

Abstract
Vulval differentiation in Caenorhabditis elegans is controlled by a conserved signal transduction pathway mediated by Ras and a kinase cascade that includes Raf, Mek and MAPK. Activation of this cascade is positively regulated by a number of proteins such as KSR (kinase suppressor of Ras), SUR-8/SOC-2, SUR-6/PP2A-B and CDF-1. We describe the functional characterization of sur-7 and several genes that regulate signaling downstream of ras. We identified sur-7 by isolating a mutation that suppresses an activated ras allele, and showed that SUR-7 is a divergent member of the cation diffusion facilitator family of heavy metal ion transporters that is probably localized to the endoplosmic recticulum membrane and regulates cellular Zn2+ concentrations. Genetic double mutant analyses suggest that the SUR-7-mediated effect is not a general toxic response. Instead, Zn2+ ions target a specific step of the pathway, probably regulation of the scaffolding protein KSR. Biochemical analysis in mammalian cells indicates that high Zn2+ concentration causes a dramatic increase of KSR phosphorylation. Genetic analysis also indicates that PP2A phosphatase and PAR-1 kinase act downstream of Raf to positively and negatively regulate KSR activity, respectively.
Keywords: C. elegans, CTAK1, Ras, Raf, sur-6, sur-7, vulval induction
Introduction
The small GTPase Ras is an important regulator of eukaryotic cell proliferation and differentiation. Genetic and biochemical studies have defined a well-characterized linear pathway in which Ras functions as a molecular switch for transducing extracellular signals to the nucleus via activation of a canonical mitogen-activated protein kinase (MAPK) cascade (reviewed in Vojtek and Der, 1998). Continued genetic and biochemical studies argue that multiple regulatory inputs converge on the RTK/Ras/MAP kinase cascade to ensure appropriate levels, timing and location of the signaling activity mediated by this pathway.
Genetic modifier screens in both Drosophila and Caenorhabditis elegans have identified several key players in the central linear pathway. Such screens also identified both positive and negative regulators of Ras-mediated signaling, including a number of factors that appear to regulate Raf and MEK activities. For example, KSR (kinase suppressor of Ras) was identified as a positive regulator of Ras signaling in both C. elegans and Drosophila suppressor screens (Kornfeld et al, 1995; Sundaram and Han, 1995; Therrien et al, 1995). Subsequent analysis suggested that kinase activity is not required for KSR function and that it may serve as a scaffold for Raf, MEK and other proteins (Morrison, 2001). The positive regulators sur-6 and sur-8 (suppressor of ras mutations) were identified in C. elegans (Sieburth et al, 1998, 1999). SUR-6 is a regulatory B/PR55 subunit of protein phosphatase 2A while SUR-8 is a novel but conserved leucine-rich repeat protein that complexes with Ras and Raf to enhance Raf activation (Li et al, 2000). Additionally, a C. elegans cation diffusion facilitator (CDF) family member, cdf-1, was identified as a positive regulator of Ras signaling (Bruinsma et al, 2002). Presumably, cdf-1(lf) results in increased cytosolic zinc concentrations, negatively affecting the pathway downstream of let-60/Ras and upstream of mek-2. There are some conflicting genetic and biochemical data however for some positive regulators of Ras signaling. Biochemical analyses indicated that KSR acts as a scaffold protein that facilitates the activation of MEK by Raf (reviewed by Morrison, 2001; Raabe and Rapp, 2002). However, previous genetic analyses in Drosophila and C. elegans suggested that KSR functions upstream of Raf in concert with SUR-6 (Therrien et al, 1995; Sieburth et al, 1999).
Here we characterize the role of a divergent CDF family member, sur-7, as a positive regulator of Ras signaling. Our analyses of interactions between sur-7 and other regulators clarify the discrepancies between genetic and biochemical studies of KSR and sur-6, and more precisely address where within the pathway intracellular zinc may exert its effect.
