

Abstract
We have identified and reconstituted a multicomponent redox-chaperone network that appears to be designed to protect proteins against stress-induced unfolding and to refold proteins when conditions return to normal. The central player is Hsp33, a redox-regulated molecular chaperone. Hsp33, which is activated by disulfide bond formation and subsequent dimerization, works as an efficient chaperone holdase that binds to unfolding protein intermediates and maintains them in a folding competent conformation. Reduction of Hsp33 is catalyzed by the glutaredoxin and thioredoxin systems in vivo, and leads to the formation of highly active, reduced Hsp33 dimers. Reduction of Hsp33 is necessary but not sufficient for substrate protein release. Substrate dissociation from Hsp33 is linked to the presence of the DnaK/DnaJ/GrpE foldase system, which alone, or in concert with the GroEL/GroES system, then supports the refolding of the substrate proteins. Upon substrate release, reduced Hsp33 dimers dissociate into inactive monomers. This regulated substrate transfer ultimately links substrate release and Hsp33 inactivation to the presence of available DnaK/DnaJ/GrpE, and, therefore, to the return of cells to non-stress conditions.
Keywords: chaperone, disulfide bond, Hsp33, oxidative stress, redox regulation
Introduction
The cytosolic heat-shock protein Hsp33 is a recently discovered molecular chaperone that is regulated by the redox conditions of the cell. Hsp33 efficiently protects proteins and cells against the otherwise lethal effects of reactive oxygen species, a function that is vital to cellular survival (Jakob et al, 1999).
Oxidizing conditions specifically activate the chaperone function of Hsp33. This post-translational regulation occurs via an unusual redox switch, which consists of four conserved cysteine residues in the C-terminal part of Hsp33. Under normal reducing conditions, these four cysteines coordinate one zinc ion (Jakob et al, 2000). In this state, Hsp33 is monomeric and the chaperone function is downregulated. Upon exposure of Hsp33 to oxidizing conditions such as H2O2, two intramolecular disulfide bonds are rapidly formed (Barbirz et al, 2000). This appears to cause major conformational rearrangements in the protein and triggers the dimerization of two oxidized Hsp33 molecules (Graumann et al, 2001). Oxidized Hsp33 dimers recognize and interact with folding intermediates of other proteins and very efficiently prevent their irreversible aggregation processes in vitro. In vivo, Hsp33 has been shown to play an important role in the defense of bacteria against oxidative stress (Jakob et al, 1999).
An important aspect in understanding how Hsp33 performs this protective task is to elucidate the fate of substrates that interact with Hsp33. Molecular chaperones have the ability to bind folding intermediates and to prevent their nonspecific aggregation (Ellis et al, 1989). The fate of the substrate proteins once bound to the chaperones depends largely on the mechanism of the individual chaperone system. Some chaperone systems such as the GroEL/ES or the DnaK/DnaJ/GrpE system have been shown to work as ‘foldases'; they actively support the folding of the proteins to their native state. Other chaperones such as DnaJ or small heat-shock proteins, on the other hand, work as ‘holdases'. They bind tightly to folding intermediates, efficiently preventing protein aggregation, but do not support the refolding of the protein (Beissinger and Buchner, 1998). To change the affinity for the substrates and allow their productive folding, foldase systems typically utilize ATP binding and hydrolysis to switch from a low- to a high-affinity binding state. Holdases, on the other hand, are usually ATP-independent chaperones and often work to deliver substrate proteins to either the proteolytic system or to chaperone foldases (Beissinger and Buchner, 1998). In contrast to the majority of ATP-dependent chaperones that continuously cycle between a high- and low-affinity binding state, Hsp33 appears to be in a high-affinity binding state only under certain stress conditions. This raised the important question of whether Hsp33 functions as a separate foldase system that is able to use changes in the redox conditions to actively support the refolding of proteins (Tsai et al, 2001), or if Hsp33 works as a chaperone holdase that binds tightly to its substrate proteins and requires foldase systems for the efficient refolding of the bound substrate proteins.
Our results show that Hsp33 is the central part of a redox-regulated chaperone machinery. Under stress conditions, activated Hsp33 works as a chaperone holdase that efficiently prevents protein aggregation. In vivo, Hsp33 is quickly reduced by the cellular glutaredoxin and thioredoxin systems. Effective dissociation of the reduced Hsp33–substrate protein complex requires the DnaK/DnaJ/GrpE system, which binds the substrate proteins and supports their refolding to the native state. This sophisticated network guarantees that unfolded substrate proteins are only released from Hsp33 when reducing conditions are restored and sufficient amounts of the heat-shock-sensing system DnaK/DnaJ/GrpE are present.
Results
Reducing conditions are not sufficient to cause substrate release from Hsp33
Oxidized Hsp33 dimers have a very high affinity for unfolded substrate proteins. Analysis of the stability of Hsp33–substrate protein complexes revealed that stoichiometric amounts of oxidized, active Hsp33 dimers prevent the aggregation of thermally unfolding proteins such as luciferase for more than 20 h at heat-shock temperatures in vitro (see supplementary data, Figure 1). Reduced Hsp33 monomers, on the other hand, show a very low affinity (Graumann et al, 2001). Therefore, it was conceivable that substrate dissociation was simply inducible by the reduction of Hsp33 dimers and their subsequent monomerization. To investigate whether the reduction of Hsp33 was indeed sufficient to induce Hsp33 to release its substrate proteins, we formed complexes between Hsp33 and thermally unfolded luciferase, added high concentrations of the reducing agent DTT (5 mM) and incubated the reaction at 43°C. At this temperature, all substrate molecules should aggregate rapidly upon release from Hsp33. As shown in Figure 1, highly sensitive light scattering measurements did not reveal any detectable increase in luciferase aggregation after addition of DTT (line b). Even extended incubation of Hsp33:luciferase complexes for 2 h in the presence of DTT failed to reveal any luciferase precipitation (data not shown), indicating that the addition of high concentrations of DTT to Hsp33:luciferase complexes is not sufficient to cause any substrate release.
Figure 1.

Reduction of Hsp33 is not sufficient to cause substrate release from Hsp33. Light scattering measurements of luciferase (140 nM) were performed either in the absence (a) or presence of 210 nM active Hsp33 dimers (b, c) at 43°C. At the time point indicated by the arrow, 5 mM DTT was added to reactions a and b, and the incubation was continued at 43°C. Inset: DTT induces the fast reduction of Hsp33's thiol groups. Dimeric oxidized Hsp33 in complex with thermally unfolded luciferase (left panel) or oxidized Hsp33 alone (right panel) was incubated with 5 mM DTT at 25°C. At the time points indicated, aliquots were withdrawn and free thiol groups were covalently modified with the 500 Da molecule AMS as described. Similar results were obtained at 43°C, albeit with significantly faster reduction rates.
