaft et al, 1994). In 2-month-old Col18a1−/− mice, we did not observe sub-RPE deposit formation. Thus, the deposit formation in Col18a1−/− mice is an age-dependent process, which is associated with the progressive attenuation of visual function.
Since collagen XVIII is a heparan sulfate proteoglycan (HSPG) (Halfter et al, 1998), we tested if the deposits are due to the lack of collagen XVIII protein or due to a reduced heparan sulfate (HS) content of Bruch's membrane, by examining sub-RPE deposit formation in mice with further depleted HS content of Bruch's membrane. These mice lack Col18a1 and also exon 3 of the perlecan gene, resulting in a loss of attachment sites for three HS side chains of perlecan (Hspg2Δ3/Δ3), but have essentially normal levels of perlecan core protein (Rossi et al, 2003). We found that sub-RPE deposits in the Col18a1−/−/Hspg2Δ3/Δ3 mice were similar in size and morphology as in Col18a1−/− mice (Figure 2G). These findings suggest that the formation of sub-RPE deposits is a consequence of the lack of collagen XVIII protein and not due to a reduced HS content of Bruch's membrane. Although the possibility of a compensatory upregulation of other HSPGs at the BM site in double-mutant mice—and therefore an HS content like in Col18a1−/− mice—cannot be entirely excluded, this seems unlikely based on the previously reported observation of more severe lens degeneration in double-mutant mice, when compared to Hspg2Δ3/Δ3 mice (Rossi et al, 2003). While the lens in Col18a1−/− mice shows no degeneration, in double-mutant mice lens degeneration appears earlier than in Hspg2Δ3/Δ3 mice, suggesting that the additional lack of HS in the lens capsule is not compensated by upregulation of other HSPGs in the BM.
An increased accumulation of electron-lucent debris and lipids within Bruch's membrane has been observed in ApoE−/− mice (Dithmar et al, 2000). To test if the extent of sub-RPE deposit formation in Col18a1−/− mice is influenced by the accumulation of debris and lipids within Bruch's membrane, we crossed Col18a1−/− mice with ApoE−/− mice, and examined the formation of sub-RPE deposits in the double-mutant mice. We found that the extent and onset of deposit formation in Col18a1−/−/ApoE−/− mice was not significantly different from Col18a1−/− mice (Figure 3). This suggests that sub-RPE deposit formation in Col18a1−/− mice is a process that is independent of the accumulation of abnormal lipid material into Bruch's membrane.
Figure 3.
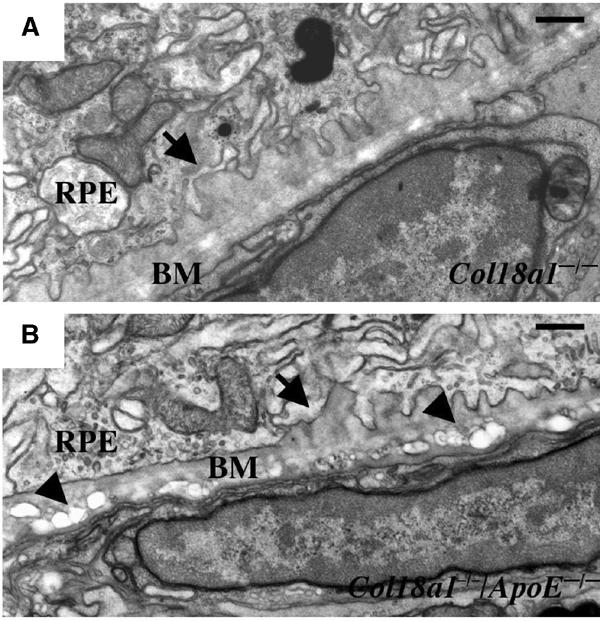
Sub-RPE deposit formation occurs independently of accumulation of lipids in Bruch's membrane. (A) Early sub-RPE deposits (arrow) in a 7-month-old Col18a1−/− mouse. Scale bar, 0.5 μm. (B) Electron-lucent material (arrowheads) in Bruch's membrane (BM) in a 7-month-old ApoE−/−/Col18a1−/− mouse that shows similar sub-RPE deposits (arrow) as seen in Col18a1−/− mice. Scale bar, 0.5 μm.
Sub-RPE deposits in Col18a1−/− mice contain excess BM material
To test if the sub-RPE deposit formation in aged Col18a1−/− mice may serve as a model for basal laminar deposits formation in early ARMD, we aimed to characterize the composition of these deposits and compare them to basal laminar deposits. It has been demonstrated by immuno-EM that basal laminar deposits in early ARMD contain excess BM material, including type IV collagen (van der Schaft et al, 1994). Based on the morphological similarities of the observed sub-RPE deposits in aged Col18a1−/− mice to basal laminar deposits, we speculated that the deposits result from abnormal accumulation of BM material produced by the RPE. We performed immuno-EM experiments with polyclonal antibodies against BM components and other proteins produced by the RPE, such as TIMP-3 or ApoE. We found that the sub-RPE deposits labeled strongly for the BM component type IV collagen (Figures 4A and B). In addition, high-magnification EM images suggested the presence of the BM component type VIII collagen in the deposits (Figure 4C), based on the observation of typical hexagonal arrays as formed by collagen VIII molecules within Descemet's membrane or in vitro (Sawada et al, 1990). In situ hybridization for Col8a1 and Col8a2 demonstrated that murine RPE cells express type VIII collagen (Figure 4D).
Figure 4.

Sub-RPE deposits in Col18a1−/− mice contain excess BM material. (A) Immuno-EM labeling of an 18-month-old Col18a1−/− mouse eye with polyclonal anti-type IV collagen antibodies. The BM (ECBM) of the endothelial cells (EC) of the choroid layer shows heavy labeling, as well as the electron-dense sub-RPE deposits (BLD). The RPE BM is indicated (arrow). Collagen fibers (CF) can be seen within Bruch's membrane. Scale bar, 250 nm. (B) Higher magnification of sub-RPE deposits that were labeled for type IV collagen. Scale bar, 250 nm. (C) Arrows indicate area of hexagonal ultrastructure observed within areas of sub-RPE deposits in Col18a1−/− mice that resemble type VIII collagen. Scale bar, 0.5 μm. (D) In situ hybridization for Col8a2 shows a positive purple stain of corneal endothelium (arrow), and also for RPE cells (line). Retina (R) and lens (L) are indicated. Magnification × 10.
