

Abstract
Collapsin response mediator proteins (CRMPs) are cytosolic phosphoproteins involved in neuronal differentiation and axonal guidance. CRMP2 was previously shown to mediate the repulsive effect of Sema3A on axons and to participate in axonal specification. The X-ray crystal structure of murine CRMP1 was determined at 2.1 Å resolution and demonstrates that CRMP1 is a bilobed ‘lung-shaped' protein forming a tetrameric assembly. Structure-based mutagenesis of surface-exposed residues was employed to map functional domains. As a rapid assay for CRMP, we exploited a reconstituted Sema3A signaling system in COS-7 cells expressing the receptor components Neuropilin1 and PlexinA1 (NP1/PlexA1). In these cells, CRMP and PlexA1 form a physical complex that is reduced in amount by NP1 but enhanced by Sema3A/NP1. Furthermore, CRMP accelerates Sema3A-induced cell contraction. Alanine substitutions in one domain of CRMP1 produce a constitutively active protein that causes Sema3A-independent COS-7 contraction. This mutant CRMP mimics the DRG neurite outgrowth-inhibiting effects of Sema3A and reduces Sema3A-induced axonal repulsion. These data provide a structural view of CRMP function in Plex-dependent Sema3A signaling.
Keywords: CRMP, neuropilin, plexin, semaphorin
Introduction
The semaphorins constitute a major family of axon guidance cues (Raper, 2000). Sema3A repulses axons through the co-receptor proteins neuropilin-1 (NP1) (He and Tessier-Lavigne, 1997; Kolodkin et al, 1997) and Plexin A (PlexA1-3) (Takahashi et al, 1999; Tamagnone et al, 1999; Cheng et al, 2001). Of the co-receptors, NP1 represents the ligand-binding partner, while PlexA1 includes a cytoplasmic segment that transduces the Sema3A signal into cytoskeletal reorganization. Alteration of growth cone cytoskeletal dynamics appears to be mediated by the small G protein Rac1 (Jin and Strittmatter, 1997), PAK kinase (Aizawa et al, 2001), LIM kinase and its substrate, cofilin, which acts to depolymerize actin.
In addition, members of the collapsin response mediator protein (CRMP) family are also critical to Sema3A function. Antibodies to a specific region of CRMPs abolish Sema3A-mediated growth cone collapse (Goshima et al, 1995). The vertebrate branch of the family currently includes five CRMP isoforms (Nakamura et al, 2000), while the Drosophila genome appears to encode for a single CRMP. Sequence homology with unc-33 (38% identity, 61% similarity for chick CRMP2) may reflect aspects of shared function. Unc-33 mutants demonstrate abnormal axon growth, with premature termination, aberrant branching, and unusual microtubule morphology (Li et al, 1992).
CRMP family members share a remarkable sequence similarity (approximately 60% identity) with dihydropyrimidinase (DHPase), an enzyme involved in pyrimidine catabolism, although no DHPase activity has been detected for CRMP (Wang and Strittmatter, 1997). CRMP1, CRMP2, CRMP3, and CRMP4 family members are approximately 75% identical in protein sequence. CRMP5 (also known as CRAM or CRMP3 associated molecule) shares a 50% identity with other CRMPs and DHPase (Fukada et al, 2000; Inatome et al, 2000). The CRMPs share 25–40% identity with the bacterial hydantoinase proteins, several of which have known structures (Abendroth et al, 2002b; Cheon et al, 2002; Xu et al, 2003).
Like unc-33, the CRMPs appear to play a complex role in axon growth as well as microtubule dynamics and axon induction. CRMPs localize to the lamellipodia and filopodia of axonal growth cones, suggesting a role in axon guidance (Minturn et al, 1995). Moreover, CRMP2 is upregulated after axotomy (Minturn et al, 1995), and appears to increase the formation of axon-type processes from hippocampal neurons (Inagaki et al, 2001). CRMP2 has been reported to bind tubulin dimers directly and modulate microtubule assembly (Fukata et al, 2002). CRMPs have also been implicated in the pathogenesis of a paraneoplastic neurologic syndrome (Honnorat et al, 1999). Interaction studies have implicated phospholipase D2 (PLD2) (Lee et al, 2002), the cytosolic tyrosine kinase Fes (Mitsui et al, 2002), and intersectin in CRMP function (Quinn et al, 2003).
To better understand the molecular bases of CRMP function, we have determined the crystal structure of CRMP1. CRMP1 assumes a bilobed ‘lung-shaped' structure with an N-terminal β-sheet domain and a C-terminal α/β barrel domain. Four CRMP1 protomers comprise a large oligomeric structure supported by interactions among surface α-helices. Structure-based mutagenesis has revealed a constitutively active mutant CRMP1, which implicates a phylogenetically conserved loop region in CRMP1 effects on cell morphology.
Results
CRMP1 structure determination
To characterize the bases of CRMP action, we determined the X-ray structure of murine CRMP1. Limited proteolysis of CRMP1 (residues 8–572) demonstrated that the C-terminal segment (approximately 75 amino acids) is proteolytically susceptible (data not shown). Recombinant CRMP1 (residues 8–525) yielded high-quality crystals with two protomers/asymmetric unit. The structure was determined at 2.1 Å resolution. Phases were determined by comparison of diffraction data of three crystals: a sulfur methionine protein crystal used as native, a selenomethionine protein crystal with data collected at three wavelengths, and a second selenomethionine protein crystal. The current refinement model has an R-factor of 20.4% and a free R-value of 23.3% (Brunger, 1992) with excellent stereochemistry (Table I).
Table 1.
Statistics of the crystallographic analysis
| Structure determination (22 Se sites) | ||||||
|---|---|---|---|---|---|---|
| Data set | Resolution (Å) | Reflections, measured/unique | Completeness (%), overall/outer shell | Rsqym (%), overall/outer shell | Phasing power, isoacentric/centric | Rcullis, ano/iso |
| Crystal 1—native | ||||||
| λ=0.9840 Å | 24.0–2.10 | 460410/84627 | 99.6/99.9 | 9.8/31.0 | — | — |
| Crystal 2—Se-Met | ||||||
| λ1=0.9792 Å | 24.0–2.35 | 746069/115452 | 99.8/99.9 | 7.3/29.8 | 3.09/2.05 | 0.89/0.53 |
| λ2=0.9794 Å | 24.0–2.35 | 670003/108347 | 100.0/99.9 | 8.4/28.8 | 2.83/1.31 | 0.86/0.52 |
| λ3=0.9641 Å | 24.0–2.35 | 644465/104178 | 100.0/100.0 | 9.1/29.9 | 2.70/1.29 | 0.85/0.54 |
| Crystal 3—Se-Met | ||||||
| λ=0.9840 Å | 24.0–2.12 | 753778/78239 | 93.4/76.8 | 6.3/31.3 | 2.43/1.23 | —/0.58 |
| Overall figure of merit=0.68 | ||||||
| Refinement—crystal 3 | ||||||
| |
Resolution (Å) |
Completeness (%) |
R-factor |
Free R-factor |
||
| Data with ∣F∣>2σ∣F∣) | 24.0–2.12 | 90.4 | 0.204 | 0.233 | ||
| R.m.s.d. |
Bond lengths, 0.0127 Å |
Bond angles, 1.53° |
|
|
|
|
| Rsym=Σ∣I−〈I〉∣/ΣI, where I=observed intensity, 〈I〉=average intensity obtained from multiple observations of symmetry-related reflections; phasing power=r.m.s.d.; (∣FH∣/E), ∣FH∣=heavy atom structure factor amplitude; E=residual lack of closure. r.m.s.d. bond lengths and r.m.s.d. bond angles are the respective root mean square deviations from ideal values. Free R-factor was calculated with 5% of data omitted from the structure refinement. | ||||||
Structural overview
The structure of CRMP1 is illustrated in Figure 1A. Electron density was visible for residues 15–490, consistent with the proteolysis results described above. The polypeptide chain consists of two domains in the form of a ‘molecular lung' of dimensions 65 × 75 × 50 Å. Structure-based sequence alignments of vertebrate and invertebrate CRMP homologues show significant pairwise identities (33–76%) and demonstrate that the hydrophobic core is conserved, whereas insertions map to surface loops (Figure 2). We conclude, therefore, that the phylogenetically conserved regions of all known members of the CRMP share the three-dimensional structure illustrated in Figure 1 (Sander and Schneider, 1991).
Figure 1.