We considered that this might be due to the slow and incomplete reduction of Hsp33's thiol groups when unfolded substrate proteins are bound. To investigate this possibility, we performed thiol-trapping experiments with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), a reagent that adds a 500 Da AMS moiety to each free thiol group. This leads to a differential migration of proteins in SDS–PAGE, depending on the number of free or disulfide-bonded cysteines. As shown in the inset to Figure 1, after the addition of 5 mM DTT, all four cysteines of Hsp33 were quickly reduced (t1/2<1 min). This process appeared to be only slightly slower in the presence of unfolded luciferase (compare the right and left panels). This demonstrated that active Hsp33 dimers are quickly reduced by DTT, but that this reduction is not sufficient for the inactivation of Hsp33's substrate binding activity. In vivo, this lack of inactivation by simple reduction would certainly be beneficial under conditions that are reducing yet nonpermissive for protein folding because dissociation of Hsp33–substrate complexes under these conditions would inevitably lead to the immediate aggregation of the substrate proteins.
Identification of a redox-regulated chaperone holdase–foldase interaction
To investigate the fate of Hsp33's substrate proteins in more detail, we reconstituted the thermal unfolding processes in vitro using thermally unfolding luciferase as a model substrate protein. Luciferase was incubated in the absence of Hsp33 or in the presence of a five-fold molar excess of functional Hsp33 dimers at 43°C. Within 10 min of incubation, luciferase was completely inactive whether or not Hsp33 was present (compare the solid diamonds in Figure 2A and the inset). We then shifted the temperature to 25°C and added high concentrations of DTT (5 mM). As shown in Figure 2A, only very minor amounts of luciferase were reactivated, and this was independent of the presence of Hsp33 (open circles), revealing that neither DTT addition nor lowering the incubation temperature was sufficient to induce any substantial refolding of luciferase.
Figure 2.
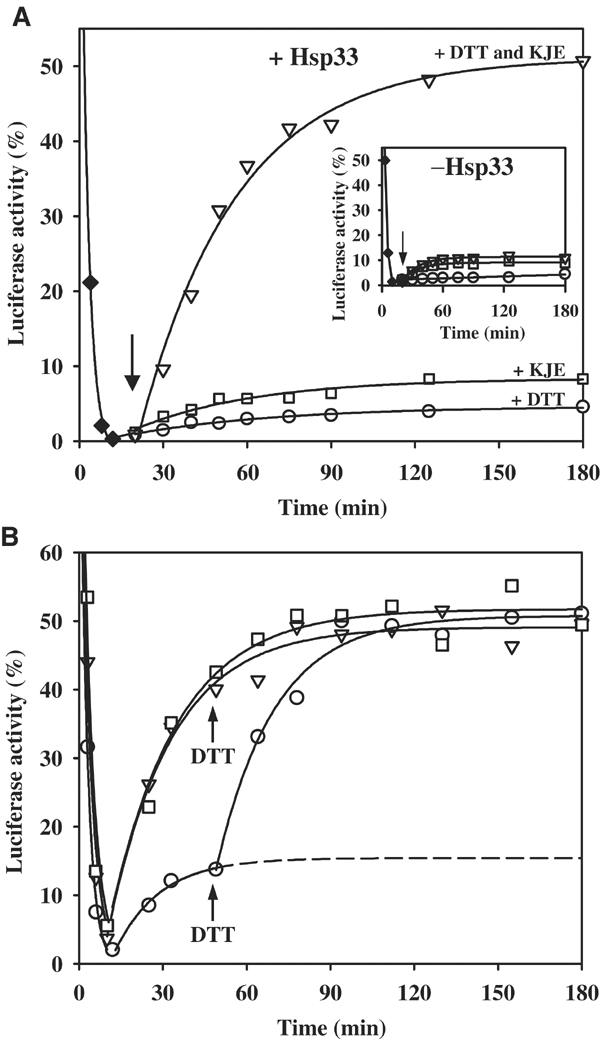
Substrate shuttling from the Hsp33 holdase to the DnaK/DnaJ/GrpE foldase. (A) Hsp33 transfers substrates to the DnaK/DnaJ/GrpE system for refolding. Luciferase (80 nM) was thermally unfolded in the presence of a five-fold molar excess of active Hsp33 dimers for 10 min at 43°C (⧫). Then, the temperature was shifted to 25°C. After 10 min of incubation at 25°C (as indicated by the arrow), either (○) 5 mM DTT, (□) DnaK/DnaJ/GrpE in a 5:2:5 molar ratio of the monomers or (▿) a combination of 5 mM DTT and DnaK/DnaJ/GrpE (5:2:5) was added to the reaction. The reactivation reaction of luciferase was monitored following luminescence. Inset: Substrate transfer and luciferase refolding requires the presence of active Hsp33 during inactivation. Luciferase (80 nM) was thermally unfolded for 10 min at 43°C without any chaperones (⧫). At 10 min after shift of the temperature to 25°C, either (○) 5 mM DTT, (□) DnaK/ DnaJ/GrpE in a 5:2:5 molar ratio of the monomers or a combination of (▿) 5 mM DTT and DnaK/DnaJ/GrpE (5:2:5) was added to the reaction and the reactivation reaction was monitored. (B) Reduction of Hsp33 and substrate transfer to the DnaK/DnaJ/GrpE system is not rate limiting for luciferase refolding. Luciferase (80 nM) was thermally unfolded (43°C) with DnaK/DnaJ/GrpE (molar ratio 5:2:5) either in the (□) presence of 5 mM DTT and a 10-fold molar excess of reduced, monomeric Hsp33, or in the (▿) absence of Hsp33 or (○) in the presence of a five-fold molar excess of active Hsp33 dimers. After 10 min at 43°C, the temperature was shifted to 25°C. At the time point indicated, 5 mM DTT was added to the reactions that did not contain DTT.
We decided to then investigate the possibility that substrate proteins bound to Hsp33 require an additional foldase system for refolding. One potential foldase system was the DnaK/DnaJ/GrpE machinery because this system is known to support the refolding of thermally unfolded proteins both in vitro and in vivo (Mogk et al, 1999). We, therefore, analyzed whether the Hsp33 holdase is able to cooperate with the DnaK/DnaJ/GrpE foldase in the refolding of luciferase. Luciferase was inactivated in the presence of active Hsp33 at 43°C and potential refolding conditions were established by shifting the incubation reaction to 25°C. Then, DnaK/DnaJ/GrpE was added in the absence or presence of 5 mM DTT. As shown in Figure 2A (inverted triangles), the simultaneous addition of DTT and the DnaK folding system to the Hsp33:luciferase complex resulted in the refolding of more than 50% of luciferase molecules. This dramatic result showed that luciferase, when thermally inactivated in the presence of Hsp33, retained its ability to refold but that the refolding could only be triggered by reducing Hsp33 in the presence of the DnaK/DnaJ/GrpE system. When active Hsp33 was not present during the inactivation reaction of luciferase, addition of the DnaK/DnaJ/GrpE system with or without DTT during the recovery period allowed very little reactivation (Figure 2A inset, inverted triangles and squares). This inability to recover luciferase activity is probably due to the formation of irreversible luciferase aggregates that form when Hsp33 is absent from the thermal unfolding reaction. Similarly, the absence of DTT during the recovery period resulted in equally low refolding yields, presumably because luciferase molecules remained bound to Hsp33 and, thus, remained inaccessible to the DnaK/DnaJ/GrpE foldase system (Figure 2A, open squares). That the complete DnaK/DnaJ/GrpE system is essential for high yields of refolded luciferase became evident when individual components of the DnaK/DnaJ/GrpE system were omitted from the recovery reaction. Here, the same low refolding yields were observed as when the complete DnaK/DnaJ/GrpE system or MgATP was omitted (data not shown). This indicated that the MgATP-dependent foldase activity of the complete DnaK/DnaJ/GrpE system is required to obtain the release and refolding of substrate proteins. These results clearly demonstrated that the presence of active Hsp33 during the inactivation of luciferase is essential for keeping luciferase in a folding competent state. Only upon reduction of Hsp33 can luciferase molecules be shuttled to the DnaK/DnaJ/GrpE system for refolding.