Results
sur-7(ku119) suppresses an activated ras allele
Previous studies have shown that the C. elegans let-60/Ras gene plays critical roles in a number of developmental processes (reviewed by Sternberg and Han, 1998). The best understood role for let-60 is its requirement for appropriate induction of a subset of hypodermal cells to generate the vulva, the hermaphrodite egg-laying and copulatory structure (reviewed in Kornfeld, 1997; Sternberg and Han, 1998). Reduction of let-60 activity by mutations leads to a failure in vulva precursor cell (VPC) induction, while gain-of-function (gf) alleles result in VPC overinduction causing a multivulva (Muv) phenotype characterized by numerous ventral protrusions along the length of the animal. To identify genes acting downstream of let-60/Ras, screens were previously carried out in our laboratory and others to isolate alleles that suppress this Muv phenotype (e.g. Wu and Han, 1994; Kornfeld et al, 1995; Sieburth et al, 1998). One allele, ku119, was identified and mapped genetically to the right arm of LG X.
In wild-type animals, three of six VPCs are induced to a vulva fate. let-60(gf) mutant animals are 76% Muv and on average 4.6 VPCs are induced. When ku119 is homozygous in a let-60(n1046) background, the Muv percentage and induction are reduced to nearly wild-type levels (Table I). In an otherwise wild-type background however, ku119 exhibits no obvious vulva underinduction defects. The locus defined by ku119 was named sur-7.
Table 1.
Double and triple mutant analyses of sur-7 and other genes for their roles in vulval induction
| Genotype | %M UV (n) | Average #VPC induced (n) |
|---|---|---|
| let-60(n1046) | 76 (128) | 4.62 (27) |
| let-60(n1046);sur-7(ku119) | 2 (634) | 3.03 (38) |
| let-60(kuIs12) | 86 (22) | 4.68 (22) |
| let-60(kuIs12);sur-7(ku119) | 17 (35) | 3.21(35) |
| lin-15(n765) | 91 (47) | 5.37 (51) |
| lin-15(n765) sur-7(ku119) | 41 (49) | 3.36 (37) |
| lin-31(n301) | 90 (49) | 3.81(20) |
| lin-31(n301);sur-7(ku119) | 95 (65) | 3.96 (20) |
| lin-1(ar147) | 100 (>100) | 5.91 (21) |
| lin-1(ar147);sur-7(ku119) | 100 (>100) | 5.85 (27) |
| Hsp-dRaf(gf) | 35 (60) | 3.84 (30) |
| Hsp-dRaf(gf);sur-7(ku119) | 35 (62) | 3.63 (28) |
| Hsp-dRaf(gf)a | 47 (30) | 3.57 (30) |
| Hsp-dRaf(gf);sur-6(ku123)a | 47 (17) | 3.75 (17) |
| Hsp-dRaf(gf);sur-8(ku167)b | ND | 3.75 (28) |
| Hsp-dRaf(gf);ksr-1(ku68)a | 46 (28) | 3.51(28) |
| Hsp-dRaf(gf);mpk-1(ku1)b | ND | 2.97 (28) |
| lin-45(gf) | 77 (334) | 4.44 (20) |
| lin-45(gf);sur-7(ku119) | 2 (435) | 3.06 (20) |
| lin-45(gf);sur-8(ku167) | 70 (246) | 3.81 (25) |
| Sur-6(ku123) lin-45(gf) | 0 (334) | 3.00 (23) |
| lin-45(gf);ksr-1(ku68) | 1 (98) | 3.00 (25) |
| Mek-2(ku114);lin-45(gf) | 0 (104) | 3.00 (25) |
| Hsp-mpk-1(gf) hsp-Dmek(gf) | 97 (240) | ND |
| Hsp-mpk-1(gf) hsp-Dmek(gf);sur-7(ku119) | 95 (273) | ND |
| Hsp-mpk-1(gf) hsp-Dmek(gf);sur-2(lf)c | 0 (25) | ND |
| Rol-4(sc8); let-60(n1046) | 86 (217) | ND |
| Par-1(b274);let-60(n1046) | 100 (112) | ND |
| Sur-6(ku123);let-60(n1046) | 6 (240) | ND |
| Sur-6(ku123);par-1(b274);let-60(gf) | 65 (48) | ND |
| let-60(n1046);par-1(b274);sur-7(ku119) | 94 (54) | ND |
| Sur-8(ku167) let-60(n1046) | 4 (333) | ND |
| Sur-8(ku167) let-60(n1046);par-1(b274) | 65 (111) | ND |