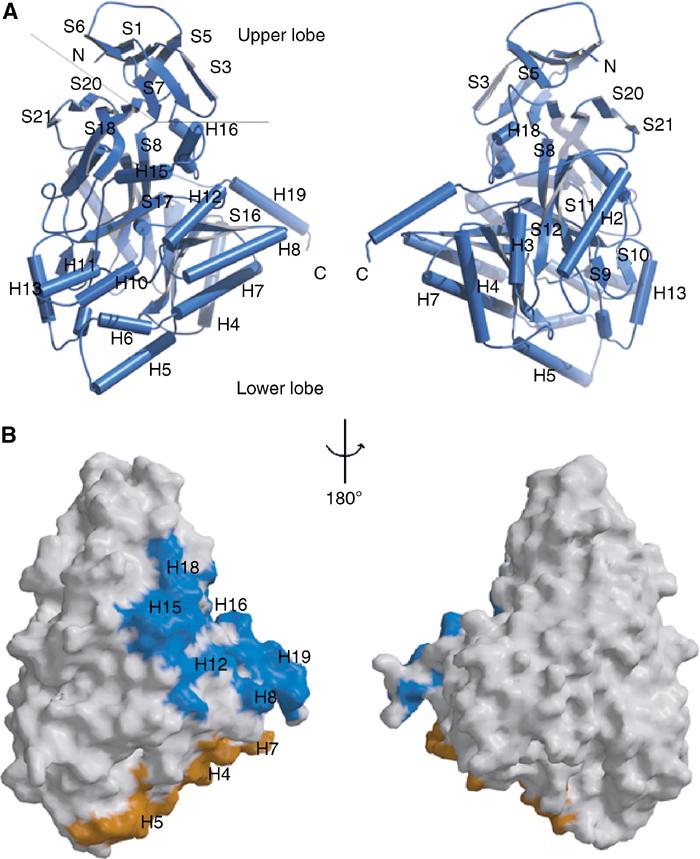
Three-dimensional structure of CRMP1 monomer. (A) BOBSCRIPT (Esnouf, 1999) drawing of the front and back views of CRMP1. α-Helices are labeled H1–H19 and β-strands are labeled S1–S21. The positions of the N- and C-termini are indicated. A thin line separates the N-terminal upper lobe from the C-terminal lower lobe. (B) GRASP (Nicholls et al, 1991) representations of the solvent-accessible surface of CRMP1 calculated using a water probe radius=1.4 Å. The surface overlying residues contributing to tetramer interfaces 1 and 2 are colored blue and gold, respectively. The orientations are identical to those in (A). Secondary structure elements contributing to the tetramer interfaces are labeled.
Figure 2.

Sequence alignment of CRMPs and related proteins. Secondary structural elements were obtained from the X-ray structure. Gray circles denote disordered residues or residues not included in the construct (1–8, 526–572). Sequence homology is encoded by a gray → purple color gradient (40–100% identity). Functional classifications: 1, tetramer interface 1; 2, tetramer interface 2. Sequence numbering for mouse CRMP1 is provided in green above the secondary structure elements.
Figure 2 shows a comparison of the amino-acid sequence of five mammalian CRMP family members, Drosophila CRMP, unc-33, and the related human DHPase protein with the CRMP1 secondary structure highlighted above. The N-terminal domain, which forms the ‘upper lobe' or ‘apex' of the lung, is a seven-stranded antiparallel β-sheet (residues 15–69; dimensions 32 × 26 × 22 Å), formed by β-strands S1-S2-S3-S4-S5-S6-S7. The C-terminal domain, which forms the ‘lower lobe' or ‘base', is an α/β barrel (residues 70–490; dimensions 60 × 65 × 49 Å) with secondary structure elements arranged in the order S8-S9-S10-H1-S11-H2-S12-H3-S13-H4-S14-H5-H6-H7-S15-H8-S16-H9-H10-H11-H12-S17-H13-H14-H15-H16-H17-H18-S18-S19-S20-S21-H19. Underlined secondary structure elements comprise an eight-fold α/β ‘TIM barrel' (pairwise α-carbon root mean square deviation (r.m.s.d.) of 3.5 Å for 162 residues of CRMP1 versus Leishmania mexicana triose phosphate isomerase), which forms the core of CRMP1. The remaining secondary structure elements surround the core, with some elements contributing to the oligomerization interface of CRMP1 (see below). An extended 15-residue linker (length 39 Å) connects β-strand S21 to α-helix H19. The lower lobe interacts with the upper lobe via hydrogen bonding between β-strands S18 and S7, and packing of α-helices H16, H17, H18, and the H18–S18 loop against β-strands S1–S7.
CRMP1 forms a tetramer
The two CRMP1 protomers comprising the asymmetric unit are related by a noncrystallographic two-fold axis of symmetry. The resulting interface (interface 1, shown between the blue and yellow or between the green and red monomers in Figure 3) buries a total of 2275 Å2 of solvent-accessible surface area. In addition, each CRMP1 monomer packs against a crystallographic symmetry-related molecule (interface 2, shown between blue and red, or green and yellow monomers) to bury an additional 2416 Å2 of solvent-accessible surface area, giving a tetrameric structure for CRMP1. There are no contacts between diametrically opposed monomers. The observation of a CRMP1 homotetramer, burying a total of 9382 Å2 of solvent-accessible surface area, is consistent with prior yeast two-hybrid data and gel-filtration studies, which show that CRMP family members oligomerize and exist as tetramers when purified from brain (Wang and Strittmatter, 1997). Rat DHPase also occurs as a tetramer (Kikugawa et al, 1994) as does D-hydantoinase, a homologous bacterial protein (Figure 3B, see below).
Figure 3.
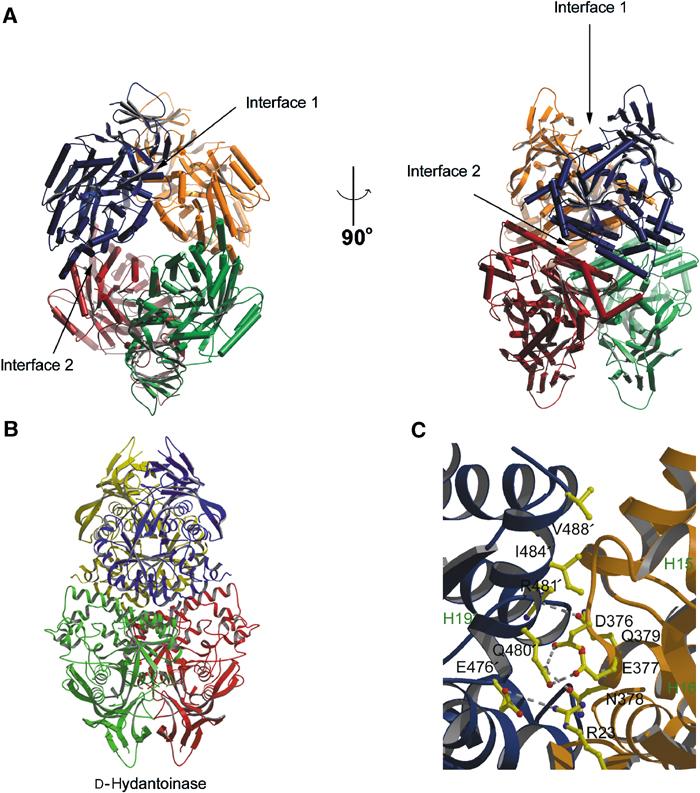
CRMP forms a D-hydantoinase-like tetramer. (A) BOBSCRIPT (Esnouf, 1999) drawing of CRMP1 tetramer. Individual protomers are colored blue, yellow, green, and red. The two intermolecular interfaces contributing to the tetramer are denoted by arrows. (B) MOLSCRIPT (Kraulis, 1991) drawing of D-hydantoinase tetramer. Individual protomers are colored blue, yellow, green, and red. (C) Detailed MOLSCRIPT (Kraulis, 1991) representation of a portion of CRMP-CRMP interface 1. Residues contributing to the interface are depicted as ball-and-stick figures, and hydrogen bonds are shown as dashed gray lines. Secondary structure elements are labeled in green and numbered according to Figure 1. Residues and secondary structure elements of the blue protomer are denoted with an apostrophe. Backbone atoms, which play a significant role in the H15–H16 loop interaction with helix H19′, have been omitted for clarity.
Interface 1 of the CRMP1 tetramer is stabilized by salt bridges, hydrogen bonds, and van der Waals contacts between residues in the S2–S3 segment, α-helix H8, and the H15–H16 segment of one monomer, and α-helices H18, H12, and H19, respectively, of a second monomer (Figures 1B and 3). Interface 2, at the base of the lung, is stabilized by interactions between α-helices H4 and H7 of one monomer, and α-helices H5 and H7, respectively, of its partner.