Thus, sub-RPE deposits in Col18a1−/− mice resemble basal laminar deposits not only in their morphology but also in their molecular composition. Based on these findings, we suggest that Col18a1−/− mice may serve as a model for studying mechanisms of basal laminar deposits formation.
Aged Col18a1−/− mice have reduced retinyl esters in the RPE and decreased rhodopsin content in the retina
Sub-RPE deposit formation in early ARMD is believed to interfere with transport processes and metabolism of the RPE, such as with the uptake or processing of vitamin A. Vitamin A is modified in the RPE in order to provide 11-cis retinal to the photoreceptors, which is required for vision. We speculated that the abnormal deposit formation in aged Col18a1−/− mice might interfere with vitamin A uptake or processing. In order to test this hypothesis, we measured retinyl ester content in the RPE of mutant and control mice, these esters being RPE storage forms of vitamin A, and found significantly reduced retinyl ester content in the RPE of aged Col18a1−/− mice with values of less than 20% compared to those of wild-type littermates (Figure 5A). In 2-month-old mutant mice, no deposits were detected, and consistent with this observation, the retinyl ester content was not significantly reduced in the RPE of these mice. Serum retinoid levels showed no difference between mutant and wild-type mice (data not shown), excluding a systemic retinoid abnormality in Col18a1−/− mice as the reason for the reduced RPE retinyl esters.
Figure 5.

Aged Col18a1−/− mice have reduced retinyl esters in the RPE, decreased rhodopsin contents in the retina, and reduced RPE65 protein. (A) Total endogenous retinyl ester content is reduced in 16-month-old Col18a1−/− mice when compared to wild-type littermates, but not in young mutant mice (2-month-old). The contents of retinyl esters are indicated as pmol/eye. Values are mean±s.e.m. of 2–3 separate experiments. We confirmed in a series of independent HPLC analyses with C57Bl/6 mice that the observed age-dependent increase of retinyl ester contents in the RPE of C57Bl/6 control mice is normal. (B) Measurements of rhodopsin contents (pmol/eye) in 2- and 16-month-old wild-type and mutant mice. Aged mutant mice show a significant reduction of rhodopsin contents in the retina. Values are mean±s.e.m. of 3–4 separate experiments. (C) Western blot of whole-eye homogenates of 18-month-old Col18a1−/− mice (KO) and matched wild-type control animals (WT). Numbers indicate densitometric measurements for β-actin (loading control) and RPE65 labeling, demonstrating a decrease of RPE65 protein in KO mice. Recombinantly produced RPE65 protein (RPE65) served as control sample.
Based on these data, we hypothesized that the reduction of retinyl esters in the RPE in aged Col18a1−/− mice might be associated with an insufficient supply of 11-cis retinal to photoreceptors, and thus result in reduced rhodopsin concentrations in the retina. We measured rhodopsin content in the eyes of dark-adapted Col18a1−/− mice and wild-type littermates, and observed a significant reduction of rhodopsin in the retinas of aged Col18a1−/− mice (Figure 5B).
Reduced levels of RPE65 protein in the RPE of aged Col18a1−/− mice
We performed high-dose vitamin A administration experiments with aged Col18a1−/− and control mice, and measured the effect of vitamin A on visual sensitivity by ERGs. If the sub-RPE deposits are the rate-limiting factor for vitamin A uptake into the RPE, one would expect that high systemic concentrations of vitamin A would increase vitamin A uptake into the RPE and consequently lead to increased ERG amplitudes in the mutant mice. Systemic administration of five times higher doses of vitamin A than were sufficient to increase ERG amplitudes in vitamin A-deprived wild-type animals (Katz et al, 1993) did not lead to a significant increase in ERG amplitudes in aged Col18a1−/− mice. B-wave amplitudes of baseline ERGs in a group of 16-month-old mutant mice were 397±112 μV, and 48 h after intramuscular administration of 40 μg vitamin A, b-wave amplitudes had not significantly changed (450±192 μV). Administration of higher doses of vitamin A and ERG measurements up to 96 h after injections failed to detect a significant increase of b-wave amplitudes in aged Col18a1−/− mice. These findings suggest that the reduced retinyl esters in the RPE and rhodopsin levels in the neural retina are, at least in part, a consequence of an RPE/neural retina dysfunction. To provide further evidence for this hypothesis, we measured RPE65 protein levels in the eyes of aged mutant and control mice. RPE65 is essential for the formation of 11-cis retinal (Redmond et al, 1998), and has recently been demonstrated to be a major membrane-associated retinoid binding protein of the RPE (Jahng et al, 2003). In western blot experiments, we found in the eyes of aged Col18a1−/− mice a reduction of RPE65 protein levels to about a third when compared with age-matched controls (Figure 5C). Thus, lack of collagen XVIII results in RPE dysfunction with an abnormal vitamin A metabolism, associated with decreased levels of RPE65 and retinyl esters, and in reduced retinal rhodopsin levels with attenuation of visual function.
Abnormalities in the neural retina of Col18a1−/− mice
Photoreceptors depend on a proper function of the RPE. Retinal abnormalities have been described in ARMD eyes, with an increased expression of GFAP in Müller cells (Guidry et al, 2002). Based on the observed morphological changes of the distal photoreceptors and the RPE abnormalities in Col18a1−/− mice, we examined if the retina showed further pathological changes. We found an increased expression of GFAP in the retina of Col18a1−/− mice (Figure 6). GFAP expression was highest at local areas in the retina where photoreceptors appeared disorganized.
Figure 6.