| let-60(n1046);ksr-1(ku68) | 0 (>100) | ND |
| let-60(n1046);par-1(b274);ksr-1(ku68) | 13 (23) | ND |
| Mpk-1(ku1);let-60(n1046) | 0 (>100) | ND |
|
Mpk-1(ku1);let-60(n1046);par-1(b274) |
2 (41) |
ND |
| %Muv, percentage of worms with a Multivulva phenotype examined under a dissecting scope. #VPC induced, number of vulval precursor cells induced to vulval cells examined under Nomarski optics on a compound microscope. (n), number of animals examined. ND, not determined. kuIs12 is an integrated array carrying multiple copies of wild-type let-60. For lin-15(n765) experiments, animals were grown at 18.2°C. hsp-dRaf(gf) is described in Materials and methods and Sieburth et al, (1999). lin-45(gf) is described in the text and Materials and methods. The hsp-mek/mpk-1(gf) strain has an integrated array containing gain-of-function mutant genes of both mpk-1 and d-mek (Lackner and Kim, 1998) (see Materials and methods for assay conditions. par-1 was marked with rol-4 in all lines tested. sur-8, sur-6 and ksr-1 were marked with dpy-20, unc-29 and lon-2, respectively. sur-7(ku119) and mek-2(ku114) homozygotes were identified by sequence analysis. Induction data were not collected for par-1 experiments in order to monitor embryonic phenotype to score for homozygous par-1(lf)). | ||
| a Sieburth et al (1999). | ||
| b Sieburth and Han (1998). | ||
| c Lackner and Kim (1996). | ||
To determine if ku119 is a loss-of-function mutation, we performed RNAi against sur-7 in wild-type, let-60(n1046) and let-60(n1046);sur-7(ku119) animals (Materials and methods). RNAi against sur-7 caused no overt phenotype in wild-type or let-60(n1046);sur-7(ku119) animals and strongly suppressed the Muv phenotype to 3.2% (n=947) in let-60(n1046). As there were no obvious phenotypic differences between sur-7(ku119) and sur-7(RNAi), we conclude that sur-7(ku119) is a strong lf allele.
SUR-7 cloning and classification as a CDF family member
To identify the sur-7 locus, ku119 was mapped using genetic markers and single-nucleotide polymorphisms (SNPs) to an LGX map position of approximately 23.9 between the SNPs pk6169 and F38E9.1 (Materials and methods). One cosmid in this interval (F01G12) and a subclone containing the single open reading frame F01G12.2 conferred robust rescuing activity to sur-7(ku119). The predicted F01G12.2 ORF was sequenced and a G to A transition was identified at position +5 in the 5′ splice site in the third of six introns (Figure 1). We performed RT–PCR to confirm the gene structure and that the ku119 mutation results in intron 3 splicing failure. Sequence analysis of wild-type RT–PCR product confirmed that the predicted F01G12.2 gene structure is correct. Using an exon three specific primer, we identified two RT–PCR products in ku119 animals, one containing the third intron, which would result in premature translational stop, while the second corresponded to the predicted wild-type mRNA. Thus, sur-7(ku119) may not be a complete lf allele.
Figure 1.
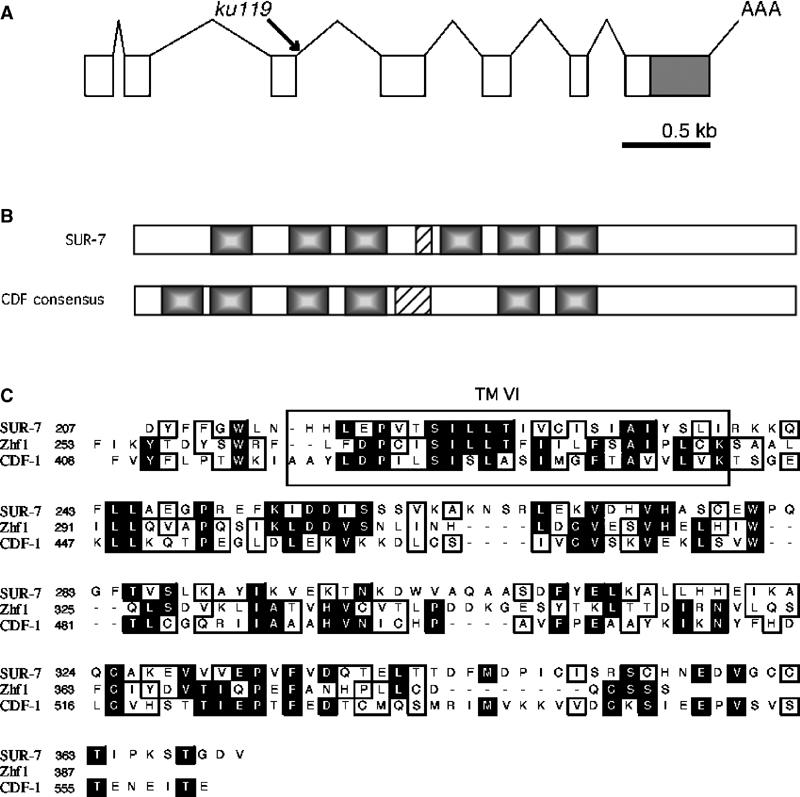