We knew from earlier studies that the DnaK/DnaJ/GrpE system, when present during the thermal inactivation reaction, is also able to maintain unfolding luciferase molecules in a folding competent state (Mogk et al, 1999). In the case of the DnaK/DnaJ/GrpE system, however, simply changing the temperature to permissive folding conditions is sufficient to cause the refolding of luciferase. Here, no significant differences in the refolding kinetics or yields of luciferase were observed when DTT was omitted from the refolding reaction and added later during the recovery period, indicating that neither the luciferase refolding process nor the action of the DnaK/DnaJ/GrpE system is very DTT sensitive (Figure 2B, compare squares to triangles). That both Hsp33 and the DnaK/DnaJ/GrpE system can interact with unfolding luciferase and that the DnaK/DnaJ/GrpE system can support the refolding of luciferase molecules that are released from Hsp33 suggested that the two systems might have overlapping substrate specificity. This should result in a competition for luciferase binding when both systems were simultaneously present during the inactivation reaction. To analyze this potential competition between the two chaperone systems, luciferase unfolding was performed in the presence of both active Hsp33 and the DnaK/DnaJ/GrpE system. Assuming that there is no substrate protein exchange between these systems under oxidizing conditions, the extent of binding to the foldase DnaK/DnaJ/GrpE system should be reflected by the amount of luciferase that can be refolded just by lowering the temperature. The extent of binding to the holdase Hsp33, on the other hand, should be reflected by the amount of luciferase that is specifically refolded when DTT is added to induce the substrate transfer to the DnaK/DnaJ/GrpE system for refolding. As shown in Figure 2B (open circles), a simple shift of the temperature to permissive folding conditions in the absence of DTT caused the refolding of only about 10% of luciferase molecules. This suggested that oxidized Hsp33 dimers are able to out-compete the DnaK/DnaJ/GrpE system for luciferase binding. Subsequent addition of DTT then allowed nearly 50% of the luciferase molecules to recover their activity, indicating that the reduction of Hsp33 allowed the shuttling of almost 40% of luciferase molecules to the DnaK/DnaJ/GrpE system for refolding. Importantly, the reactivation kinetics of luciferase were very similar and were not influenced by the prior association with Hsp33. This indicated that neither reduction of Hsp33 nor the substrate transfer from Hsp33 onto the DnaK/DnaJ/GrpE system was rate limiting in the refolding of luciferase. The overlapping substrate specificity and the efficient substrate transfer between the two systems are the main prerequisites for a well-functioning substrate relay between a holdase system that protects proteins under stress conditions and a foldase system that supports the refolding of those substrate proteins under nonstress conditions.
Identification of the physiological thiol reductants of Hsp33
DTT is a strong but nonphysiological reductant. In order to elucidate which physiological system(s) might be responsible for reducing Hsp33, components of the two major redox systems of Escherichia coli were tested. We investigated the influence of reduced glutathione (GSH) as well as the thioredoxin system on the reduction process of Hsp33. We thermally unfolded luciferase in the simultaneous presence of active Hsp33 and the DnaK/DnaJ/GrpE system and then shifted the temperature to folding-permissive temperatures. As shown previously (Figure 2B), no significant refolding of luciferase was observed unless 5 mM DTT was added (Figure 3A, compare open circles and inverted triangles). In the presence of 5 mM GSH, the refolding reaction of luciferase was significantly slower than in the presence of 5 mM DTT (Figure 3A, diamonds). This suggested that physiological concentrations of GSH were less effective in reducing Hsp33 than DTT and that reduction of Hsp33 by GSH became rate limiting in the refolding of luciferase. This was confirmed by thiol trapping experiments that showed that physiological concentrations of GSH were indeed very slow to reduce Hsp33's thiol groups (Figure 3A, inset).
Figure 3.
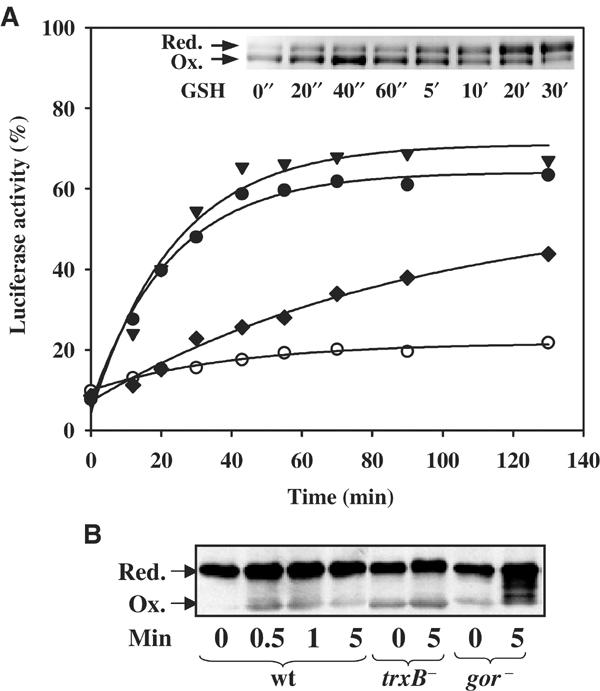
Identification of the physiological reductants of Hsp33. (A) Reduction of Hsp33 is catalyzed by thioredoxin in a TrxB- and NADPH-dependent manner in vitro. Luciferase (140 nM) was thermally unfolded in the presence of DnaK/DnaJ/GrpE (molar ratios 5:2:5) and a five-fold molar excess of active Hsp33 dimer for 8 min at 43°C. After 12 min of incubation at room temperature, the reaction was shifted to 30°C (t=0 min) and the incubation reaction was continued in the (○) absence of any additives or in the presence of either (▾) 5 mM DTT, (⧫) 5 mM GSH or (•) 2.8 μM TrxA, 70 nM TrxB, 50 μM NADPH. Only the recovery period at 30°C is shown. Inset: Physiological concentrations of reduced glutathione are not capable of quickly reducing oxidized Hsp33 dimers. Oxidized Hsp33 dimers (3 μM) were incubated in the presence of 5 mM GSH at 30°C. At the time points indicated, aliquots were removed and thiol trapping with AMS was performed as described. (B) Glutaredoxin and thioredoxin systems reduce Hsp33 in vivo. DHB4 wild-type, WM93 (ΔtrxB) or WP840 (Δgor) cells were grown at 30°C until an OD600 of 0.5 was reached. Then, the cells were shifted to 45°C. At 1 min after the shift, a sample was taken (t=0) and H2O2 was added (4 mM). Further samples were taken at the indicated time points. In vivo thiol trapping with AMS was performed and the samples were processed as described.