The yeast two-hybrid studies also demonstrated some specificity in hetero-oligomerization. Specifically, the CRMP1–CRMP5 interaction is very weak (Fukada et al, 2000), and CRMP1, CRMP2, and CRMP3 prefer hetero-oligomerization to homo-oligomerization (Wang and Strittmatter, 1997). These preferences can be partially explained by an analysis of sequence conservation at the dimer interfaces. Of the residues involved in tetramer contacts, 71% (32/45) are conserved among all five vertebrate CRMP isoforms and human DHPase, suggesting that the CRMP homo- and heterotetramers and the DHPase tetramer resemble the oligomeric structure of CRMP1 shown in Figure 3. Among the four highly homologous CRMP isoforms (1–4), 84% of residues contributing to tetramerization are conserved (Figure 2). Significant differences in sequence are, however, likely to affect the intermolecular interaction of α-helix H19 with the H15–H16 loop.
These differences fall into two categories. The first involves the variation of polar residues (Figures 2 and 3C). Gln480 in α-helix H19 interacts with residues Asp376 (OD2–NE2=3.1 Å), Glu377 (OE2–OE1=3.0 Å), and Asn378 (OD1–NE2=2.8 Å) from the H15–H16 loop of CRMP1. Although residues in H15–H16 are conserved among all CRMP isoforms, Gln480 is Arg or Lys in CRMPs 2–5. Interaction of these basic residues with the acidic residues in the H15–H16 loop may explain the preference for hetero-oligomeric over homo-oligomeric CRMP interactions. Two more polar residues undergo potentially significant variations. Gln379 in CRMP1, which forms a weak hydrogen bond with Arg481 (OE1–NE=3.4 Å), is Glu in CRMP3, which may participate in stronger salt-bridge interactions. Arg211 in CRMP1, which forms an unfavorable electrostatic interaction with Arg245, is Gln in CRMPs 2–4, but not in CRMP5. CRMP5 also includes a number of variations in residues involved in hydrogen bonding or salt bridges in the CRMP1 tetramer. The most significant is an Arg in the place of Gln379, which would lead to an unfavorable repulsion with Arg481 (see above). Additionally, Glu208, which is involved in a favorable salt bridge with Arg245 (OE1–NH1=3.3 Å), is Gly in CRMP5. Lys269, which forms a hydrogen bond with Tyr316 (ND–OH=3.2 Å), is Met in CRMP5. All three of these residue differences should weaken the affinity of CRMP5 for CRMP1.
The second category of tetramer interface variations that may influence oligomerization specificity includes interchanges of small and large hydrophobic residues. Ala371 in the H15–H16 loop makes a van der Waals contact with Ile484 (CA–CG2=4.8 Å). In CRMP2, Ala371 is Val, which should make a more significant van der Waals interaction with Ile484 of CRMP1, thus favoring hetero-oligomeric interactions of CRMP1 with CRMP2. Additional ‘small to large' and ‘large to small' changes are seen at hydrophobic residues Ile484, Val488, and Phe489, which could potentially relieve or introduce detrimental steric clashes and favor hetero-oligomeric interactions.
Chimeric analysis has previously been used to identify segments of CRMP2 and CRMP3 that mediate hetero-oligomeric specificity (Wang and Strittmatter, 1997). These experiments demonstrated that segments across the entire protein contribute to the CRMP2–CRMP3 interaction, with a predominant role for the N-terminal 435 amino acids. Since CRMP2 and CRMP3 share a high degree of identify across interfaces 1 and 2 (Figure 2), it is plausible that the actual three-dimensional structures of the two proteins have evolved to provide optimal distances for numerous van der Waals interactions, thus favoring a hetero-oligomeric interaction.
It has been proposed that as a result of weak affinity of CRMP1 for CRMP5, these proteins would not be found in the same tetramer (Fukada et al, 2000). Since the diametrically opposed proteins in the CRMP1 tetramer do not interact, it is possible that two CRMP family members that demonstrate only a weak direct interaction can still be included in the same tetramer. With regard to Drosophila and Caenorhabditis elegans CRMP, only a subset of residues found at the tetramerization interfaces are conserved. Given low sequence identities between invertebrate and vertebrate CRMP family members, it is not clear whether these residues contribute to similar homo-oligomeric interactions.
CRMP1 resembles metal-dependent amidohydrolases
The DALI 3D web-based server (Holm and Sander, 1993) employs quantitative pairwise geometrical comparisons between structures to identify proteins that share similar three-dimensional folds. A search using the DALI server revealed strong similarities between the structure of the CRMP1 monomer and a number of proteins in the Protein Data Bank (PDB; www.rcsb.org/pdb/), including D-hydantoinase (PDB ID 1K1D: Z-score=57.1, r.m.s.d.=1.4 Å, sequence identity=39%, 456 α-carbon pairs; PDB ID 1GKQ: r.m.s.d.=1.4 Å, sequence identity=36%, 456 α-carbon pairs; PDB ID 1NFG: r.m.s.d.=1.4 Å, sequence identity=35%, 454 α-carbon pairs) (Abendroth et al, 2002b; Cheon et al, 2002; Xu et al, 2003); L-hydantoinase (PDB ID 1GKR: Z-score=48.9, r.m.s.d.=2.4 Å, sequence identity=26%, 438 α-carbon pairs) (Abendroth et al, 2002a); dihydroorotase (PDB ID 1J79: Z-score=18.2, r.m.s.d.=2.8 Å, sequence identity=14%, 325 α-carbon pairs) (Thoden et al, 2001); urease alpha subunit (PDB ID 1UBP: Z-score=21.8, r.m.s.d.=3.2 Å, sequence identity=16%, 356 α-carbon pairs) (Benini et al, 1996); phosphotriesterase (PDB ID 1PSC: Z-score=13.7, r.m.s.d.=3.2 Å, sequence identity=14%, 252 α-carbon pairs) (Benning et al, 1995); cytosine deaminase (PDB ID 1K6W: Z-score=13.2, r.m.s.d.=4.1 Å, sequence identity=15%, 341 α-carbon pairs) (Ireton et al, 2002); and adenosine deaminase (PDB ID 1A4M: Z-score=10.4, r.m.s.d.=3.6 Å, sequence identity=7%, 255 α-carbon pairs) (Wang and Quiocho, 1998). These nine proteins are members of a ‘superfamily' of metal-dependent amidohydrolases, predicted to share a similar three-dimensional structure (Holm and Sander, 1997). Despite resemblance at the structural level, CRMP1 demonstrates very low to modest sequence conservation with the nine amidohydrolase proteins, with pairwise sequence identities ranging from 6 to 39% for the structurally homologous segments.
The structural similarity between the CRMP1 and D-hydantoinase monomers merits further elaboration. For the 460 residues N-terminal to helix α-H19 (CRMP1 nomenclature), the two proteins have nearly identical tertiary structures (r.m.s.d.=1.4 Å2), which is consistent with the 39% sequence identity over this region. Moreover, both are tetrameric (Figure 3A and B). The two structures differ, however, in that the D-hydantoinases have no structural counterpart to the H19 α-helix. This relatively minor difference at the monomer level leads to significant differences in tetramer assembly. While D-hydantoinase predominantly uses interactions between the N-terminal upper lobes (including the S5–S6 loop, see mutation analysis below) of different monomers to form one of the two tetramer interfaces (Figure 3B), the corresponding segment of CRMP1 does not substantially participate in homomeric interactions. Instead, CRMP1 forms its ‘upper' tetramer interface via interactions between the unique H19 α-helix and the H15–H16 loop (Figures 1, 3A and C). (The second tetramer interface, involving α-helices H4–H8, is similar for the two proteins.) These differences in quaternary structure lead to significantly greater solvent accessibility for the upper lobe of CRMP relative to that of procaryotic hydantoinases.
Although the chemical reactions catalyzed by the amidohydrolase superfamily members differ widely, the proteins share a group of five residues that comprise the active site and are essential for catalytic function. CRMP1 lacks features essential for amidohydrolytic activity and we do not believe that CRMP1 uses this cleft as an enzyme active site. Four of the five residues involved in metal binding and catalysis in other super family members are absent within CRMP1.
Heterologous cellular assay for CRMP function in Sema3A signaling
In order to explore the relationship between surface domains of the CRMP structure and function, a simple cellular assay amenable to testing a number of CRMP mutants was required. Non-neuronal COS-7 cells expressing PlexA1/NP1 complexes on the cell surface contract in response to Sema3A (Takahashi et al, 1999). This system was employed to examine the effects of CRMPs on Sema3A signaling. Cells expressing PlexA1 and NP1 alone or in combination with different isoforms of CRMP were incubated with AP-Sema3A fusion protein or the NP-1 antagonist AP-Sema3F. As shown previously, when PlexA1 and NP1 are expressed alone, the COS-7 cells have a spread morphology after 60 min Sema3F and 10 min Sema3A treatments. The cell area decreases after 60 min of Sema3A treatment as the cells contract (Figure 4). Cells expressing PlexA1, NP1, and CRMP1 show a reduction in cell area after only 10 min of Sema3A treatment (Figure 4A and B). CRMPs1-4 are equally capable of accelerating Sema3A-induced contraction (Figure 4B). Without Sema3A or after 60 min of Sema3A treatment, there is no effect of CRMP on cellular spreading.