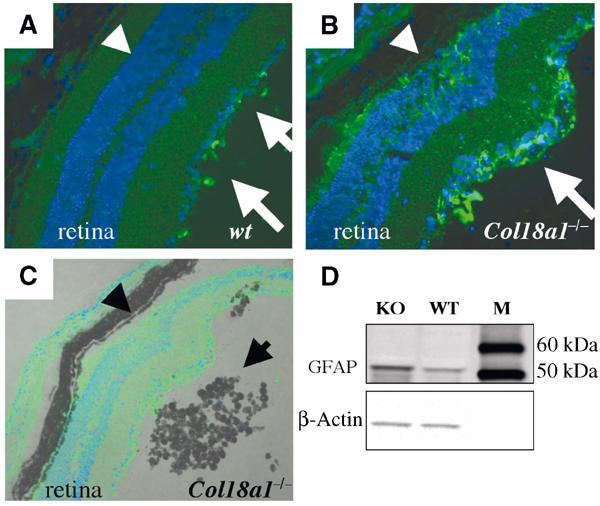
Increased expression of GFAP in the neural retina of Col18a1−/− mice. (A) Immunofluorescence labeling of GFAP (using an FITC-conjugated secondary antibody) in a section of an 18-month-old wild-type mouse. Some labeling for GFAP can be seen at the inner limiting membrane region of the retina (arrows), but no GFAP was detected in the photoreceptor layer (arrowhead). Magnification × 40. (B) Increased GFAP labeling is found in the photoreceptor layer (arrowhead) and the inner limiting membrane region (arrow) in an 18-month-old Col18a1−/− mouse eye. Local disorganization of photoreceptors can be observed as well (region between arrow and arrowhead). Magnification × 40. (C) Overlay image of the same region as in (B). Pigmented cells (arrow) accumulate at regions of photoreceptor disorganization (arrowhead) at the retinal–vitreal interface. Magnification × 20. (D) Western blot showing increased GFAP protein in an 18-month-old Col18a1−/− mouse eye (densitometric value: 1436) in comparison to the wild-type control sample (densitometric value: 471). β-Actin was used as a loading control (KO densitometric value: 605; WT densitometric value: 504). M: marker lane.
We previously described F4/80-positive macrophage-like pigmented cells that migrate out of the iris in aged Col18a1−/− mice (Marneros and Olsen, 2003), but not in young mutant mice. What leads to the migration of these cells in aged mutant mice is unclear. Here we find that these macrophage-like cells accumulate at the retinal/vitreous border at areas of heavily increased GFAP expression and local photoreceptor disorganization (Figure 6). This observation suggests that the age-dependent RPE dysfunction and retinal changes attract these macrophage-like cells.
In conclusion, lack of collagen XVIII leads to functional RPE abnormalities with the formation of excess BM-like material as basal laminar-like deposits under the RPE, an altered vitamin A metabolism of the RPE, and neural retina changes with reduced rhodopsin levels that result in an attenuation of visual function with pathological ERGs (Figure 7). The observed RPE abnormalities and loss of visual function in aged mutant mice, most likely due to the effect of the lack of collagen XVIII/endostatin in the underlying Bruch's membrane, suggest that the absence of this collagen might cause altered properties of Bruch's membrane and induce functional changes in the RPE. To better understand how collagen XVIII/endostatin may function within Bruch's membrane and other BMs, we determined the ultrastructural organization of collagen XVIII molecules in BMs.
Figure 7.
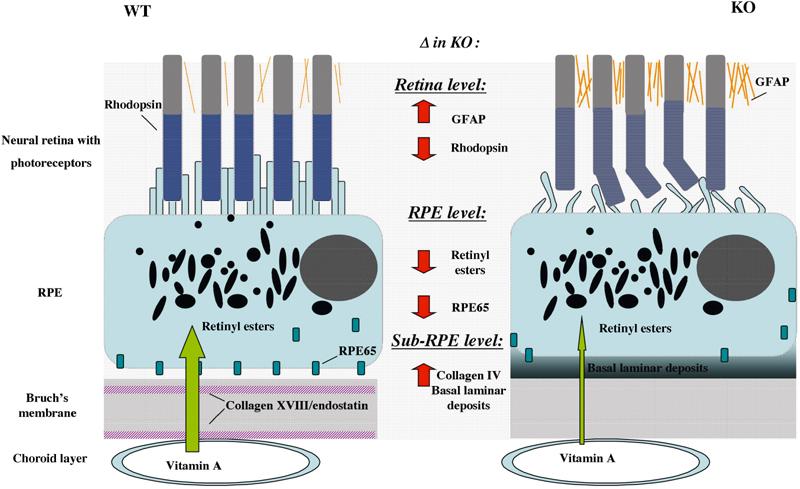
Schematic model of RPE and retinal abnormalities in Col18a1−/− mouse eyes in comparison to normal eyes. Sub-RPE deposits in mutant mice are associated with reduced RPE65 protein and reduced retinyl esters in the RPE, reduced retinal rhodopsin content, photoreceptor abnormalities, and increased retinal GFAP expression.
Ultrastructural localization of collagen XVIII/endostatin in Bruch's membrane and other BMs
We performed immuno-EM of Bruch's membrane with antibodies against distinct collagen XVIII domains (Figure 8A). To distinguish between the N-terminal and the C-terminal region of collagen XVIII, we generated affinity-purified polyclonal antibodies against the N-terminal NC11(XVIII) and the C-terminal endostatin domains. Additionally, we used antibodies against the central NC7(XVIII) domain and against a peptide sequence within the NC1(XVIII) domain, located N-terminal to the protease cleavage site for endostatin release. We found labeling with the anti-NC11(XVIII) antibodies (Figure 8B) in the regions of the sublamina densa of the RPE BM and of the endothelial BM of the choroid layer. In contrast, labeling for endostatin in Bruch's membrane was closer to the RPE and endothelial cell basal cell membrane (Figure 8C). Thus, collagen XVIII molecules in Bruch's membrane are oriented in a polarized fashion, with the C-terminal endostatin domain facing the RPE/endothelial cell and the N-terminal NC11(XVIII) domain facing the inner/outer collagenous layer of Bruch's membrane.