SUR-7 is a cation diffusion facilitator protein. (A) Structure of sur-7 and lesion identified in sur-7(ku119). A G to A transition was identified in the 5′ splice site of the third intron. (B) Comparison between SUR-7 and a consensus for the CDF family of proteins. SUR-7 contains six transmembrane domains (shaded regions). Five of six domains align physically with those in other CDF proteins. Most CDF proteins have a histidine-rich loop with as many as 13 histidine residues between TMs IV and V. SUR-7 has a short H-rich region between TMs III and IV. (C) Alignment between SUR-7, CDF-1 and S. pombe Zhf-1. SUR-7 shares the greatest degree of identity with other CDF proteins from TM VI to its C-terminus. In this region, SUR-7 is 16% identical and 36% similar to CDF-1, and 17% identical and 32% similar to Zhf1. CDF-1 and Zhf1 are 24% identical and 44% similar over the same region. Black backgrounds represent identity and outlines represent similarity.
An NCBI BLAST search using the SUR-7 protein sequence produced no significant homology. However, motif analysis using the PROSITE database suggested similarity to the CDF family of proteins (Falquet et al, 2002). This similarity was primarily recognized due to the presence of six transmembrane domains, a feature common to most CDF proteins. CDF proteins are a class of ubiquitous metal transporters that facilitate metal ion traffic across the plasma membrane and the membranes of organelles. Another C. elegans CDF family member, cdf-1, was recently identified as a positive regulator of Ras signaling (Bruinsma et al, 2002). cdf-1(lf) alleles similarly suppress let-60(n1046) Muv to nearly wild-type levels.
CDF proteins are found in both prokaryotes, and eukaryotes and vary greatly in size and sequence. In general, CDF proteins possess six transmembrane domains and a histidine-rich loop between membrane spanners IV and V (Palmiter and Findley, 1995; Paulsen and Saier, 1997). This H-rich loop, thought to be important for metal ion binding, and the termini of the protein presumably face cytoplasmically. There are several examples of deviation from these family characteristics. Yeast Msc2 and human hZTL1 have 12 membrane spanners (Li and Kaplan, 2001; Cragg et al, 2002). Additionally, the termini of hZTL1 are predicted to face extracellularly and hZnt6 has no H-rich loop (Huang et al, 2002). Therefore, this is a loosely defined family of proteins, which share the one specific trait of conferring either resistance or hypersensitivity to various heavy metals such as Zn2+, Cd2+, Cu2+, Co2+ and Ni2+ when their expression is altered (Gaither and Eide, 2001). SUR-7 has no H-rich loop between membrane spanners IV and V. Instead there is a cluster of five histidine residues between spanners III and IV (Figure 1). We compared the sequences of five representative CDF proteins (yeast cot1, yeast zhf1, human Znt1, human Znt5 and C. elegans CDF-1) and found the greatest sequence similarities in their C-termini and six transmembrane domains. SUR-7 displays the greatest sequence similarity to these proteins from membrane spanner VI through the C-terminus, with sequence identities ranging from 16 to 23% (Figure 1). While SUR-7 lacks similarity to other CDF family members in the remainder of its sequence, the N-terminal half of SUR-7 and the other proteins primarily define the other five transmembrane domains. When aligned against the other five CDF proteins, all but one of the predicted SUR-7 membrane spanners aligned precisely with those of the other protein family members.