Figure 4.
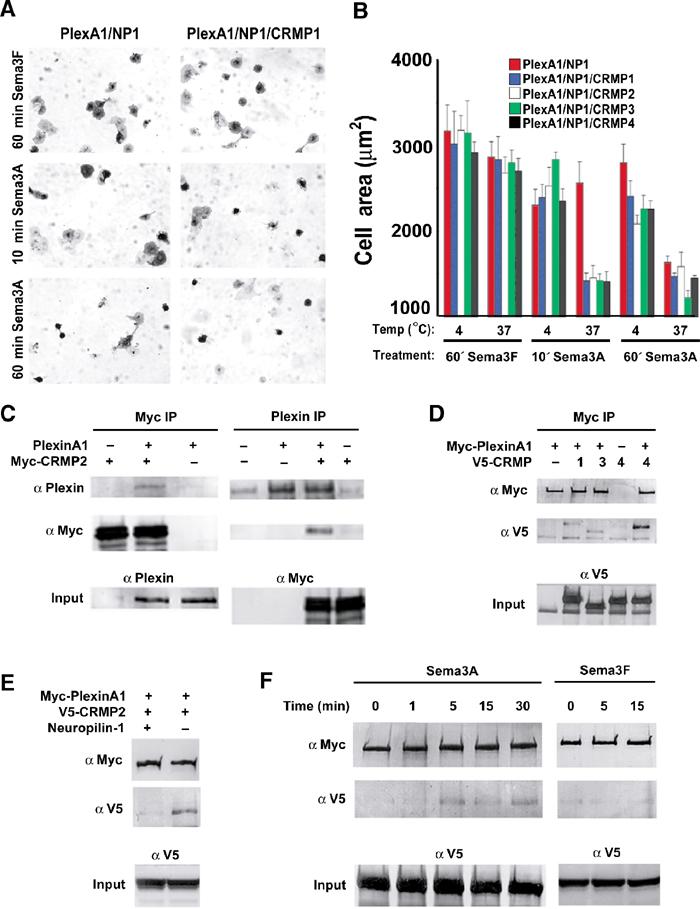
CRMPs accelerate the NP1/PlexA1-mediated Sema3A-induced COS7 cell contraction. (A) COS-7 cells expressing Myc-PlexA1/ NP1 alone or with CRMP1 were treated with AP-Sema3A or AP-Sema3F for the indicated times. Whereas Sema3F-treated cells do not exhibit any morphological changes, cells treated with Sema3A contract within 10 min in the presence of CRMP1. In contrast, cells expressing only PlexA1/NP1 shrink after 60 min Sema3A incubation. (B) Quantification of cellular areas (mean±s.e.m.) of COS-7 cells expressing PlexA1/NP1 and different CRMP isoforms and treated with Sema3F or Sema3A at 4 or 37°C. CRMPs 1–4 are designated as C1–4. All four CRMP isoforms were able to facilitate COS-7 contraction after Sema3A treatment for 10 min at 37°C. (C) HEK293T cells were transfected with vector or untagged full-length PlexA1 with Myc-tagged CRMP2, or vector or Myc-CRMP2 with PlexA1. Lysates were immunoprecipitated with anti-Myc or anti-Plex antibodies and immunoblotted with anti-Plex and anti-Myc antibodies. Note that PlexA1 co-precipitated with Myc-CRMP2 (left panel) and Myc-CRMP2 co-precipitated with anti-Plex (right panel). Input represents preimmune lysate. (D) HEK293T cells were transfected with vector or Myc-PlexA1 with V5-tagged CRMP1, CRMP3, or CRMP4. Lysates were immunoprecipitated with anti-Myc antibody, and the eluates were probed with anti-Myc or anti-V5 antibodies. All three CRMPs co-precipitate with Myc-PlexA1. (E) HEK293T cells were transfected with V5-CRMP4 and with either Myc-PlexA1 or NP1/Myc-PlexA1 dual-expression vector. Cell lysates were immunoprecipitated with anti-Myc antibody and blotted with anti-Myc and anti-V5 antibody. V5-CRMP4 co-precipitated with Myc-PlexA1 only in the absence of NP1. (F) V5-CRMP4- and NP1/Myc-PlexA1-transfected HEK293T cell lysates were treated with Sema3A or Sema3F at room temperature for the indicated periods of time. Lysates were immunoprecipitated with anti-Myc antibody and blotted with anti-Myc and anti-V5 antibodies. Co-precipitated V5-CRMP4 is detected after 5 min Sema3A treatment and can still be seen after 30 min Sema3A treatment. Sema3F does not increase the ability of Myc-PlexA1 to co-precipitate V5-CRMP4.
To provide further evidence of CRMP participation in Plexin-dependent signaling, interactions between PlexA1 and CRMPs were explored by co-immunoprecipitation. Myc-tagged CRMP2 and wild-type (WT) PlexA1 were expressed either alone or together, and one or the other protein was immunoprecipitated. Anti-Myc-CRMP immunoprecipitates contain anti-PlexA1 immunoreactivity only from doubly transfected cells (Figure 4C, left panel). Similarly, anti-PlexA1 immunoprecipitates of co-expressing cell lysates exhibit Myc-reactive CRMP (Figure 4C, right panel). Myc-PlexA1 interacted with V5-CRMP-1, -2, or -4 (Figure 4D). Direct complex formations between recombinant purified PlexA1 and CRMP2 from Escherichia coli were not detectable (data not shown). This may reflect a requirement for additional protein(s) present in HEK293T cells and neurons, or an issue with bacterially expressed PlexA1 protein folding.
Further evidence for CRMP participation in PlexA1 COS cell signaling derives from modulation of the interaction by NP1 and Sema3A. When NP1 is co-expressed with PlexA1, V5-CRMP is virtually undetectable in anti-Myc immunoprecipitates (Figure 4E). However, when the cells are treated with Sema3A for 5 min, a significant amount of V5-CRMP4 immunoprecipitates with PlexA1, and this effect persists for at least 30 min (Figure 4F). Therefore, Sema3A binding to PlexA1–NP1 complexes attenuates the ability of NP1 to interfere with PlexA1–CRMP interactions. The NP1 antagonist, Sema3F, fails to increase the amount of V5-CRMP4 detected in anti-Myc immunoprecipitates, verifying the specificity of these ligand effects on CRMP–PlexA1 interactions in COS-7 cells (Figure 4F).
Effect of CRMP1 mutants on COS-7 contraction
In order to identify functional domains of CRMP1, a number of mutant proteins were generated based on information gained from the crystal structure. Alanine substitution mutations of 1–7 residues were performed to disrupt several regions of the protein (Figures 5A and 6A). Mutation sites map to solvent-accessible areas that are not buried in the tetrameric structure plus residues found within the CRMP1 monomer–monomer interaction interfaces 1 and 2. Residues were chosen on the basis of high sequence conservation (Figure 2) and/or correspondence to structural features involved in homo- and hetero-oligomeric interactions for the amidohydrolase family members described above. In addition, one truncation mutant was constructed by deleting the highly conserved C-terminal 12 residues (8–560).
Figure 5.