Figure 8.

Ultrastructural localization of collagen XVIII and endostatin in Bruch's membrane. (A) Diagram of the domain structure of type XVIII collagen chains. Each chain contains 10 triple-helical (COL) domains (black boxes) flanked and interrupted by 11 non-triple-helical (NC) domains. The C-terminal NC1 domain contains the endostatin domain at the C-terminus. The locations of sequences used for antibody generation are indicated by vertical arrows. (B) Immuno-EM labeling of mouse Bruch's membrane with an anti-NC11(XVIII) antibody. Labeling is seen in the region of the sublamina densa of the RPE BM (black arrows) or of the endothelial BM of the choroid layer (black arrowheads). The white arrow indicates RPE basal cell membrane, and the white arrowhead indicate endothelial basal cell membrane. Scale bar, 150 nm. (C) Immuno-EM labeling of mouse Bruch's membrane with an anti-endostatin antibody. Labeling is seen within the lamina densa of the RPE BM (black arrows) or of the endothelial BM of the choroid layer (black arrowhead). Scale bar, 150 nm. (D) Rotary shadowing EM of recombinant collagen XVIII. Kinks and bends are visible (black arrow). The N-terminal myc tag is labeled with an anti-myc tag antibody (white arrow). Scale bar, 17 nm. (E) Rotary shadowing EM of collagen XVIII purified from chicken vitreous. Kinks and bends are indicated (black arrow). Scale bar, 17 nm.
We further investigated if the distribution of labeling with these antibodies is different in Bruch's membrane when compared to other BMs, such as epidermal and vascular endothelial BMs. We found a similar distribution of labeling, suggesting that the localization of collagen XVIII domains in BMs shows no major variation in endothelial and epithelial BMs. In skin epidermal BMs, the C-terminal NC1(XVIII) and endostatin domains were found within the lamina densa of the BM (Figure 9A), colocalized with perlecan (not shown). In contrast, the N-terminal NC11(XVIII) domain was localized in the matrix subjacent to the lamina densa of the BM (Figures 9B and C); antibodies against the intermediate NC7(XVIII) domain showed labeling of regions in between (not shown). The average distance between gold particles using anti-endostatin and anti-NC11(XVIII) antibodies was about 70 nm (Figure 9D), and that between endostatin and NC7(XVIII) was about 30 nm.
Figure 9.
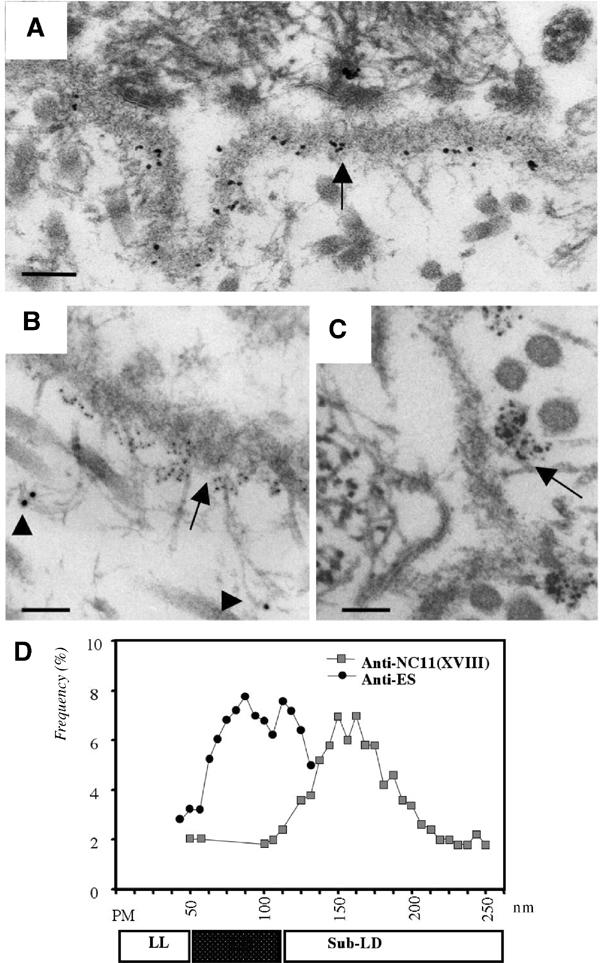
Ultrastructural localization of collagen XVIII and endostatin in the skin BM. (A) Immuno-EM labeling of mouse skin with an anti-endostatin antibody. Labeling is seen within the lamina densa of the epithelial BM. Scale bar, 130 nm. (B) Double immunogold labeling of human skin with antibodies against NC1(VII) (large gold particles, arrowheads) and NC11(XVIII) (small gold particles, arrow) domains. Loop structures connecting large gold particles at the lamina densa are likely to be anchoring fibrils containing type VII collagen. Scale bar, 90 nm. (C) Immunogold labeling of human skin with polyclonal antibodies against the NC11(XVIII) common region. Note clusters of gold particles along the sublamina densa of the epidermal BM. Most label is seen on the matrix side of the lamina densa. Scale bar, 70 nm. (D) Frequency distribution of distances between gold particles and basal plasma membrane of epithelial cells. Distances (in nm) between basal plasma membrane (PM) of basal keratinocytes in skin and gold particles after labeling with polyclonal anti-NC11(XVIII) (shaded squares) and anti-endostatin (filled circles) antibodies are shown. For each antibody, 100 particles were measured; the frequency of particles at different distances is plotted along the Y-axis. The stippled box below the diagram indicates the position of the lamina densa (LD) and open boxes indicate the regions of lamina lucida (LL) and sublamina densa (Sub-LD).
Rotary shadowing EM of recombinant full-length collagen XVIII and purified collagen XVIII from chicken vitreous (Figures 8D and E) showed that collagen XVIII molecules have flexible or kinky regions, likely due to the presence of the non-triple-helical domains that interrupt the triple-helix.