To test whether the thioredoxin system is capable of efficiently reducing Hsp33, 2.8 μM of reduced thioredoxin (TrxA), catalytic amounts of thioredoxin reductase (TrxB) and saturating amounts of the electron donor NADPH were added instead of DTT (Figure 3A, closed circles). Here, the refolding of luciferase was nearly as fast as in the presence of 5 mM DTT, indicating that micromolar concentrations of the thioredoxin system reduce Hsp33 as quickly as millimolar concentrations of DTT. This was also confirmed by thiol trapping experiments, which showed that the TrxA/TrxB/NADPH-mediated reduction of Hsp33's thiol groups proceeded with a rate that was very similar to the rate that was observed in the presence of 5 mM DTT (data not shown). This reduction of Hsp33 by the thioredoxin system allowed the efficient transfer of the substrate from Hsp33 to the DnaK/DnaJ/GrpE system and the refolding of luciferase.
In vivo thiol trapping studies of Hsp33 revealed that E. coli strains that have mutations in both thioredoxin and glutaredoxin systems (ΔtrxA Δgor) accumulate more than 60% of Hsp33 in the oxidized form (Jakob et al, 1999). To analyze which of the two systems is the physiological reductant of Hsp33, we exposed wild-type E. coli and various mutant strains to a combination of heat and oxidative stress treatment and analyzed the in vivo redox state of Hsp33 using the AMS thiol trapping technique. The mutant strains were either solely defective in the thioredoxin system based on a mutation in the thioredoxin reductase gene (ΔtrxB), or in the glutaredoxin system based on a mutation in the glutathione reductase gene (Δgor). As shown in Figure 3B, both redox-deficient strains accumulated more oxidized Hsp33 than stress-treated wild-type cells, but still significantly less than cells that are defective in both redox systems simultaneously. These findings indicated that thioredoxins and glutaredoxins are both able to quickly reduce Hsp33 during the stress treatment in vivo.
Identification of a third Hsp33 species: the reduced Hsp33 dimer
Hsp33 was found to be only very transiently oxidized in wild-type cells that are treated with a combination of oxidative stress and heat shock (Figure 3B). This finding agreed well with the observation that the cytosolic redox systems are quickly restored and often play autoregulatory roles in reducing key players such as the oxidative stress transcription factor OxyR in E. coli (Zheng et al, 1998). In contrast to OxyR, however, where reduction leads to the inactivation of OxyR and to the termination of the oxidative stress response, reduction of Hsp33's cysteines did not appear to cause the release of substrate proteins or a significant change in substrate affinity. This became especially evident when we compared the apparent stability of oxidized and reduced Hsp33–substrate protein complexes. For this experiment, we formed Hsp33–luciferase complexes during incubation at elevated temperatures and either reduced them with the TrxA/TrxB/NADPH system or left them untreated for extended periods of time at 30 or 43°C. Then, the substrate transfer was induced by the addition of either DnaK/DnaJ/GrpE to the reduced Hsp33–luciferase complexes or by the addition of both TrxA/TrxB/NADPH and DnaK/DnaJ/GrpE to the oxidized Hsp33–luciferase complexes. The luciferase reactivation reactions were then monitored at 30°C and found to be nearly indistinguishable in terms of both refolding kinetics and reactivation yields (data not shown), supporting the evidence that reduced Hsp33 dimer–substrate protein complexes do not dissociate unless the DnaK/DnaJ/GrpE system is present. These data suggested the existence of a reduced Hsp33 species, presumably Hsp33 dimers, which has sufficiently high affinity for substrate proteins to maintain them in a stable Hsp33–substrate complex but quickly release the substrate proteins when functional DnaK/DnaJ/GrpE is present.
To first investigate whether reduced Hsp33 dimers are stable intermediates in Hsp33's mechanistic life cycle, fluorescence anisotropy measurements were conducted. These measurements allow one to monitor changes in the oligomerization state of proteins due to differences in the rotational mobility of monomeric and dimeric species. Comparison of the anisotropy signal of oxidized, dimeric Hsp33 with reduced, monomeric Hsp33 revealed a small but significant and reproducible signal difference, which we used to monitor directly the dissociation of Hsp33 dimers into the monomers (Figure 4A). We analyzed the rate at which oxidized Hsp33 dimers are reduced and dissociate in the absence of substrate proteins by incubating oxidized Hsp33 dimers with 5 mM DTT. Defined time points after the start of the incubation reaction, fluorescence anisotropy measurements were conducted or samples were taken for thiol trapping experiments. As previously noted, reduction of Hsp33's thiol groups proceeded very rapidly with a half-time of about 0.6 min at 20°C (Figure 4A, closed circles). The dissociation of Hsp33 dimers into reduced Hsp33 monomers, however, was found to be significantly slower and proceeded with a half-time of more than 22 min at this temperature (Figure 4A, open circles). This was independent of the Hsp33 concentration used (data not shown), suggesting that the dissociation of reduced Hsp33 dimers is an irreversible and rate-determining process in the inactivation process of Hsp33. These results were also confirmed by ultracentrifugation assays (see supplementary material).
Figure 4.
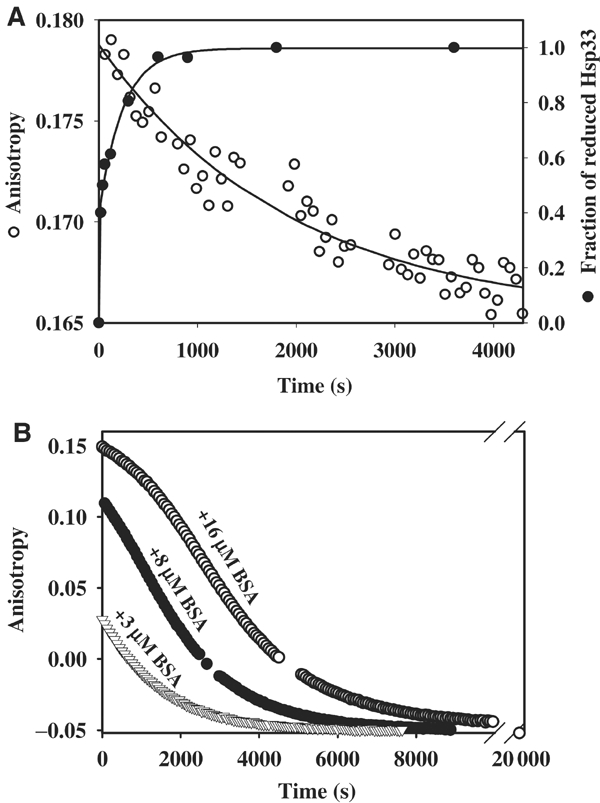
Identification of a stable Hsp33 intermediate: the reduced, active Hsp33 dimer. (A) Reduced Hsp33 dimers are kinetically stable. Oxidized Hsp33 dimers (3 μM) were incubated in 40 mM HEPES-KOH, 20 mM KCl, pH 8.0, at 20°C. A stable anisotropy signal at λex=280 nm and λem=350 nm was detected. To monitor (○) dissociation of reduced Hsp33 dimers, 5 mM DTT was added to the reaction. The anisotropy signal decreased and approached the lower signal of monomeric, reduced Hsp33. The curve was fitted according to a pseudo-first-order reaction and a rate constant of 5.2 × 10−4 s−1 was determined. A very similar rate constant was obtained when 15 μM Hsp33 dimers were used instead (4.7 × 10−4 s−1). To monitor the (•) reduction of Hsp33's thiol groups under these conditions, aliquots were taken and AMS trapped as described. The proteins were separated on 14% SDS–PAGE and the fraction of reduced Hsp33 protein was determined using a densitometer. A rate constant of 0.018 s−1 was determined for the reduction process of Hsp33. (B) Substrate proteins stabilize reduced Hsp33 dimers and delay inactivation of Hsp33. Oxidized Hsp33 dimers (3 μM) labeled with the fluorescent dye Oregon-green were incubated in 40 mM HEPES-KOH, 20 mM KCl, pH 8.0, at 20°C in the presence of (▿) 3 μM BSA, (•) 8 μM BSA or (○) 16 μM BSA. To reduce the dimeric Hsp33–BSA complexes, 5 mM DTT was added to the reaction, and the change in anisotropy signal was followed at λex=506 nm and λem=524 nm. Thiol trapping experiments showed that the rate of Hsp33 reduction was not significantly influenced by the presence of 16 μM BSA. Analysis of the apparent dissociation rate of fluorescently labeled Hsp33 dimers in the absence of BSA revealed a rate constant that was very similar to unlabeled Hsp33 (5.6 × 10−4 s−1).