sur-7(ku119) animals are sensitive to high Zn2+ concentrations
Like several human and yeast CDF proteins, mutations in C. elegans cdf1 caused hypersensitivity toward increased Zn2+ concentrations (Palmiter and Findley, 1995; Bruinsma et al, 2002). To investigate a similar response by sur-7(ku119) animals, we performed a Zn2+ sensitivity assay (Bruinsma et al, 2002; Materials and methods). Expectedly, sur-7(ku119) animals were more sensitive to increasing concentrations of Zn2+ than wild type (Figure 2). The progeny of animals placed on metal-supplemented growth media showed increasing sensitivity to Zn2+ concentrations at and above 0.25 mM as assayed by their rate of developmental maturation. These differences were found to be significant at and above 1.0 mM Zn2+ (student's t-test: α 0.05). However, sur-7(ku119) animals were more tolerant to increased Zn2+ than cdf-1(n2527) controls. The difference in tolerance between cdf-1(n2527) and wild type as well as sur-7(ku119) was significant at 0.25 and 0.05 mM Zn2+ (Tukey–Kramer HSD: α 0.05); however, Zn2+ tolerance was not statistically different between sur-7(ku119) and cdf-1(n2527) at and above 1.0 mM Zn2+. While these observations may reflect functional differences between the two proteins, they suggest that like CDF-1, SUR-7 functions as a heavy metal ion transporter, and specifically, but perhaps not exclusively, mediates cytoplasmic efflux of Zn2+ ions. As further evidence for similar functions of SUR-7 and CDF-1, we found that alleles in both genes conferred statistically significant (student's t-test: α 0.05) increased tolerance—rather than heightened sensitivity as was the case for Zn2+—to Cu2+ (at 0.5 mM but not above) compared to wild-type controls, but showed little or no difference in sensitivity for other heavy metal ions tested (Co2+ and Cd2+) (Materials and methods; data not shown). CDF proteins are not thought to function as copper transporters; therefore, the increased resistance to Cu2+ levels exhibited by both sur-7(ku119) and cdf-1(n2527) may reflect physiological conditions resulting from excess Zn2+ accumulation. Nonetheless, these results support the view that SUR-7 and CDF-1 similarly affect heavy metal ion homeostasis in C. elegans.
Figure 2.
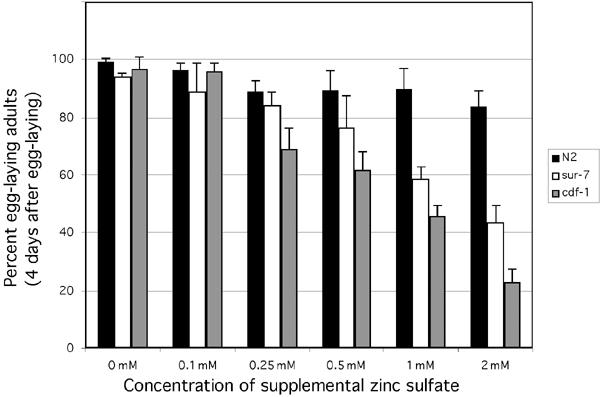
cdf-1(lf) and sur-7(lf) mutants display heightened toxicity toward increasing concentrations of Zn2+. Animals grown on metal supplemented media were scored for percentage reaching egg-laying maturity after 4 days (Materials and methods). Each bar represents the mean of three independent experiments and shows the calculated standard deviation. sur-7(ku119) and cdf-1(n2527) responses were found to be significantly different from wild type (but not one another) at and above 1.0 mM Zn2+ (Tukey–Kramer: α 0.05).