Collagen XVIII/endostatin is a component of BM networks
To further examine the presence of collagen XVIII and endostatin within networks of major BM components, BM preparations from skin homogenates were used for double-labeling immuno-EM experiments. These preparations were generated by the mechanical separation of superficial layers of human dermis after high-salt epidermolysis, and double-labeled using combinations of antibodies against endostatin, NC7(XVIII), NC11(XVIII), and antibodies against major BM components. Distinct colocalization of collagen XVIII and endostatin with perlecan (Figures 10A and B) was found in these preparations. This observation is consistent with our immuno-EM results showing colocalization of the endostatin domain with perlecan within the lamina densa. In summary, the immuno-EM experiments suggest that collagen XVIII/endostatin is part of perlecan-containing BM networks in vivo.
Figure 10.

Collagen XVIII is anchored into perlecan-containing BM networks. (A) Immunogold labeling of BM preparations from human dermis with antibodies against NC11(XVIII) domain and perlecan. Large (18 mm) gold particles (arrowhead) indicate labeling with a polyclonal antibody against NC11(XVIII). Small (12 mm) gold particles (arrow) indicate labeling with a monoclonal antibody against perlecan. Collagen fibril diagonally across field. Scale bar, 120 nm. (B) Immunogold labeling of BM preparations from human dermis with antibodies against endostatin and perlecan. Large (12 nm) gold particles indicate labeling with a polyclonal antibody against endostatin (arrowhead). Small (6 nm) particles indicate labeling with a monoclonal antibody against perlecan (arrow). Scale bar, 120 nm. (C) Immunogold labeling of BM preparations from human dermis treated with heparitinase and immunogold labeled with antibodies against NC11(XVIII) and endostatin domains. Large (18 nm) gold particles (black arrow) indicate labeling with a polyclonal anti-NC11(XVIII) antibody. Small (12 nm) gold particles (arrowhead) indicate labeling with an anti-endostatin antibody. Colocalization of NC11(XVIII) and endostatin labeling in dermis preparations without cathepsin L treatment can be seen. Scale bar, 130 nm. (D) No colocalization of NC11(XVIII) and endostatin labeling in dermis preparations after cathepsin L incubation can be seen. Only large (18 nm) gold particles (black arrow) are detected, which indicate labeling for NC11(XVIII), but no endostatin is detected (small gold particles). Scale bar, 130 nm. (E) Western blot of BM preparations from human dermis with an anti-endostatin antibody. Lane 1: a high-molecular-weight collagen XVIII band containing endostatin and a 20 kDa endostatin band is detected after limited proteolysis with cathepsin L. Lane 2: a high-molecular-weight collagen XVIII band (∼200 kDa) containing endostatin, but no 20 kDa endostatin band, is detected when cathepsin L incubation is omitted. Lane 3: control sample of human recombinant endostatin as a 20 kDa band. (F) Model for the organization of collagen XVIII in the BM.
To examine whether endostatin is part of collagen XVIII molecules in the BM and whether it can be released from collagen XVIII through proteolytic processing, we treated the BM preparations with highly purified cathepsin L, which we showed previously to release endostatin from recombinant NC1(XVIII) in vitro (Felbor et al, 2000). After enzyme treatment, we used this material for double-labeling experiments and western blots. Colocalization of endostatin was observed with NC7(XVIII) (not shown) and NC11(XVIII) (Figure 10C) in double-labeling experiments with these BM preparations. After enzyme treatment, only labeling for NC7(XVIII) or NC11(XVIII) (Figure 10D), but not for endostatin, could be detected, implying that cathepsin L treatment removed endostatin from full-length collagen XVIII in the BM preparations. These BM preparations were exposed to high-salt epidermolysis and subsequent washing steps with nondenaturing solutions, conditions eliminating free endostatin from these preparations, as demonstrated by the lack of a 20 kDa endostatin band in western blots of these BM preparations (Figure 10E). However, anti-endostatin antibodies revealed a high-molecular-weight band corresponding to full-length collagen XVIII, detectable also with anti-NC7(XVIII) antibodies (not shown). In western blots of the cathepsin L-treated BM preparations, a 20 kDa endostatin band became apparent (Figure 10E, lane 1), demonstrating proteolytic release of endostatin from full-length collagen XVIII. In conclusion, the tissue form of collagen XVIII in BMs comprises, at least in part, the full-length protein including the C-terminal endostatin domain. Endostatin can be released from intact collagen XVIII by limited proteolysis with cathepsin L.
Discussion
BMs are not only selective barriers and scaffolds to which cells adhere, but are also regulators of cell function and cell survival. Our data presented here demonstrate that the extracellular matrix component collagen XVIII/endostatin is essential for the maintenance of the RPE and imply an important role of this collagen for Bruch's membrane function.
Aged Col18a1−/− mice show massive accumulation of electron-dense amorphous material with membranous debris between the RPE and Bruch's membrane, which is similar in appearance and composition to basal laminar deposits in early ARMD (van der Schaft et al, 1994) and contains excess BM material. This suggests that the absence of collagen XVIII leads to altered properties of Bruch's membrane, which either cause the RPE to produce excess BM material or interfere with the clearance of such BM material, eventually resulting in a progressive accumulation of basal laminar deposits-like material under the RPE with age. We did not observe sub-RPE deposit formation in young Col18a1−/− mice, which showed normal visual function in electroretinography experiments and normal retinal rhodopsin contents. In aged mutant mice that had extensive deposits throughout the entire sub-RPE space of the eye, we found a dramatic attenuation of visual function in electroretinography experiments with a reduced retinal rhodopsin content. Thus, the abnormal age-dependent sub-RPE deposit formation is associated with the progressive loss of vision in Col18a1−/− mice. The accumulation of sub-RPE deposits due to the lack of collagen XVIII/endostatin in the BM leads to functional changes of the RPE and subsequently to a reduced retinal rhodopsin content. A significant reduction of retinyl esters was found in the RPE of aged mutant mice, whereas young animals with no sub-RPE deposits had normal retinyl ester contents. Furthermore, the level of RPE65 protein, a major retinoid binding protein of the RPE that is essential for the generation of 11-cis retinal, was reduced in the RPE of aged Col18a1−/− mice. Thus, aged Col18a1−/− mice have functional RPE abnormalities that affect their retinoid metabolism, associated with reduced rhodopsin levels and loss of visual function.