Substrate binding prevents the inactivation of Hsp33
To test directly the activity of reduced Hsp33 dimers and their stability in the presence of substrate proteins, we labeled oxidized Hsp33 dimers with the fluorescent dye Oregon green. The fluorescently labeled oxidized Hsp33 dimers aggregated at elevated temperatures and could not be functionally evaluated with thermal unfolding substrate proteins. When tested on the aggregation of chemically unfolding luciferase at 20°C (Graumann et al, 2001), however, we found that the fluorescence label did not significantly alter the chaperone action of Hsp33 (data not shown). Importantly, the dissociation rate of reduced Hsp33 dimers was also not altered by the presence of the fluorescence label (app. t1/2=21 min).
Substrate binding studies of chaperones are usually conducted with small peptides or soluble proteins and not with aggregation-prone folding intermediates such as chemically denatured luciferase (Deuerling et al, 2003). This is mainly because of the molar excess of chaperones that is needed to suppress protein aggregation successfully, which leads to the presence of a substantial amount of substrate-free chaperones in solution. These substrate-free chaperones are then able to interact with additional low-affinity binding sites of soluble substrate proteins and/or protein microaggregates. This interferes with the determination of binding constants and substrate binding studies. We, therefore, decided to investigate soluble proteins that have been found to interact with chaperones such as DnaK as potential substrate proteins for Hsp33. We discovered bovine serum albumin (BSA) to be a soluble substrate protein of oxidized Hsp33 dimers (see supplementary data, Figure 2). Anisotropy measurement showed that fluorescently labeled oxidized Hsp33 dimers quickly associate with BSA, leading to an increased anisotropy signal of Hsp33. Titration experiments with BSA revealed that Hsp33 dimers form specific complexes with BSA in a 1:1 stoichiometry of one molecule of oxidized Hsp33 dimer to one molecule of BSA. We determined the KD of this binding to be 0.67 μM (see supplementary data, Figure 2). The affinity of Hsp33 for BSA was higher than the affinity of DnaK for BSA (KD=3 μM) (Crouy-Chanel et al, 1999), but presumably several magnitudes lower than Hsp33's affinity for unfolded substrate proteins.
To analyze directly the effects of DTT incubation on the substrate binding activity of Hsp33, we fully saturated fluorescently labeled Hsp33 with BSA and conducted thiol trapping and anisotropy measurements. Importantly, thiol trapping experiments showed that the reduction of Hsp33's thiol groups was not influenced by the presence of saturating amounts of BSA (data not shown). We, therefore, considered that if simple reduction of Hsp33 caused a loss in Hsp33's substrate binding affinity, then the decrease in anisotropy signal that reflects the dissociation of Hsp33–BSA complexes should parallel the rather rapid kinetics of Hsp33's thiol reduction. If, however, reduced Hsp33 dimers were an active Hsp33 species, and if the dissociation of reduced Hsp33 dimers was the rate-limiting step in substrate release, then the decrease in anisotropy signal should proceed with the same rate as the dissociation of substrate-free reduced Hsp33 dimers (Figure 4A). If bound substrate proteins led to the stabilization of reduced Hsp33 dimers, on the other hand, then substrate release should become the new rate-limiting step in the dissociation and inactivation of Hsp33. As shown in Figure 4B, increasing concentrations of BSA significantly stabilized the Hsp33–BSA complex, thus decelerating the monomerization of reduced Hsp33 dimers. An apparent half-time of more than 50 min was found when 16 μM of BSA was present as substrate protein. This clearly showed that reduced Hsp33 dimers are active as molecular chaperones, and revealed that substrate proteins stabilize reduced Hsp33 dimers. This efficiently delays Hsp33 dissociation and inactivation as long as substrate proteins are present.
Kinetics analysis of the dissociation reaction in the presence of high concentrations of BSA suggested a rather complex process, possibly involving additional Hsp33 intermediates. Addition of the complete DnaK/DnaJ/GrpE/MgATP system did not affect the rate of Hsp33–BSA dissociation, presumably because of the high concentrations of BSA that are necessary to saturate Hsp33 and the lower affinity of DnaK to BSA (Crouy-Chanel et al, 1999). Based on our refolding studies with thermally unfolded luciferase, however, this situation appears to be different when unfolded substrate proteins are bound to Hsp33. Here, DnaK/DnaJ/GrpE seems to be able to take over the substrate proteins as soon as Hsp33 dimers are reduced. This substrate transfer is a necessary prerequisite for Hsp33's dissociation into inactive monomers, and therefore links the inactivation of Hsp33 to the presence of a functional DnaK/DnaJ/GrpE system.
In vitro reconstitution of a redox-regulated chaperone network
DnaK/DnaJ/GrpE and GroEL/ES are the two main chaperone foldase systems in the prokaryotic cell. To investigate whether GroEL/GroES is equally capable of interacting with substrate proteins that are bound to Hsp33 and promote their refolding, we investigated the influence of GroEL/GroES alone and in concert with the DnaK/DnaJ/GrpE system on the refolding of substrate proteins that are complexed with Hsp33. We chose chemically unfolded substrate proteins, because the GroEL/ES system has not been found to be an effective chaperone system in protecting cytosolic proteins from thermal unfolding in vivo (Mogk et al, 1999). We used citrate synthase (CS) as the model substrate because it can use either the GroEL/GroES system (Buchner et al, 1991) or the DnaK/DnaJ/GrpE system (our studies) for refolding. We denatured CS in guanidinium hydrochloride, and initiated the refolding of CS by dilution into buffer either in the absence or presence of active Hsp33 dimers. In the absence of Hsp33, about 8% of CS refolded (Figure 5, inset). This refolding was almost completely suppressed when active Hsp33 dimers were present, indicating that stable complexes were formed between Hsp33 dimers and very early refolding intermediates of CS (Figure 5). After 20 min of reactivation in the absence (Figure 5, inset) or presence of Hsp33 (Figure 5), various combinations of reduced TrxA, DnaK/DnaJ/GrpE and GroEL/GroES were added and the reactivation yields of CS were determined 180 min after the start of the reactivation reaction. Significant reactivation of CS was only observed in reactivation reactions that minimally contained active Hsp33 at the beginning of the incubation reaction, and which were then supplemented with reduced TrxA to quickly reduce Hsp33 and the DnaK/DnaJ/GrpE system to take over and refold the substrate proteins. This agreed very well with the experiments performed with luciferase and showed that our redox-regulated chaperone network works equally well with different substrate proteins. The addition of the GroEL/GroES system supported the refolding of CS even more, suggesting a synergistic effect between the two foldase systems. With this Hsp33/TrxA/DnaK/DnaJ/GrpE/GroEL/GroES combination, up to 70% of all CS molecules were reactivated. In the absence of the DnaK/DnaJ/GrpE system, however, the GroEL/GroES system was unable to facilitate significant refolding, suggesting that GroEL/GroES does not have the ability to efficiently induce substrate release from Hsp33. This set of experiments suggested that Hsp33 is a central redox-regulated holdase. The DnaK/DnaJ/GrpE system is specifically suited to take over substrate proteins once Hsp33 is reduced, and appears to be able to relay a subset of unfolding intermediates to the GroEL/GroES system for their efficient refolding to the native state.