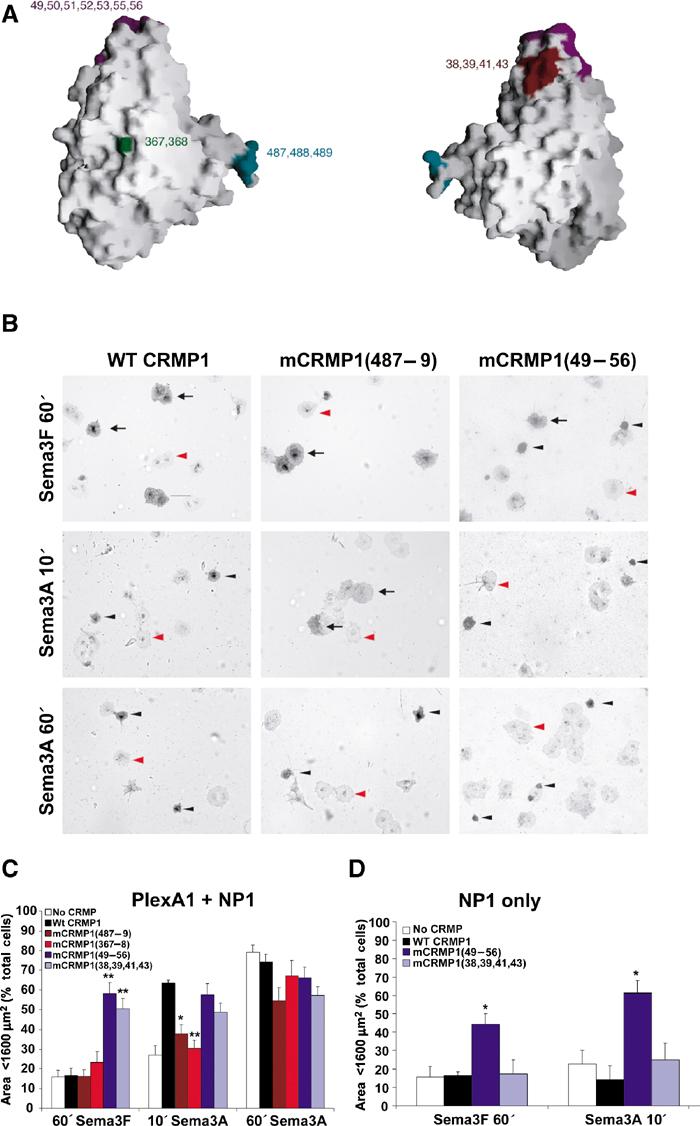
Effect of mCRMP1 mutants on COS-7 contraction. (A) GRASP (Nicholls et al, 1991) representation of CRMP1 monomer highlighting the surface regions affected by the four mCRMP1 mutants: mCRMP1(49–56) and mCRMP1(38,39,41,43) on the upper lobe, mCRMP1(367–368) in the central portion, and mCRMP1(487–489) on interface 1. (B) COS-7 cells expressing PlexA1 and NP1 with WT CRMP1, mCRMP1(487–489), or mCRMP1(49–56) were treated with Sema3A or Sema3F for the indicated times. The left column shows that cells expressing WT CRMP1 exhibit facilitated contraction after 10 min of Sema3A treatment but no contraction following 60 min with Sema3F. The middle column shows cells expressing mCRMP1(487–489) that fail to contract after 10 min Sema3A treatment, but are fully contracted after 60 min (similar to NP1/PlexA1 only expressing cells in Figure 1). The right column shows that cells expressing mCRMP1(49–56) are contracted in all treatment groups. Black arrows, black arrowheads, and red arrowheads indicate spread AP-stained cells, shrunk AP-stained cells, and unlabeled cells, respectively. (C) Quantification (mean±s.e.m.) of cell area of COS-7 cells expressing PlexA1 and NP1 with either WT CRMP1, or indicated mCRMP1 mutants. Mutant mCRMP1(487–489) and mCRMP1(367–368) failed to facilitate Sema3A-induced contraction, while mCRMP1(49–56) and mCRMP1(38,39,41,43) showed significant contraction with Sema3F treatment. (D) Quantification of cell area (mean±s.e.m.) of COS-7 cells expressing only NP1 and indicated CRMP1 construct, and treated with Sema3F for 60 min or Sema3A for 10 min. Cells expressing mCRMP1(49–56) were still contracted in the absence of PlexA1, while cells expressing mCRMP1(38,39,41,43) were spread (*P<0.005; **P<0.001).
Figure 6.
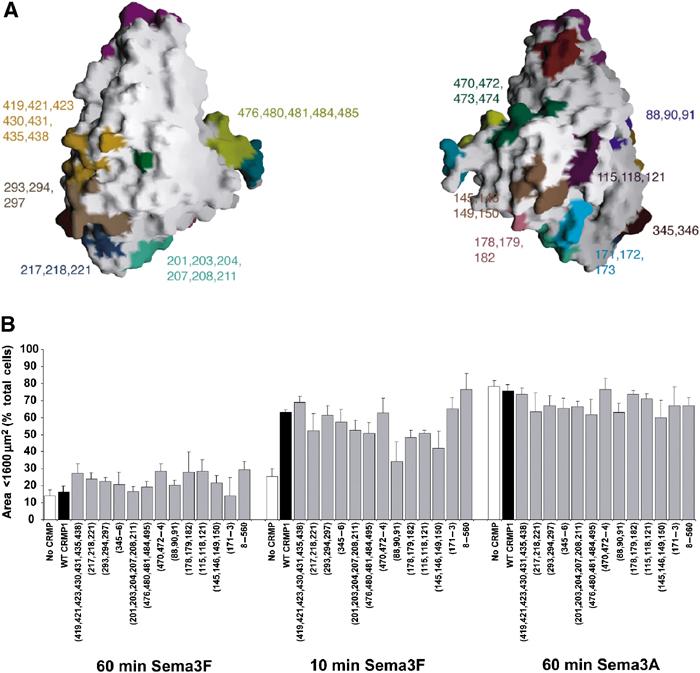
Summary of mCRMP1 mutants that had no effect on Sema3A-induced COS-7 cell contraction. (A) GRASP (Nicholls et al, 1991) representation of CRMP1 monomer highlighting the sites of 12 mutations with phenotypes identical to WT CRMP1 in the COS-7 contraction assay. (B) Quantitation of cell area (mean±s.e.m.) of COS-7 cells expressing PlexA1, NP1, and indicated mCRMP1 constructs.
CRMP1 mutants were screened in the COS-7 contraction assay to identify regions important for CRMP-mediated facilitation of Sema3A-induced contraction. COS-7 cells expressing NP1, PlexA1, and WT, mutant, or no CRMP1 were treated with Sema3A or Sema3F for 60 min or Sema3A for 10 min at 37°C. Cell area was measured and the degree of cell contraction for each group was calculated as the percentage of cells with an area below 1600 μm2 (Takahashi et al, 1999). WT CRMP1 (amino acids 8–572) facilitates cell shrinkage after 10 min Sema3A treatment when compared to cells expressing no CRMP1 (Figure 5B and C), as shown above (Figure 4A). Ala substitutions of residues 367–368 (designated as mCRMP1(367–368)) or residues 487–489 (mCRMP1(487–489)) significantly reduce CRMP1-mediated facilitation of cell contraction after 10 min Sema3A treatment, although cells are contracted after 60 min. Therefore, the response of cells expressing PlexA1, NP1, and mCRMP1(367–368) or mCRMP1(487–489) resembles that of cells expressing PlexA1 and NP1 alone, suggesting that these mutants are inactive. Sema3F does not cause contraction in any of the groups.
Mutations of residues 49, 50, 51, 52, 53, 55, 56 (within the S5–S6 linker; hereafter designated as 49–56) or 38, 39, 41, 43 (within S4 and S5), have an intriguing effect in the COS-7 contraction assay. Each cluster of residues is located within the mCRMP1 upper lobe (Figure 5). mCRMP1(49–56) or mCRMP1(38,39,41,43), when co-expressed with PlexA1 and NP1, leads to a significant increase in the percent of contracted cells after Sema3F treatment (58.4±5.4 and 50.5±5.1, respectively) as compared to WT CRMP1 (16.5±3.7; Figure 5). Cells expressing mCRMP1(49–56) and mCRMP1(38,39,41,43) do not differ from cells expressing WT CRMP1 after 10 or 60 min Sema3A treatment (Figure 5). In addition, cells expressing PlexA1, NP1, and either mCRMP1(49–56) or mCRMP1(38,39,41,43) have a contracted morphology when immunostained with an anti-NP1 antibody in the absence of ligand (data not shown). The cell contraction is not due to adverse effects of the mutant proteins on cell health; these mutations did not alter the number of transfected cells per well, and the nuclei of transfected cells appear to be healthy by Hoechst nuclear staining (data not shown). Therefore, mCRMP1(49–56) and mCRMP1(38,39,41,43) are constitutively active in COS-7 cells.
To test whether PlexA1 was necessary for the constitutive activity of these mutants, the contraction assay was performed on cells expressing only NP1 and mCRMP1(49–56), mCRMP1(38,39,41,43), or WT CRMP1. Interestingly, only mCRMP1(49–56) is able to induce contraction of COS-7 cells in the absence of PlexA1 (Figure 5D). However, the size of cells expressing NP1 and mCRMP1(38,39,41,43) does not significantly differ from those expressing NP1 and WT CRMP1 in response to Sema3A or Sema3F treatment. These results identify one region of the upper lobe of CRMP1 as being required for both upstream regulation by Plexin and downstream signaling to cell contraction.
A total of 12 other mCRMP1 surface mutants and one truncation mutant were screened for functional deficits in their ability to facilitate Sema3A-induced contraction of COS-7 cells. However, none of them differed significantly from WT CRMP1 (Figure 6). Although such results do not rule out the possibility that those regions of CRMP1 are necessary for the activity of the protein, they do emphasize the specificity and importance of the four regions that, when mutated, alter normal protein function.
Molecular interactions and subcellular localization of mCRMPs
One potential explanation for the altered phenotype of cells expressing the four mCRMPs is an aberrant subcellular localization of these mutants, with secondary alterations in function. The subcellular localization of overexpressed V5-tagged WT CRMP1 versus mCRMP1 protein was visualized by immunoflourescence in COS7 cells and in embryonic day 8 (E8) chick DRG neurons. The four mutant proteins retain localizations indistinguishable from WT CRMP1 in both cell types (Figure 7A). Thus, different protein–protein interactions are a more likely cause for the altered activity of these mutants.