In the eyes of aged mutant mice with extensive sub-RPE deposits, abnormal apical villi of the RPE showed a reduced interdigitation with photoreceptor outer segments, which were abnormally bent. In contrast, regular apical villi and photoreceptors were observed in wild-type littermates that had no sub-RPE deposits. Such morphological abnormalities of the RPE and photoreceptors have been described in association with basal laminar deposits in early ARMD (van der Schaft et al, 1994). Analysis of the retina in eyes with ARMD demonstrated an increased expression of GFAP in Müller cells as a consequence of sub-RPE deposits and RPE dysfunction (Guidry et al, 2002). Similarly, aged Col18a1−/− mice had an increase of GFAP expression in their retina.
The observed abnormalities in the eyes of aged mutant mice demonstrate that collagen XVIII is essential for the maintainance of the RPE and for proper function of Bruch's membrane. However, it is unlikely that the accumulation of excess BM material under the RPE is a specific consequence of the lack of collagen XVIII in Bruch's membrane. Instead, we suggest that the RPE forms basal laminar deposits as a reaction to cell stress or damage, caused by a structurally altered Bruch's membrane (as in Col18a1−/− mice) or by direct damage to the RPE (as in experiments with mice where laser photochemical injury of the RPE induced the formation of basal laminar deposits; Dithmar et al, 2001).
Knobloch syndrome patients with inactivating collagen XVIII mutations show progressive retinal degeneration with age-dependent loss of vision. Fundoscopic examination of the retina of Knobloch syndrome patients suggested RPE abnormalities (Passos-Bueno et al, 1994), consistent with our observations in Col18a1−/− mice that collagen XVIII/endostatin is essential for RPE function. It is likely that aged Col18a1−/− mice are a model for early abnormalities of the RPE in Knobloch syndrome patients, and that our data provide a pathogenetic mechanism for the progressive loss of vision in patients with this syndrome.
In order to assess the role of collagen XVIII/endostatin in BM organization, we determined the in vivo ultrastructural location of collagen XVIII and endostatin in BMs. The colocalization of NC1(XVIII)/endostatin with perlecan in the lamina densa and in isolated BM preparations is consistent with the results of previous in vitro experiments, showing interactions of endostatin with laminin and perlecan (Sasaki et al, 1998), and demonstrates the in vivo significance of these interactions. We further demonstrate that endostatin is at least in part found as the C-terminal domain of full-length collagen XVIII in BMs, from where it can be released by limited proteolysis (Figure 10F).
Since we did not observe differences in the structural organization of collagen XVIII in Bruch's membrane and in other ocular, epithelial, and endothelial BMs, the question arises as to why primarily eye abnormalities are found in Col18a1−/− mice and Knobloch syndrome patients, and no pathological abnormalities are seen in other organs. We speculate that the function of collagen XVIII/endostatin might not be different in Bruch's membrane and in other BMs, and that this collagen may have an important role in maintaining normal function of adjacent epithelial and endothelial cells in all BMs. However, the requirements for proper interaction with the extracellular matrix might differ between the RPE and other epithelial cells. In most epithelial tissues, regeneration occurs, but the RPE does not undergo mitosis once differentiated and has no comparable regenerative potential. The high metabolic activity of the RPE, maintained throughout life, may make it more susceptible to subtle changes in the extracellular matrix, and induce the formation of sub-RPE deposits over time.
Our findings link the lack of collagen XVIII/endostatin to morphological and functional RPE changes, and suggest a pathogenetic mechanism for the reduced visual function in Col18a1−/− mice by showing an abnormal retinoid metabolism of the RPE and a decreased rhodopsin content in their retinas. The absence of collagen XVIII/endostatin might cause subtle functional and structural changes of the highly complex Bruch's membrane, eventually resulting in an altered RPE function with abnormal basal laminar-like sub-RPE deposit formation with age, and in changes of the retina that lead to a progressive attenuation of visual function. The results highlight the importance of BM components for RPE function and for the formation of pathological sub-RPE deposits. They further suggest that Col18a1−/− mice are a model for basal laminar deposits formation in early forms of age-related retinal degenerations where sub-RPE deposits precede the retinal defects, like in ARMD.
Materials and methods
Animals
The generation of Col18a1−/− mice has been described (Fukai et al, 2002). To ensure uniformity of genetic backgrounds of littermate wild-type and homozygous mice, Col18a1−/− mice were backcrossed with C57Bl/6 mice for 15 generations. Mice lacking Col18a1 and exon 3 of perlecan have been described (Rossi et al, 2003). ApoE−/− (C57Bl/6 strain) mice were purchased from Jackson Labs (Banghor, Maine). ApoE−/−/Col18a1−/− mice and ApoE+/+/Col18a1−/− littermates were generated by crossing ApoE−/+/Col18a1−/− breeder pairs. Mice were fed a regular chow diet.
ERGs and vitamin A administration experiments
After overnight dark adaptation, full-field ERGs were elicited with flashes of white light (Li et al, 1998). ERGs were performed on a group of 2- and 16-month-old mutant (n=10) and control animals (n=6).
All-trans retinol (Sigma, St Louis, MO) was prepared as previously reported (Katz et al, 1993). Single intramuscular injections of a 40 μg dose of all-trans retinol were administered to the groups of 16-month-old wild-type and knockout mice. Prior to all-trans retinol injections, baseline ERGs were measured. At 48 and 96 h after the first injection, ERGs were measured, and a second injection was administered 48 h after the first injection.