SUR-7 is likely localized to the endoplosmic recticulum membrane
We next generated a translational green fluorescent protein (GFP) fusion construct to determine the expression pattern and subcellular distribution of SUR-7 (Materials and methods). The sur-7∷GFP construct is functional since the expression of this construct rescues sur-7(ku119);let-60(n1046) (data not shown). We integrated an extrachromosomal array carrying this construct into the genome. SUR-7∷GFP was expressed in most tissues with the notable exception of intestinal cells. Under a UV-light dissecting microscope, GFP was seen to be strongly expressed in the pharynx, some tail cells and the vulva (data not shown). Under higher magnification, GFP was observed in many if not all neurons, muscles and hypodermal cells. Strikingly, SUR-7∷GFP was not localized to the plasma membrane, as was the case for a CDF-1∷GFP construct (Bruinsma et al, 2002), but was enriched at the nuclear periphery and throughout the cytoplasm in a fenestrated pattern (Figure 3A and B). We suggest that this localization is endoplasmic reticular for two reasons. First, this expression pattern is indistinguishable from that of a C. elegans YFP∷TRAM construct (data not shown; TRAM is an endoplosmic recticulum (ER) transmembrane protein) (Rolls et al, 2002). Secondly, several CDF family members are known to be ER localized (Li and Kaplan, 2001; Clemens et al, 2002). We suggest that rather than functioning to transport heavy metals extracellularly like CDF-1, SUR-7 is localized to the membrane of cellular organelles, likely the ER, and functions to sequester intracellular Zn2+ into this structure, likewise reducing cytoplasmic levels of this metal ion. The two proteins function redundantly however, as RNAi of sur-7 in the cdf-1(lf) mutant background resulted in slow growth and pleiotrophic developmental defects (data not shown).
Figure 3.
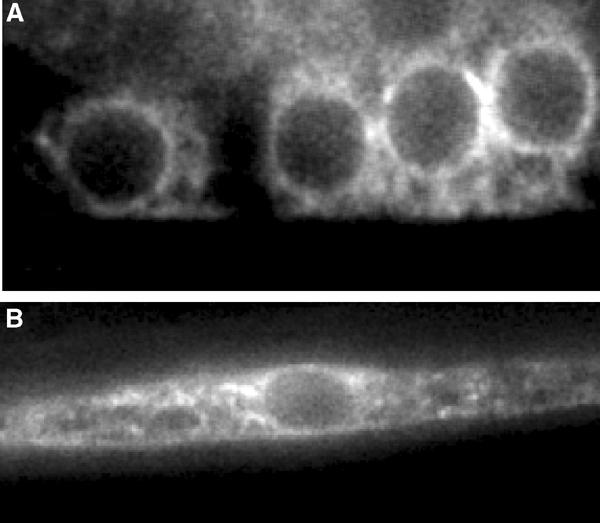
A functional translational SUR-7∷GFP construct is expressed in most tissues and is probably localized to the ER. (A) High-powered magnification of SUR-7∷GFP in four descendents of the vulva precursor cell P6.p and (B) a body wall muscle cell. SUR-7∷GFP is enriched at the nuclear envelope and expressed in a reticular pattern throughout the cytoplasm.
sur-7, ksr-1 and sur-6 likely act on factors downstream of Raf
The observations that loss-of-function mutations in sur-7, ksr-1 and sur-6 suppress activated ras suggest that these genes act on factors downstream of ras. To understand the mechanism of their functions, it is critical to know whether they also act downstream of raf. Previous genetic analyses of sur-6, sur-8 and ksr-1 suggested that each functions upstream of lin-45/Raf, as lf mutations in none of these three genes can suppress the Muv phenotype associated with an overexpressed raf gain-of-function (gf) construct (Sieburth et al, 1998, 1999) (Table I). Similarly, a Drosophila ksr lf allele could not suppress the rough eye phenotype caused by overexpression of the same Raf construct (Therrien et al, 1995). This raf(gf) construct contains a Drosophila raf transcript fused to a transmembrane domain of the Torso receptor and is under transcriptional control of a heat shock promoter (Dickson et al, 1992). Like mutations in the other positive regulators, sur-7(ku119) could not suppress the Muv phenotype associated with this construct (Table I). However, biochemical work has indicated that KSR directly interacts with both Raf and MEK and therefore ksr-1 should function genetically downstream of lin-45/Raf. These conflicting observations have been puzzling and have raised concerns about assays using this raf(gf) construct. To address this concern, we took advantage of another, likely more physiologically relevant, raf(gf) construct. The phosphorylation state of Raf is a major mechanism of its activity modulation (Brtva et al, 1995; Hu et al, 1995; Chong et al, 2001). Basal phosphorylation of putative 14-3-3 binding sites is inhibitory to Raf activity. Relief of this inhibition may be mediated by protein phosphatases 1 and 2A (Abraham et al, 2000; Jaumot and Hancock, 2001). Two consensus 14-3-3 binding sites were identified in LIN-45, homologous to those in vertebrate B-Raf, and were shown to inhibit LIN-45 activity when phosphorylated (Chong et al, 2001). Furthermore, expression of a lin-45 construct encoding alanine substitutions for these conservative serine residues caused a penetrant Muv phenotype in wild-type worms. We integrated this lin-45(gf) construct producing a strain with a strong Muv phenotype (Materials and methods and Table I). In contrast to observations using hsp-dRaf(gf), we observed that both sur-6(lf) and ksr-1(lf), but not sur-8(lf), robustly suppressed the Muv phenotype