Figure 7.
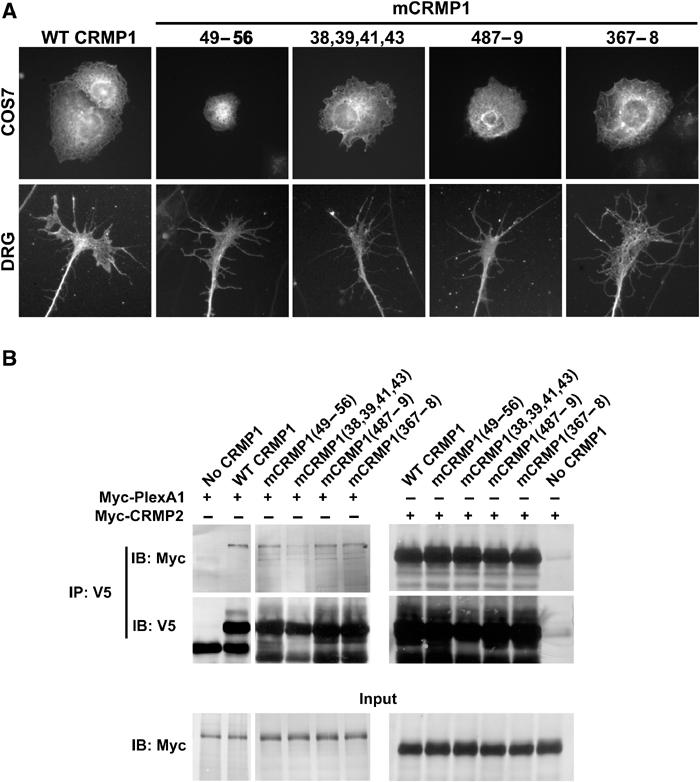
mCRMP1s are correctly localized in the cell and interact with both PlexA1 and CRMP2. (A) The subcellular localization of overexpressed mCRMP1s was examined in either transfected COS7 cells (upper row) or HSV-infected E8 chick DRG explants (lower row). All mCRMP1s showed a similar localization to overexpressed WT CRMP1. (B) HEK293T cells were transfected with either vector, V5-tagged WT CRMP1, or a V5-mCRMP1, and either Myc-tagged PlexA1 or CRMP2. Lysates were then immunoprecipitated with anti-V5 antibody and blotted with anti-Myc or anti-V5. Both Myc-PlexA1 and Myc-CRMP2 were able to co-immunoprecipitate with WT CRMP1 and all four mCRMP1s.
The altered function of the four mutant CRMPs might be due to changed interactions with Plexin, other CRMP monomers, or with downstream effector proteins. Complex formation with PlexA1 and oligomerization with other CRMPs was examined by co-immunoprecipitation of V5 and Myc epitope-tagged proteins. For CRMP–CRMP interactions, this allows an assessment of monomer versus oligomer status, but does not distinguish dimer versus tetramer status. Myc-PlexA1 is retained by each of the mutant V5-CRMP1 proteins (Figure 7B, left). Similarly, WT Myc-CRMP2 is detected in each anti-V5 immunoprecipitate of a mutant CRMP (Figure 7B, right). Therefore, the behavior of the COS7 cells expressing these mutant CRMPs is likely to result from altered interactions with cellular proteins other than Plexin or CRMP; this may include downstream effector proteins.
Overexpression of mCRMP1 in embryonic chick dorsal root ganglion neurons
Since the cell contraction assay utilizes a heterologous system, the apparent constitutive activity of mCRMP1(49–56) was examined in the physiologically relevant DRG outgrowth assay. DRG neurons were dissociated from E8 chicks and treated with a viral vector expressing EGFP, V5-tagged WT CRMP1, or V5-tagged mCRMP1(49–56). After 8 h in culture with or without Sema3A, the cells were fixed and the total neurite length/cell was measured. Transgene expression was confirmed by immunostaining with anti-V5 antibody. When compared to control neurons, neurons overexpressing WT CRMP1 show a slight reduction in neurite length (Figure 8). Inhibition of neurite outgrowth by saturating Sema3A concentrations is detectable but modest under these culture conditions due to desensitization. Overexpression of mCRMP1(49–56) reduced neurite outgrowth by ∼40% (29 μm/cell) when compared to WT CRMP1-expressing cells (Figure 8). Sema3A has no inhibitory effect on mCRMP1(49–56)-expressing cells. Therefore, the constitutive activity of mCRMP1(49–56) seen in the cell contraction assay is recapitulated in neurite outgrowth.
Figure 8.
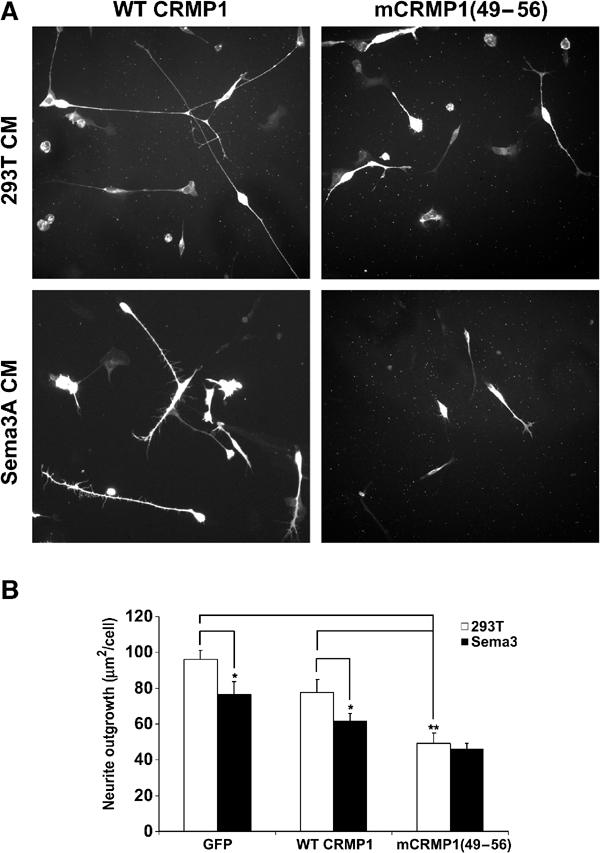
mCRMP1(49–56) reduces neurite outgrowth in DRG neurons. (A) Neurite outgrowth from dissociated E8 chick DRG neurons infected with HSV-WT CRMP1 or HSV-mCRMP1(49–56) is visualized by anti-V5 immunoflourescence. Cells were cultured with or without 100 nM Sema3A. (B) Total neurite length/cell (mean±s.e.m.) was measured for dissociated E8 chick DRG neurons infected with HSV-EGFP, or V5-tagged HSV-WT CRMP1 or HSV-mCRMP1(49–56). Neurons were cultured with or without 100 nM Sema3A. mCRMP1(49–56) significantly reduced outgrowth compared to EGFP or WT CRMP1 (**P<0.005); Sema3A reduced outgrowth only in EGFP and WT CRMP-1 cultures (*P<0.05).
To the extent that mCRMP1(49–56) activates a Sema3A pathway, axon repulsion by Sema3A should be prevented. In a collagen gel co-culture, axon growth from E8 chick DRG explants is strongly repelled from an aggregate of HEK293T cells expressing Sema3A with a four-fold reduction of growth toward the Sema3A source (Figure 9B). While DRG axons expressing WT CRMP1 exhibit a similar preference for growth away from Sema3A, DRG axons expressing mCRMP1(49–56) show much less difference between growth toward and away from Sema3A (Figure 9A–E). Thus, constitutively active mCRMP1(49–56) inhibits Sema3A-mediated axon repulsion.
Figure 9.