Retinyl ester and rhodopsin measurements
Whole eyes of 2- and 16-month-old wild-type (n=4) and mutant animals (n=4) were processed for retinyl ester measurements by HPLC with a normal-phase Lichrosphere SI-60 (Alltech, Deerfield, IL) 5 μm column and isocratic solvent as previously described (Redmond et al, 1998). Pure synthetic standards were used to confirm the identity of the peaks. Rhodopsin levels in the retina of dark-adapted mutant (n=6) and wild-type (n=7) mice were measured by microspectrophotometry. Rhodopsin was quantitated by the difference spectrum obtained from subtracting the absorbance spectrum of the pigment after exposure to white light from the data obtained before exposure; samples were solubilized in 1% dodecylmaloside.
Western blots for RPE65 and GFAP
Preparation of eye tissue from mutant (n=4) and wild-type (n=4) mice, generation of recombinant RPE65, and western blot experiments for RPE65 have been described (Ma et al, 2001). A polyclonal anti-mouse GFAP antibody was used for the detection of GFAP in western blots of whole-eye homogenates (Chemicon International Co., Temecula, CA). For loading controls, polyclonal anti-β-actin antibodies were used (Sigma).
Morphological examination of mouse eyes
For histological examination, eyes from Col18a1−/− and wild-type littermates in the age range between 1 week and 22 months were fixed in 1.25% formaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4). After postfixation in 4% osmium tetroxide, and dehydration steps, the eyes were embedded in TAAB epon (Marivac Ltd, Halifax, Canada) and used for standard transmission EM (Fukai et al, 2002).
For immunohistochemistry, we used 7 μm thick frozen sections of fixed eyes. In this study, we used antibodies recognizing collagen VII (from Dr Bruckner-Tuderman), ezrin (from Dr Arpin), a peptide of the NC1(XVIII) domain (from Dr Azar), NC7(XVIII) (from Dr Ninomiya), perlecan (from Dr Timpl), collagen IV (Chemicon), GFAP (Chemicon), and laminin (Chemicon). Primary antibodies and FITC-labeled secondary antibodies were used in serial dilutions. In situ hybridizations for Col8a1 and Col8a2 were performed using standard techniques (Fukai et al, 2002).
Generation of antibodies
We generated a polyclonal anti-NC11(XVIII) antibody against the common region (amino-acid residues 487–785, as counted from the methionine start codon) of the CR form of mouse collagen XVIII (Muragaki et al, 1995). Specific crossreactivity of this antibody with human NC11(XVIII) was demonstrated in western blot experiments. A polyclonal rabbit antibody was generated against the endostatin domain (amino-acid residues 132–315, as counted from the N-terminus of the mouse NC1 domain; Oh et al, 1994b). The clone mc3b (Oh et al, 1994a) was used as a template for PCR amplification. Antigen production, purification, immunization of rabbits, and antigen affinity purification were performed according to standard protocols. Affinity purification was performed after antibody absorption on a His–peptide column. A monoclonal anti-endostatin antibody was also generated against a recombinant human polypeptide corresponding to the mouse endostatin domain (Oh et al, 1994b). A human cDNA library was used as a template for PCR amplification. After antigen purification, a monoclonal antibody was made using standard protocols. The specificity of all the above antibodies was demonstrated by a complete lack of staining with tissues from Col18a1−/− mice.
Immuno-EM and rotary shadowing EM
Tissue pieces of wild-type and Col18a1−/− mice and of human skin were used for en bloc and section surface labeling immuno-EM as described (Fukai et al, 2002). For rotary shadowing EM, the proteins were dialyzed against 0.1 M ammonium bicarbonate, and mixed with glycerol to a final concentration of 70% glycerol (v/v) and used as described (Sakai and Keene, 1994). Recombinant chicken collagen XVIII was immunolabeled with an anti-myc tag antibody (from Dr McKeon).
Immunopurification of collagen XVIII from chicken vitreous and production of recombinant full-length chicken collagen XVIII
Collagen XVIII was isolated from chick vitreous as described (Halfter et al, 1998). Recombinant collagen XVIII was isolated from the supernatant of EBNA cells stably transfected with a full-length cDNA of chick collagen XVIII and purified by ion exchange chromatography on Q-sepharose.
BM preparations
Samples of human skin were obtained with informed consent. Isolation and immunogold labeling of BM preparations derived from the dermo-epidermal junction zone was performed as described (Kassner et al, 2003). For western blotting, skin fragments were homogenized in cathepsin L buffer and treated with heparitinase and, subsequently, highly purified cathepsin L (Brinker et al, 2000).
Acknowledgments
We thank Dr J Ma, A Kassner, S Tufa, A Clermont, M Erickson, and L Benecchi for excellent assistance. We are grateful to Drs L Bruckner-Tuderman, J-H Chang, D Azar, M Arpin, HP Bächinger, Y Ninomiya, E Weber, and R Soininen for supplying material, and Dr KC Hayes for serum retinoid analysis. We thank Drs A Milam and C Curcio for a critical reading of the manuscript. This work was supported by NIH grants AR36819, AR36820, NS33981-02, EY12231, and EY04939, Boehringer Ingelheim Fonds, EntreMed, Inc. (Rockville, MD), Shriners Hospital for Children, DFG Sonderforschungsbereich 492-grant A2, Ruth and Milton Steinbach Fund, and a grant to Storm Eye Institute from Research to Prevent Blindness.