Figure 5.
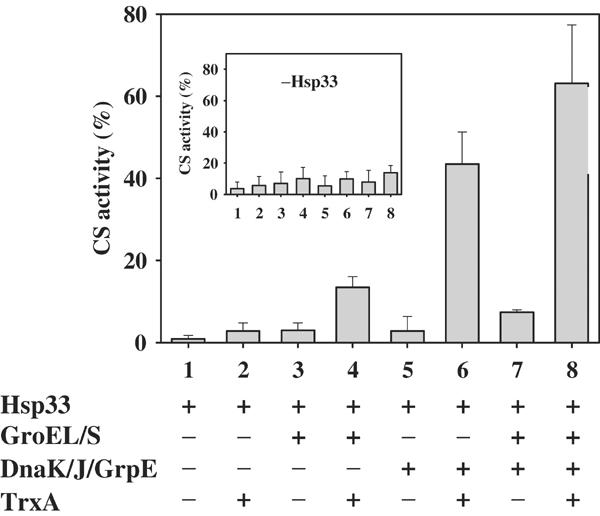
In vitro reconstitution of a redox-regulated multichaperone network. Chemically denatured CS was diluted to a final concentration of 75 nM into renaturation buffer (40 mM HEPES-KOH, pH 8.0, 10 mM KCl, 2 mM MgATP) containing a five-fold molar excess of active Hsp33 dimers. At 20 min after the start of the renaturation, various combinations of reduced TrxA (3 μM), DnaK:DnaK:GrpE (0.4 μM:0.16 μM:0.4 μM) and GroEL14:GroES7 (0.15 μM:0.5 μM) were added. The incubation reaction was continued for 160 min and the activity of CS was analyzed. The inset shows the identical experiment in the absence of dimeric Hsp33.
Discussion
In this study, we have identified a sophisticated redox-regulated chaperone network that consists of the chaperone holdase Hsp33 as the central player, the redox balancing thioredoxin and glutaredoxin systems, and the DnaK/DnaJ/GrpE foldase system. This complex network appears to protect cells against heat and oxidative stress effectively.
In analogy to ATP-dependent chaperones such as GroEL and DnaK, which cycle between low- and high-affinity binding states dependent on ATP or ADP binding, Hsp33 cycles between a low- and high-affinity binding state dependent on the redox conditions of the environment. This analogy led to the model that under oxidative stress, oxidized Hsp33 is in its high-affinity binding state and protects a number of proteins against irreversible aggregation. Upon return to nonstress conditions, the reduction-induced conformational change would then trigger the necessary substrate release and allow the substrate proteins to refold (Tsai et al, 2001). We have tested this model and discovered that substrate release and refolding are subject to much more extensive control (Figure 6). Substrate release from Hsp33 and the refolding of the substrate proteins is elegantly and very tightly coordinated with the availability of another cytosolic chaperone machinery, the DnaK/DnaJ/GrpE system. The Hsp33 and DnaK/DnaJ/GrpE systems cooperate together to maximize protein recovery after stress.
Figure 6.

A redox-regulated chaperone network. Under nonstress conditions, Hsp33 is monomeric and all four cysteines are reduced and coordinate one zinc ion. Hsp33 is in its low-affinity binding state. Upon exposure to heat and oxidative stress, two intramolecular disulfide bonds form in Hsp33, the zinc is released and Hsp33 dimerizes. Hsp33 is in its high-affinity binding state. It acts as a chaperone holdase and protects proteins against irreversible aggregation processes by tightly binding to unfolding protein intermediates. The thioredoxin system quickly reduces the dimeric Hsp33–substrate protein complexes. This does not cause any substrate protein release, but rather primes Hsp33 for fast inactivation once the DnaK/DnaJ/GrpE system is available. Upon return of cells to nonstress conditions, sufficient amounts of the DnaK/DnaJ/GrpE foldase system are available to release the substrate proteins from Hsp33. These substrate proteins are then refolded by the DnaK/DnaJ/GrpE foldase system alone or together with the GroEL/GroES system.
Under stress conditions, two intramolecular disulfide bonds form in Hsp33, which cause conformational changes and lead to the dimerization of two oxidized Hsp33 monomers (Graumann et al, 2001). In this fully active conformation, Hsp33 is able to form stable complexes with a number of different folding intermediates. In vivo, the cell's redox balancing thioredoxin and glutaredoxin systems quickly reduce the disulfide bonds in Hsp33, so that even under conditions of combined oxidative and heat stress conditions, only a very transient accumulation of oxidized Hsp33 can be detected (Figure 3B). This agreed well with the observation that the cytosolic redox systems often play autoregulatory roles by reducing key players of the oxidative stress response such as OxyR (Zheng et al, 1998). This fast reduction of Hsp33 in vivo, however, also provided evidence that the simple reduction of Hsp33 is unlikely to be sufficient to trigger substrate release. The dissociated substrate proteins would find themselves in a similarly unfavorable folding environment that was present when they first became bound to Hsp33.
OxyR protects itself against premature inactivation by using an unusually stable disulfide bond that is quite resistant against glutaredoxin-mediated reduction. This delays the termination of the oxidative stress response (Aslund et al, 1999). In the case of Hsp33, however, the premature inactivation is not prevented by unusually stable disulfide bonds, but by the formation of a kinetically stable reduced Hsp33 dimer. Fluorescence anisotropy measurements with oxidized Hsp33 dimers in the presence of DTT showed that reduced Hsp33 dimers remain stable and active for an extended period of time. The fact that the rate of dissociation was not dependent on the Hsp33 concentration showed that the dissociation of reduced Hsp33 dimers is an irreversible process. This confirmed that oxidized Hsp33 monomers, but not reduced Hsp33 monomers, are able to dimerize and become active (Graumann et al, 2001). This regulation is physiologically reasonable, because it guarantees that Hsp33's activation is limited to conditions of oxidative stress, but that the active conformation of Hsp33 is maintained well beyond the restoration of normal redox conditions.