caused by the lin-45(gf) construct (Table I). These data support a model in which SUR-8 exerts its positive regulation by facilitating the interaction between Ras and Raf (Li et al, 2000) while SUR-6 and KSR-1 function downstream of Raf possibly in a common convergent regulatory event (Sieburth et al, 1999). Moreover, PP2A has been shown to associate with Raf-1 in vertebrate cell culture and potentially mediate dephosphorylation of S259 (a putative 14-3-3 binding site), thereby contributing to Raf activation (Abraham et al, 2000). Our results suggest that such potential regulation of Raf by PP2A at S259 is not mediated by the SUR-6/PR55 regulatory subunit, since sur-6(lf) can suppress LIN-45 activation caused by mimicking dephosphorylation of this residue, which is presumably targeted by PP2A. Lastly, our data argue that sur-7 exerts its influence downstream of lin-45 as sur-7(ku119) also suppresses lin-45(gf) (Table I).
Our genetic analysis also indicates that intercellular zinc concentration, regulated by SUR-7 activity, likely affects this pathway upstream of MEK. sur-7(ku119) does not suppress the Muv phenotype of a previously described strain containing gain-of-function mutant genes in both mpk-1 and the Drosophila mek gene (Lackner and Kim, 1998) (Table I). The Muv phenotype of this mpk-1(gf)/Dmek(gf) strain has been shown to be epistatic to mutations in genes acting upstream of mek but suppressed by mutations in factors known to act downstream of mpk-1 (Lackner and Kim, 1998). Consistent with SUR-7 acting upstream of MEK, ku-119 also failed to suppress the Muv phenotype of loss-of-function mutations in lin-1 and lin-31, two transcription factors that are targets of MAPK in the VPCs. These observations are consistent with the study of CDF-1 in Xenopus oocytes (Bruinsma et al, 2002). Together, these results suggest that Zn ions negatively affect this signaling pathway at the level of Raf activation of MEK. Protein interaction analyses and the data we present here place SUR-6 and KSR-1 as the only other known positive regulators at this point in the pathway.
sur-7 and sur-6 antagonize par-1 in regulating ksr activity
The kinase C-TAK1 (Cdc-25C-associated kinase 1) has recently been shown to act as a negative regulator of Ras signaling through the MAPK cascade (Muller et al, 2001). C-TAK1 was shown to phosphorylate murine KSR and inhibit its translocation to the plasma membrane. This prevents KSR from associating with activated Raf, thereby abrogating transduction of the signal to MEK and MAPK. The C. elegans homolog of C-TAK1 is PAR-1, a kinase previously defined for its role in early embryonic cell polarity (Guo and Kemphues, 1995). No role for C. elegans par-1 in Ras-mediated VPC induction has previously been described, but mutations in par-1 have recently been shown to alter vulval morphology (Hurd and Kemphues, 2003). Thus, we investigated a role for par-1 in Ras-mediated VPC induction. par-1 lf mutant animals exhibit a 100% maternal embryonic lethal phenotype as a result of failed partitioning of early embryonic cytoplasmic determinants. We used a balanced par-1(b274) strain to generate par-1(b274);let-60(n1046) double mutants as well as triple mutants containing lf in various positive regulators. The par-1(b274);let-60(n1046) strain exhibited a 100% Muv phenotype (compared to 76% for let-60(n1046) alone) (Table I). This increase in Muv percentage suggests that PAR-1 does play a negative role in VPC induction and may function similarly to its murine homolog by regulating KSR-1. If this is true, then par-1(lf) may revert the suppression of the ras(gf) phenotype by positive regulators upstream of ksr-1 but not regulators downstream. Indeed, this is what we observed (Table I). We used sur-8 and mpk-1 as controls since genetic characterization of these genes placed them respectively upstream and downstream of lin-45/Raf. An mpk-1 partial lf allele robustly suppressed the Muv phenotype of let-60(n1046) in the presence of par-1(b274); however, sur-8(ku167) suppression was overridden and the resulting triple mutant was 65% Muv. The suggested role for par-1 also predicts that ksr-1(lf) should still suppress let-60(n1046) with par-1(b274) in the background. Again, this is what we observed. Importantly, neither sur-6(lf) nor sur-7(lf) could suppress let-60(n1046) with par-1(b274) in the background, suggesting that the loss-of-function mutant phenotype of par-1 is epistatic to that of both sur-6 and sur-7. Given that PAR-1 most likely acts upstream of KSR based on biochemical data on the mammalian homologs (Muller et al, 2001) and genetic epistatic relationships between them (Table I), this result also suggests that SUR-6 and SUR-7 act upstream of KSR-1 to regulate the signaling activity.