References
- Brinker A, Weber E, Stoll D, Voigt J, Muller A, Sewald N, Jung G, Wiesmuller KH, Bohley P (2000) Highly potent inhibitors of human cathepsin L identified by screening combinatorial pentapeptide amide collections. Eur J Biochem 267: 5085–5092 [DOI] [PubMed] [Google Scholar]
- Curcio CA, Millican CL (1999) Basal linear deposit and large drusen are specific for early age-related maculopathy. Arch Ophthalmol 117 (3): 329–339 [DOI] [PubMed] [Google Scholar]
- Dithmar S, Curcio CA, Le NA, Brown S, Grossniklaus HE (2000) Ultrastructural changes in Bruch's membrane of apolipoprotein E-deficient mice. Invest Ophthalmol Vis Sci 41 (8): 2035–2042 [PubMed] [Google Scholar]
- Dithmar S, Sharara NA, Curcio CA, Le NA, Zhang Y, Brown S, Grossniklaus HE (2001) Murine high-fat diet and laser photochemical model of basal deposits in Bruch membrane. Arch Ophthalmol 119 (11): 1643–1649 [DOI] [PubMed] [Google Scholar]
- Felbor U, Dreier L, Bryant RA, Ploegh HL, Olsen BR, Mothes W (2000) Secreted cathepsin L generates endostatin from collagen XVIII. EMBO J 19: 1187–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, Niemela M, Ilves M, Li E, Pihlajaniemi T, Olsen BR (2002) Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J 21 (7): 1535–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidry C, Medeiros NE, Curcio CA (2002) Phenotypic variation of retinal pigment epithelium in age-related macular degeneration. Invest Ophthalmol Vis Sci 43 (1): 267–273 [PubMed] [Google Scholar]
- Halfter W, Dong S, Schurer B, Cole GJ (1998) Collagen XVIII is a basement membrane heparan sulfate proteoglycan. J Biol Chem 273: 25404–25412 [DOI] [PubMed] [Google Scholar]
- Jahng WJ, David C, Nesnas N, Nakanishi K, Rando RR (2003) Cleavable affinity biotinylating agent reveals a retinoid binding role for RPE65. Biochemistry 42: 6159–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner A, Hansen U, Bruckner P (2003) Supramolecular association of collagen XVI to different fibrillar systems in human skin and cartilage. Matrix Biol 22: 131–143 [DOI] [PubMed] [Google Scholar]
- Katz ML, Chen D, Stientjes HJ, Stark WS (1993) Photoreceptor recovery in retinoid-deprived rats after vitamin A replenishment. Exp Eye Res 56: 671–682 [DOI] [PubMed] [Google Scholar]
- Li T, Sandberg MA, Pawlyk BS, Rosner B, Hayes KC, Dryja TP, Berson EL (1998) Effect of vitamin A supplementation on rhodopsin mutants threonine-17 → methionine and proline-347 → serine in transgenic mice and in cell cultures. Proc Natl Acad Sci USA 95: 11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Zhang J, Othersen KL, Moiseyev G, Ablonczy Z, Redmond TM, Chen Y, Crouch RK (2001) Expression, purification, and MALDI analysis of RPE65. Invest Ophthalmol Vis Sci 42: 1429–1435 [PubMed] [Google Scholar]
- Marneros AG, Olsen BR (2003) Age-dependent iris abnormalities in mice lacking collagen XVIII/endostatin with similarities to human pigment dispersion syndrome. Invest Ophthalmol Vis Sci 44: 2367–2372 [DOI] [PubMed] [Google Scholar]
- Muragaki Y, Timmons S, Griffith CM, Oh SP, Fadel B, Quertermous T, Olsen BR (1995) Mouse Col18a1 is expressed in a tissue-specific manner as three alternative variants and is localized in basement membrane zones. Proc Natl Acad Sci USA 92: 8763–8767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SP, Kamagata Y, Muragaki Y, Timmons S, Ooshima A, Olsen BR (1994a) Isolation and sequencing of cDNAs for proteins with multiple domains of Gly-X-Y repeats identify a novel family of collagenous proteins. Proc Natl Acad Sci USA 91: 4229–4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SP, Warman ML, Seldin MF, Cheng SD, Knoll JH, Timmons S, Olsen BR (1994b) Cloning of cDNA and genomic DNA encoding human type XVIII collagen and localization of the a1(XVIII) collagen gene to mouse chromosome 10 and human chromosome 21. Genomics 19: 494–499 [DOI] [PubMed] [Google Scholar]
- O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88: 277–285 [DOI] [PubMed] [Google Scholar]
- Passos-Bueno MR, Marie SK, Monteiro M, Neustein I, Whittle MR, Vainzof M, Zatz M (1994) Knobloch syndrome in a large Brazilian consanguineous family: confirmation of autosomal recessive inheritance. Am J Med Genet 52 (2): 170–173 [DOI] [PubMed] [Google Scholar]
- Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet 20 (4): 344–351 [DOI] [PubMed] [Google Scholar]
- Rossi M, Morita H, Sormunen R, Airenne S, Kreivi M, Wang L, Fukai N, Olsen BR, Tryggvason K, Soininen R (2003) Heparan sulfate chains of perlecan are indispensable in the lens capsule but not in the kidney. EMBO J 22 (2): 236–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai LY, Keene DR (1994) Fibrillin: monomers and microfibrils. In Methods in Enzymology, Ruoslahti E, Engvall E (eds) Vol. 245, pp 29–52. New York: Academic Press [DOI] [PubMed] [Google Scholar]
- Sasaki T, Fukai N, Mann K, Gohring W, Olsen BR, Timpl R (1998) Structure, function and tissue forms of the C-terminal globular domain of collagen XVIII containing the angiogenesis inhibitor endostatin. EMBO J 17: 4249–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarks SH (1976) Ageing and degeneration in the macular region: a clinicopathological study. Br J Ophthalmol 60: 324–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada H, Konomi H, Hirosawa K (1990) Characterization of the collagen in the hexagonal lattice of Descemet's membrane: its relation to type VIII collagen. J Cell Biol 110 (1): 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sertie AL, Sossi V, Camargo AA, Zatz M, Brahe C, Passos-Bueno MR (2000) Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum Mol Genet 9 (13): 2051–2058 [DOI] [PubMed] [Google Scholar]
- Van der Schaft TL, Mooy CM, Bruijn WC, Bosman FT, de Jong PTVM (1994) Immunohistochemical light and electron microscopy of basal laminar deposits. Graefe's Arch Clin Exp Ophthalmol 232: 40–46 [DOI] [PubMed] [Google Scholar]