So, while oxidative stress appears to be essential for the activation of Hsp33, reducing conditions are not sufficient for its inactivation. Efficient substrate release from Hsp33 is specifically linked to reducing conditions and the presence of the DnaK/DnaJ/GrpE foldase system. We assume that oxidized Hsp33 dimers undergo conformational rearrangements upon reduction, which appear to be essential for the DnaK/DnaJ/GrpE system to take over Hsp33's substrate proteins. As a result of substrate release, reduced Hsp33 dimers are then able to dissociate, thereby terminating the activation cycle of Hsp33.
The DnaK/DnaJ/GrpE system has been identified to be the major protective chaperone system under thermal stress conditions, and has recently been identified as a system that protects cells against oxidative stress (Mogk et al, 1999; Echave et al, 2002). It is available under nonstress conditions, where it downregulates the activity of the E. coli heat-shock σ factor. Upon accumulation of unfolded proteins, its free concentration decreases, which in turn is thought to increase the concentration of the heat-shock σ factor and to induce ultimately the heat-shock response (Straus et al, 1990). Upon return to nonstress conditions, sufficient amounts of DnaK/DnaJ/GrpE are available again to downregulate the 32σ factor and to terminate the heat-shock response. Now it appears that the DnaK/DnaJ/GrpE system also triggers the inactivation of the molecular chaperone Hsp33. This provides an elegant means of linking the substrate release and inactivation of an extremely efficient chaperone holdase to the availability of the DnaK/DnaJ/GrpE chaperone foldase machine, and therefore to the return to nonstress conditions.
The GroEL/GroES complex is the second major foldase system in the cell. We found that it is unable to trigger substrate release substantially from reduced Hsp33 dimers. The presence of both GroEL/GroES and DnaK/DnaJ/GrpE foldase systems, however, significantly increased the amount of CS that could be refolded upon release from Hsp33. Interestingly, a similar preference for the DnaK/DnaJ/GrpE system has been observed in the release of substrate proteins from the small heat-shock proteins (sHsp) (Veinger et al, 1998; Mogk et al, 2003). In the case of sHsps, however, it appears that it is the disaggregating power of the DnaK/DnaJ/GrpE system that makes this foldase system essential for the extraction of substrate proteins from small heat-shock proteins and for their subsequent refolding (Mogk et al, 2003). Once the DnaK/DnaJ/GrpE system has disaggregated the soluble sHsp–substrate protein microaggregates, however, the addition of GroEL/GroES increases the reactivation yields as well (Veinger et al, 1998; Mogk et al, 2003). Substrate transfer between small Hsps and DnaK/DnaJ/GrpE becomes, therefore, a rate-limiting step in the refolding of substrate proteins, depending on the size of aggregates that need to be disaggregated. In contrast, Hsp33 dimers appear to form distinct 1:1 complexes with substrate proteins (see supplementary material, Figures 1 and 2), and neither reduction of Hsp33 nor substrate transfer was found to be a rate-limiting step in the refolding of thermally unfolded luciferase molecules or chemically unfolded CS molecules. This suggests that either reduced Hsp33 dimers show a significantly lower affinity for substrate proteins than the DnaK/DnaJ/GrpE system, or that the DnaK/DnaJ/GrpE system might be specifically involved in the fast dissociation of reduced Hsp33 dimer–substrate protein complexes. In either case, once reducing conditions are restored and DnaK/DnaJ/GrpE is available, the substrate proteins are transferred onto the DnaK/DnaJ/GrpE system and quickly refolded.
Hsp33 efficiently protects cells against a combination of heat and oxidative stress (Jakob et al, 1999). This suggested that Hsp33 plays its most important role during severe stress conditions. Our discovery that substrate release and, therefore, Hsp33's inactivation is regulated by the DnaK/DnaJ/GrpE system now suggests that Hsp33 is able to sequester unfolded and oxidatively damaged proteins during severe stress conditions while the other chaperone systems such as the DnaK/DnaJ/GrpE system might be saturated with protein folding intermediates. When the stress subsides and the DnaK/DnaJ/GrpE system becomes reavailable, Hsp33's task is fulfilled. The substrate proteins are released from Hsp33 for refolding and Hsp33 returns to its inactive conformation, but remains primed to jump into action if oxidative stress returns.
Materials and methods
Proteins
Firefly luciferase, pig heart mitochondrial CS and BSA were obtained from Hoffmann LaRoche. Hsp33 was overexpressed and purified as described (Jakob et al, 1999). Reduced and oxidized Hsp33 was prepared as described (Graumann et al, 2001). To fluorescence label Hsp33, 17.5 μM of oxidized, dimeric Hsp33 was incubated with 200 μM Oregon Green® 514 carboxylic acid, succinimidyl ester (Molecular Probes) in 40 mM HEPES-KOH, pH 7.2, under continuous stirring for 60 min at room temperature. Excess dye was removed by extensive dialysis against the same buffer. The degree of labeling was found to be 1.8 molecules of label per oxidized Hsp33 monomer. DnaK, DnaJ, GrpE, GroEL and GroES were purified as described (Buchberger et al, 1994). Dr Charles Williams (University of Michigan) kindly provided purified TrxA and TrxB. TrxA was reduced with 10 mM DTT for 90 min at 37°C. DTT was removed using PD10 gel filtration columns (Amersham Pharmacia).
Thermal aggregation of luciferase
Luciferase (140 nM) was incubated in 40 mM HEPES-KOH, pH 7.5, at 43°C either in the absence or presence of dimeric Hsp33 (Jakob et al, 1999). Light scattering was determined using a fluorescence spectrophotometer (Hitachi F4500) with λex/em set to 350 nm and slit widths set to 2.5 (ex) and 5.0 (em). To investigate the effects of reducing conditions on the complex formation, 5 mM DTT was added.
Thiol trapping of Hsp33
In vitro: Complex formation between dimeric Hsp33 (2.5 μM) and luciferase (1 μM) was induced by incubating the two proteins in 40 mM HEPES-KOH, pH 8.0, 20 mM KCl for 20 min at 43°C. To reduce dimeric Hsp33 (2.5–3 μM), 5 mM DTT or 5 mM GSH was added. Aliquots (100 μl) were taken and the reaction was stopped by the addition of TCA to a final concentration of 10% w/v and incubation on ice. Samples were spun down (13 000 rpm, 4°C, 30 min) and the pellets were resuspended in 20 μl 15 mM AMS (Molecular Probes) in a buffer containing 6 M urea, 200 mM Tris–HCl, pH 9.0, 10 mM EDTA, 0.5 % w/v SDS. Samples were incubated at 37°C for 60 min in the dark and analyzed on nonreducing SDS–PAGE.
In vivo: DHB4 wt strains, WM93 (ΔtrxB) and WP840 (Δgor) strains were grown to an OD600 of 0.5 at 30°C in LB (Prinz et al, 1997; Jakob et al, 1999). To induce oxidative stress under heat-shock conditions, cells were shifted to 45°C and 4 mM H202 was added after 1 min. At various time points, 0.8 ml of cell suspension was added to 0.2 ml ice-cold 50% TCA (w/v) and incubated on ice. The AMS trapping was then performed as described above.