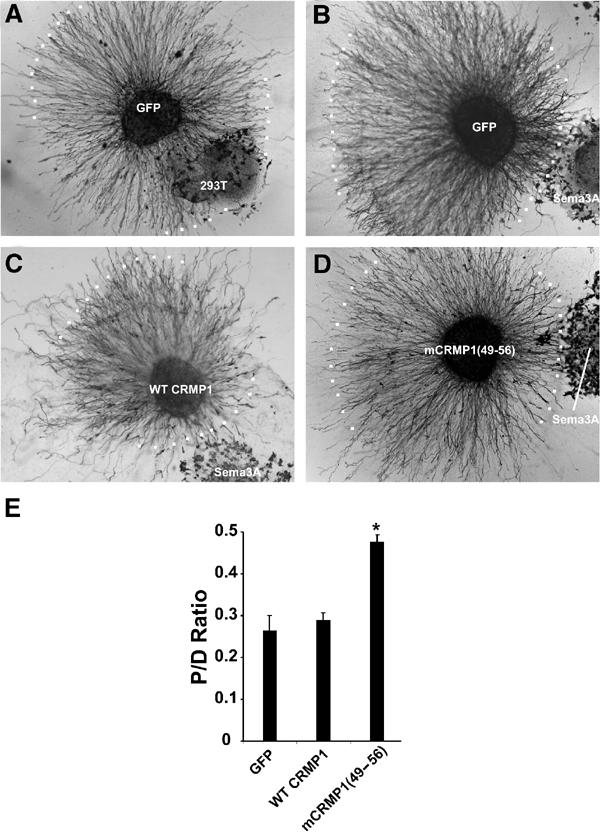
mCRMP1(49–56) attenuates Sema3A-mediated axon repulsion. (A–D) E8 chick DRG explants were infected with either HSV-GFP (A,B), or V5-tagged HSV-WT CRMP1 (C) or HSV-mCRMP1(49–56) (D), and grown in a collagen gel with Sema3A-expressing HEK293T cells (B,D) on control HEK293T (A) cells and visualized with GFP or V5 immunostaining. White dots represent the borders of axon growth. (E) Sema3A-mediated repulsion was quantitated by determining the ratio of axonal outgrowth proximal (P) and distal (D) to the Sema3A-expressing 293T cells (P/D ratio; mean±s.e.m.). Overexpression of mCRMP1(49–56) significantly reduced Sema3A-mediated axon repulsion (*P<0.005).
Discussion
Structural analysis
The determination of the X-ray structure of CRMP1 permits reinterpretation of previously published experimental data and suggested further experiments to investigate the molecular bases for CRMP action. With the exception of the H19 α-helix, the CRMP1 monomer structure closely resembles that of a number of metal-dependent amidohydrolases, including the bacterial hydantoinases. However, the precise manner in which CRMP assembles as a tetramer, which may be critical to its function (see below), could not have been predicted on the basis of sequence similarity to any known protein. An examination of monomer–monomer interfaces permitted an explanation for previous homo- and hetero-oligomerization preferences.
CRMP1 mutants and functional assays
Mutating various residues on the structure of CRMP1 resulted in a number of proteins that behaved differently from WT CRMP1 in the COS-7 contraction assay. The crystal structure of the CRMP1 tetramer was used to target mutation sites to those residues that are exposed (and therefore more likely to have biological function), and residues that fall within monomer–monomer interaction interface 1 or 2. When residues in the N-terminal ‘upper lobe' region were mutated (mCRMP1(49–56) and mCRMP1(38,39,41,43)), the resulting proteins had constitutive activity in the COS-7 contraction assay. In addition, mCRMP1(49–56) reduced neurite outgrowth in dissociated DRG neurons and Sema3A-mediated repulsion in DRG explants. We suggest that this region is involved in the negative regulation of CRMP in the basal state. Specifically, these residues appear to inhibit CRMP activation of the downstream effectors that result in cell shape changes.
Additional support for our contention that the S5–S6 domain of CRMP plays a regulatory role comes from the observation that antibodies raised against a peptide created from an overlapping region, residues 30–48, block Sema3A-induced growth cone collapse (Goshima et al, 1995). The fact that substitution of side chains in this surface patch generates constitutive activity suggests that a conformational change may occur to allow CRMP1 to activate downstream signaling proteins. In the simplest model, WT CRMP1 would require interaction with a molecular complex including PlexA1 to induce the same conformational shift. Blocking this region with a bound antibody would then be predicted to prevent the conformational change from occurring. Both mutant proteins do retain their ability to interact with PlexA1. It is remarkable that mCRMP1(49–56) retained constitutive activity in the absence of PlexA1, while activity of mCRMP1(38,39,41,43) required PlexA1. Therefore, it appears that mutation of residues 38, 39, 41, and 43 increases the propensity for the conformational shift, but an interaction with the basal state of PlexA1 is required for full activation of CRMP.
Plexin-independent action of mCRMP1[49–56] demonstrates that CRMP can activate a set of downstream effectors without PlexA1. Similarly, the slow COS-7 cell contraction induced by Sema3A/NP-1/PlexA1 without CRMP indicates that Plexin can act weakly without CRMP. For intact neuronal growth cones, Sema3A signals are thereby transduced through independent but synergistic activities of CRMP and Plexin. Signal amplification occurs via recruitment and activation of CRMP at sites of Sema3A-dependent Plexin activation. This CRMP-mediated amplification appears critical for rapid cell contraction in reconstituted systems and for dose-limited Sema3A growth cone regulation in DRG neurons. Plexin and CRMP may share overlapping and nonoverlapping sets of downstream effector proteins.
Two CRMP1 mutants, involving residues 367–368 and 487–489, respectively, were found to result in functionally inactive proteins in the COS-7 contraction assay. These mutants do not have dominant negative activity in COS or DRG assays (data not shown) and retain their ability to interact with PlexA1. The sites of these mutations are separated by over 100 amino acids; however, they both are found within or near tetramer interface 1. mCRMP1(367–368) spans α-helix H15 and includes Lys368, which is conserved across all five mammalian CRMPs, invertebrate CRMPs, DHPase, and unc-33. mCRMP1(487–489) involves mutations of the last three amino acids of α-helix H19, including Lys487, which is conserved as Lys or Arg across the five mammalian CRMPs. While these residues do not alter CRMP dimerization, they might regulate tetramerization.
In conclusion, we present the X-ray crystal structure of CRMP1, and delineate residues required for protomer interactions. Structure-based mutagenesis has elucidated regions of the protein that are required for normal function. A constitutively active form of CRMP1 was identified. It is now clear that the S5–S6 linker region of the upper lobe of CRMP participates in signal amplification, by regulating the activation of downstream effectors in a Plexin-dependent motility-regulating cascade.
Materials and methods
Protein preparation and crystallization
Murine CRMP1s (residues 8–572, 8–525, and 8–511) were expressed in E. coli as glutathione-S-transferase fusion proteins and purified to homogeneity by affinity and Q-sepharose chromatographies. S-Met and Se-Met CRMP1 (8–525) crystals were obtained by hanging drop vapor diffusion against 30% isopropanol, 100 mM Tris pH 8.0, 200 mM ammonium acetate at 4°C, using a protein concentration of 20 mg/ml. Rectangular prism-shaped crystals grew in space group C2221 (a=123.4 Å, b=152.1 Å, c=157.2 Å) with two protomers/asymmetric unit, and diffracted to 2.1 Å resolution. Crystal cryoprotection was achieved by adding glycerol to a final concentration of 20% (v/v).
Data collection, structure determination, and refinement
All diffraction data were collected at Beamline X9A at Brookhaven National Laboratory National Synchrotron Light Source under standard cryogenic conditions (see Table I for complete crystallographic statistics). A multiwavelength anomalous dispersion experiment was conducted by recording diffraction data at three X-ray wavelengths (Se absorption edge inflection point (λ=0.9792 Å), peak (λ=0.9794 Å), and high-energy remote (λ=0.9641 Å)) with one CRMP1 crystal. An additional low-energy remote wavelength data set (λ=0.9840 Å) was obtained with a second Se-Met crystal. A native data set (λ=0.9840 Å) was obtained with an S-Met crystal. Data were processed using DENZO/SCALEPACK (Otwinowski and Minor, 1997). A total of 14 Se sites were identified using SNB (Weeks and Miller, 1999) and an additional eight sites were obtained by isomorphous and anomalous difference Fourier calculations with FFT using phases estimated with MLPHARE (Dodson et al, 1997). For phasing, the S-Met crystal was treated as a ‘native data set' and the other two crystals as ‘derivatives', as this strategy appeared to provide the highest quality electron density maps for automated model building. Experimental phases were estimated at 2.12 Å resolution using MLPHARE (Dodson et al, 1997) (overall figure of merit=0.68), and improved by density modification and two-fold noncrystallographic symmetry averaging. Iterative rounds of model building/refinement were performed using ARP/wARP (Lamzin and Wilson, 1993), O (Jones and Kjeldgaard, 1997), and CNS (Brunger et al, 1998), yielding a nearly complete atomic model. The Se-Met crystal data from 24.0–2.12 Å, with 753 778 reflections (78 239 unique), Rsym(I)=6.3% (31.3% in the outermost shell), and overall completeness of 90.4% were used for refinement, as they offered the most detailed 2∣Fobs∣−∣Fcalc∣ electron density maps.
The current refinement model consists of CRMP residues 15–490 for both protomers in the asymmetric unit (950 residues in total), and 803 water molecules giving a working R-factor of 20.4% and a free R-value of 23.3%. Root mean square deviations on bond lengths and angles were 0.0127 Å and 1.53°, respectively. PROCHECK (Laskowski et al, 1993) showed no unfavorable (φ,ψ) combinations with main-chain and side-chain stereochemical parameters better than average (overall G-value=0.4). The Protein Data Bank accession number for atomic coordinates is 1KCX.
Protein expression and cellular assays of CRMP function
All assays utilized previously described methods (Takahashi et al, 1999; Takahashi and Strittmatter, 2001). Details are available in supplemental information.
Supplementary Material
Supplementary Information
Acknowledgments
We thank Dr KR Rajashankar (National Synchrotron Light Source) for help with Beamline X9A. This work was supported by grants to SMS and SKB from the NIH. SMS is an Investigator of the Patrick and Catherine Weldon Donaghue Medical Research Foundation. SKB is an Investigator of the HHMI. RCD was supported by the Weill Cornell/Rockefeller/Sloan-Kettering MD/PhD Program and the Andrew Mellon Foundation. Murine CRMP1 is target T-45 of the NIH-NIGMS-funded New York-Structural GenomiX Research Center (www.nysgxrc.org).
References
- Abendroth J, Niefind K, May O, Siemann M, Syldatk C, Schomburg D (2002a) The structure of L-hydantoinase from Arthobacter aurescens leads to an understanding of dihydropyrimidinase substrate and enantio specificity. Biochemistry 41: 8589–8597 [DOI] [PubMed] [Google Scholar]
- Abendroth J, Niefind K, Schomburg D (2002b) X-ray structure of a dihydropyrimidinase from Thermus sp. at 1.3 A resolution. J Mol Biol 320: 143–156 [DOI] [PubMed] [Google Scholar]
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I (2001) Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci 4: 367–373 [DOI] [PubMed] [Google Scholar]
- Benini S, Ciurli S, Nolting HF, Mangani S (1996) X-ray absorption spectroscopy study of native and phenylphosphorodiamidate-inhibited Bacillus pasteurii urease. Eur J Biochem 239: 61–66 [DOI] [PubMed] [Google Scholar]
- Benning MM, Kuo JM, Raushel FM, Holden HM (1995) Three-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry 34: 7973–7978 [DOI] [PubMed] [Google Scholar]
- Brunger A, Adams PD, Clore GM, Gros P, Grosse-Kuntsleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ (1998) Crystallography and NMR system: a new software system for macromolecular structure determination. Acta Crystallogr D54: 905–921 [DOI] [PubMed] [Google Scholar]
- Brunger AT (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355: 472–475 [DOI] [PubMed] [Google Scholar]
- Cheng HJ, Bagri A, Yaron A, Stein E, Pleasure SJ, Tessier-Lavigne M (2001) Plexin-A3 mediates semaphorin signaling and regulates the development of hippocampal axonal projections. Neuron 32: 249–263 [DOI] [PubMed] [Google Scholar]
- Cheon YH, Kim HS, Han KH, Abendroth J, Niefind K, Schomburg D, Wang J, Kim Y (2002) Crystal structure of D-hydantoinase from Bacillus stearothermophilus: insight into the stereochemistry of enantioselectivity. Biochemistry 41: 9410–9417 [DOI] [PubMed] [Google Scholar]
- Dodson EJ, Winn M, Ralph A (1997) Collaborative computational project, number 4: providing programs for protein crystallagrophy. In Methods in Enzymology, Carter CWJ, Sweet RM (eds). New York: Academic Press [DOI] [PubMed] [Google Scholar]
- Esnouf RM (1999) Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr D55: 938–940 [DOI] [PubMed] [Google Scholar]
- Fukada M, Watakabe I, Yuasa-Kawada J, Kawachi H, Kuroiwa A, Matsuda Y, Noda M (2000) Molecular characterization of CRMP5, a novel member of the collapsin response mediator protein family. J Biol Chem 275: 37957–37965 [DOI] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K (2002) CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4: 583–591 [DOI] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM (1995) Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376: 509–514 [DOI] [PubMed] [Google Scholar]
- He Z, Tessier-Lavigne M (1997) Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 90: 739–751 [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C (1993) Protein structure comparison by alignment of distance matrices. J Mol Biol 233: 123–138 [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C (1997) An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins 28: 72–82 [PubMed] [Google Scholar]
- Honnorat J, Byk T, Kusters I, Aguera M, Ricard D, Rogemond V, Quach T, Aunis D, Sobel A, Mattei MG, Kolattukudy P, Belin MF, Antoine JC (1999) Ulip/CRMP proteins are recognized by autoantibodies in paraneoplastic neurological syndromes. Eur J Neurosci 11: 4226–4232 [DOI] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K (2001) CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci 4: 781–782 [DOI] [PubMed] [Google Scholar]
- Inatome R, Tsujimura T, Hitomi T, Mitsui N, Hermann P, Kuroda S, Yamamura H, Yanagi S (2000) Identification of CRAM, a novel unc-33 gene family protein that associates with CRMP3 and protein-tyrosine kinase(s) in the developing rat brain. J Biol Chem 275: 27291–27302 [DOI] [PubMed] [Google Scholar]
- Ireton GC, McDermott G, Black ME, Stoddard BL (2002) The structure of Escherichia coli cytosine deaminase. J Mol Biol 315: 687–697 [DOI] [PubMed] [Google Scholar]
- Jin Z, Strittmatter SM (1997) Rac1 mediates collapsin-1-induced growth cone collapse. J Neurosci 17: 6256–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Kjeldgaard M (1997) Electron-density map interpretation. Methods Enzymol 277: 173–208 [DOI] [PubMed] [Google Scholar]
- Kikugawa M, Kaneko M, Fujimoto-Sakata S, Maeda M, Kawasaki K, Takagi T, Tamaki N (1994) Purification, characterization and inhibition of dihydropyrimidinase from rat liver. Eur J Biochem 219: 393–399 [DOI] [PubMed] [Google Scholar]
- Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD (1997) Neuropilin is a semaphorin III receptor. Cell 90: 753–762 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Lamzin VS, Wilson KS (1993) Automated refinement of protein models. Acta Crystallogr D49: 129–149 [DOI] [PubMed] [Google Scholar]
- Laskowski RJ, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check stereochemical quality of protein structures. J Appl Crystallogr 26: 283–290 [Google Scholar]
- Lee S, Kim JH, Lee CS, Kim Y, Heo K, Ihara Y, Goshima Y, Suh PG, Ryu SH (2002) Collapsin response mediator protein-2 inhibits neuronal phospholipase D(2) activity by direct interaction. J Biol Chem 277: 6542–6549 [DOI] [PubMed] [Google Scholar]
- Li W, Herman RK, Shaw JE (1992) Analysis of the Caenorhabditis elegans axonal guidance and outgrowth gene unc-33. Genetics 132: 675–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minturn JE, Fryer HJ, Geschwind DH, Hockfield S (1995) TOAD-64, a gene expressed early in neuronal differentiation in the rat, is related to unc-33, a C. elegans gene involved in axon outgrowth. J Neurosci 15: 6757–6766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui N, Inatome R, Takahashi S, Goshima Y, Yamamura H, Yanagi S (2002) Involvement of Fes/Fps tyrosine kinase in semaphorin3A signaling. EMBO J 21: 3274–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Kalb RG, Strittmatter SM (2000) Molecular basis of semaphorin-mediated axon guidance. J Neurobiol 44: 219–229 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp K, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Quinn CC, Chen E, Kinjo TG, Kelly G, Bell AW, Elliott RC, McPherson PS, Hockfield S (2003) TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. J Neurosci 23: 2815–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper JA (2000) Semaphorins and their receptors in vertebrates and invertebrates. Curr Opin Neurobiol 10: 88–94 [DOI] [PubMed] [Google Scholar]
- Sander C, Schneider R (1991) Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 9: 56–68 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM (1999) Plexin–neuropilin-1 complexes form functional semaphorin-3A receptors. Cell 99: 59–69 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Strittmatter SM (2001) Plexina1 autoinhibition by the plexin sema domain. Neuron 29: 429–439 [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, Comoglio PM (1999) Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99: 71–80 [DOI] [PubMed] [Google Scholar]
- Thoden JB, Phillips GN Jr, Neal TM, Raushel FM, Holden HM (2001) Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochemistry 40: 6989–6997 [DOI] [PubMed] [Google Scholar]
- Wang LH, Strittmatter SM (1997) Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. J Neurochem 69: 2261–2269 [DOI] [PubMed] [Google Scholar]
- Wang Z, Quiocho FA (1998) Complexes of adenosine deaminase with two potent inhibitors: X-ray structures in four independent molecules at pH of maximum activity. Biochemistry 37: 8314–8324 [DOI] [PubMed] [Google Scholar]
- Weeks CM, Miller R (1999) The design and implementation of SnB V2.0. J Appl Crystallogr 32: 120–124 [Google Scholar]
- Xu Z, Liu Y, Yang Y, Jiang W, Arnold E, Ding J (2003) Crystal structure of D-hydantoinase from Burkholderia pickettii at a resolution of 2.7 Angstroms: insights into the molecular basis of enzyme thermostability. J Bacteriol 185: 4038–4049 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information