Chaperone-mediated reactivation of thermally unfolded luciferase and chemically denatured CS
Luciferase (80 or 140 nM) was incubated either alone or in the presence of a five-fold molar excess of Hsp33 dimers in 40 mM HEPES-KOH, pH 8.0, 20 mM KCl, 2 mM MgATP, 0.03 mg/ml BSA for 8 or 10 min at 43°C. The reaction was cooled down to room temperature for 10–15 min and further incubated at 25 or 30°C, as indicated in the text. Aliquots were taken and assayed for luciferase activity (Herbst et al, 1997). DTT (5 mM), GSH (5 mM) or TrxB (70 nM), reduced TrxA (2.8 μM), NADPH (50 μM) were used to reduce Hsp33. Molar ratios (5:2:5:1) of DnaK/DnaJ/GrpE to luciferase were used to refold luciferase. CS (12 μM) was chemically denatured in 4.5 M guanidinium–HCl, 50 mM Tris–HCl, pH 8.0, for at least 2 h. Reactivation was initiated by a 1:160 dilution into 40 mM HEPES-KOH, pH 8.0, 10 mM KCl, 2 mM MgATP with or without a five-fold molar excess of Hsp33 dimers. After 20 min of incubation at 25°C, a combination of reduced TrxA (final conc. 3 μM), DnaK (0.4 μM)/DnaJ (0.16 μM)/GrpE (0.4 μM) or GroEL14 (0.15 μM)/GroES7 (0.5 μM) was added to the reactivation reactions. After 180 min of total incubation at 25°C, CS activity was assayed as described (Buchner et al, 1991).
Fluorescence anisotropy measurements
Anisotropy measurements were conducted using a Fluoromax II fluorescence spectrophotometer, equipped with computer-controlled polarizers. In total, 0.1 mg/ml or 0.5 mg/ml oxidized Hsp33 dimers were incubated in the presence of 5 mM DTT. The change in anisotropy signal upon dissociation into the monomers was analyzed at λex=280 nm and λem=350 nm. When the influence of DTT on the Hsp33–BSA complex stability was tested, 0.1 mg/ml Oregon-green labeled Hsp33 dimers were incubated in the absence or presence of increasing concentrations of BSA and changes in anisotropy were monitored at λex=506 nm and λem=524 nm. The G-factor was always set to 1.00.
Supplementary Material
Supplementary Data
Acknowledgments
We are extremely grateful to Drs Bernd Bukau and Matthias Mayer for providing us with the DnaK/DnaJ/GrpE expression system and for the purification protocols. We thank Drs James Bardwell and Jeannette Winter for a critical reading of the manuscript. The National Institute of Health Grant GM065318 and a Burroughs Wellcome Fund Career Award to UJ, and a PhD scholarship of the Boehringer Ingelheim Fonos to JHH supported this work.
References
- Aslund F, Zheng M, Beckwith J, Storz G (1999) Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol-disulfide status. Proc Natl Acad Sci USA 96: 6161–6165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbirz S, Jakob U, Glocker MO (2000) Mass spectrometry unravels disulfide bond formation as the mechanism that activates a molecular chaperone. J Biol Chem 275: 18759–18766 [DOI] [PubMed] [Google Scholar]
- Beissinger M, Buchner J (1998) How chaperones fold proteins. Biol Chem 379: 245–259 [PubMed] [Google Scholar]
- Buchberger A, Schroder H, Buttner M, Valencia A, Bukau B (1994) A conserved loop in the ATPase domain of the DnaK chaperone is essential for stable binding of GrpE. Nat Struct Biol 1: 95–101 [DOI] [PubMed] [Google Scholar]
- Buchner J, Schmidt M, Fuchs M, Jaenicke R, Rudolph R, Schmid FX, Kiefhaber T (1991) GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry 30: 1586–1591 [DOI] [PubMed] [Google Scholar]
- Crouy-Chanel A, Kohiyama M, Richarme G (1999) Interaction of DnaK with native proteins and membrane proteins correlates with their accessible hydrophobicity. Gene 230: 163–170 [DOI] [PubMed] [Google Scholar]
- Deuerling E, Patzelt H, Vorderwulbecke S, Rauch T, Kramer G, Schaffitzel E, Mogk A, Schulze-Specking A, Langen H, Bukau B (2003) Trigger factor and DnaK possess overlapping substrate pools and binding specificities. Mol Microbiol 47: 1317–1328 [DOI] [PubMed] [Google Scholar]
- Echave P, Esparza-Ceron MA, Cabiscol E, Tamarit J, Ros J, Membrillo-Hernandez J, Lin EC (2002) DnaK dependence of mutant ethanol oxidoreductases evolved for aerobic function and protective role of the chaperone against protein oxidative damage in Escherichia coli. Proc Natl Acad Sci USA 99: 4626–4631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ, van der Vies SM, Hemmingsen SM (1989) The molecular chaperone concept. Biochem Soc Symp 55: 145–153 [PubMed] [Google Scholar]
- Graumann J, Lilie H, Tang X, Tucker KA, Hoffmann JH, Vijayalakshmi J, Saper M, Bardwell JC, Jakob U (2001) Activation of the redox-regulated molecular chaperone Hsp33—a two-step mechanism. Structure (Camb) 9: 377–387 [DOI] [PubMed] [Google Scholar]
- Herbst R, Schafer U, Seckler R (1997) Equilibrium intermediates in the reversible unfolding of firefly (Photinus pyralis) luciferase. J Biol Chem 272: 7099–7105 [DOI] [PubMed] [Google Scholar]
- Jakob U, Eser M, Bardwell JC (2000) Redox switch of hsp33 has a novel zinc-binding motif. J Biol Chem 275: 38302–38310 [DOI] [PubMed] [Google Scholar]
- Jakob U, Muse W, Eser M, Bardwell JC (1999) Chaperone activity with a redox switch. Cell 96: 341–352 [DOI] [PubMed] [Google Scholar]
- Mogk A, Schlieker C, Friedrich KL, Schonfeld HJ, Vierling E, Bukau B (2003) Refolding of substrates bound to small Hsps relies on a disaggregation reaction mediated most efficiently by ClpB/DnaK. J Biol Chem 278: 31033–31042 [DOI] [PubMed] [Google Scholar]
- Mogk A, Tomoyasu T, Goloubinoff P, Rudiger S, Roder D, Langen H, Bukau B (1999) Identification of thermolabile Escherichia coli proteins: prevention and reversion of aggregation by DnaK and ClpB. Embo J 18: 6934–6949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA, Aslund F, Holmgren A, Beckwith J (1997) The role of the thioredoxin and glutaredoxin pathways in reducing protein disulfide bonds in the Escherichia coli cytoplasm. J Biol Chem 272: 15661–15667 [DOI] [PubMed] [Google Scholar]
- Straus D, Walter W, Gross CA (1990) DnaK, DnaJ, and GrpE heat shock proteins negatively regulate heat shock gene expression by controlling the synthesis and stability of sigma 32. Genes Dev 4: 2202–2209 [DOI] [PubMed] [Google Scholar]
- Tsai B, Rodighiero C, Lencer WI, Rapoport TA (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104: 937–948 [DOI] [PubMed] [Google Scholar]
- Veinger L, Diamant S, Buchner J, Goloubinoff P (1998) The small heat-shock protein IbpB from Escherichia coli stabilizes stress-denatured proteins for subsequent refolding by a multichaperone network. J Biol Chem 273: 11032–11037 [DOI] [PubMed] [Google Scholar]
- Zheng M, Aslund F, Storz G (1998) Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279: 1718–1721 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data