High Zn2+ levels increase KSR phosphorylation in mammalian tissue culture cells
In mammalian tissue culture cells, C-TAK1 negatively regulates KSR function by phosphorylating a serine residue at 392 (Muller et al, 2001). Such a modification results in KSR binding to 14-3-3 and being cytoplasmically sequestered. Our genetic data presented above suggest that increased Zn2+ concentrations in sur-7(ku119) animals may negatively affect signaling through the MAPK cascade by one of at least three means. Increased Zn2+ levels may enhance the activity of PAR-1/C-TAK1, reduce the activity of PP2A that is regulated by the B-subunit SUR-6, or reduce KSR activity via a mechanism that is independent of PAR-1 and SUR-6. To further address these possibilities, we examined the effect of increased Zn2+ concentration on KSR phosphorylation in HEK 293 cells. We observed a striking shift in mobility of KSR after treating the cell culture with 1 mM Zn2+ for 3 h (Figure 4A). This shift was due to phosphorylation of the protein since it is eliminated by treatment with lambda phosphatase (Figure 4B). Interestingly, this mobility shift of KSR is not obviously affected by alanine substitutions at S297 and S392, the two major phosphorylation sites in quiescent cells (Cacace et al, 1999; Volle et al, 1999). Since S392 is the C-TAK1 phosphorylation target, our results suggest that elevated Zn2+ concentrations do not promote KSR phosphorylation by affecting C-TAK1 activity on S392. However, we cannot exclude the possibility that elevated Zn2+ concentrations act on C-TAK1 for phosphorylation at different residues of KSR.
Figure 4.
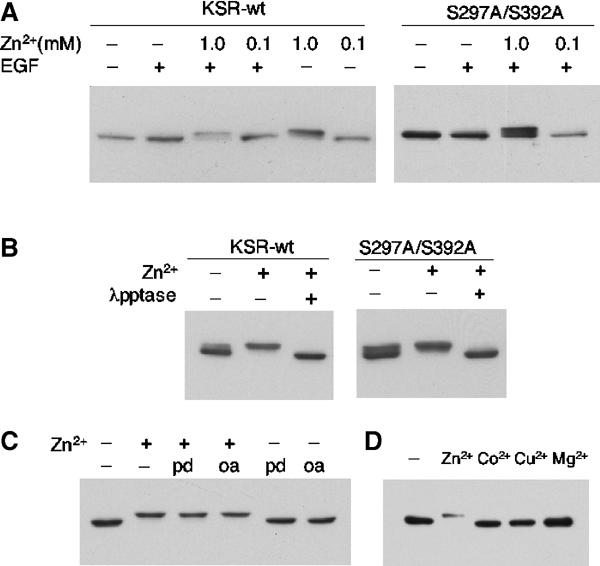
Elevated Zn2+ levels drastically increase the phosphorylation of KSR in HEK293 cells. (A) Addition of 1 mM Zn2+ causes a mobility shift of KSR. Flag-tagged KSR1 was transfected into HEK293 cells. At 24 h after transfection, cell cultures were treated with ZnSO4 for 3 h and/or 50 ng/ml EGF for 3 min. The mouse KSR protein in the cell lysates was analyzed by SDS–PAGE and Western blot using anti-M2 antibody. In a control test, a Flag-tagged Smad3 protein displayed no shift under the same condition (data not shown). On the right panel, both serine 297 and 392 were changed to alanine by in vitro mutagenesis. (B) In vitro treatment of lambda phosphatase eliminated the up-shift of KSR, indicating that the Zn2+-induced shift of the protein is due to phosphorylation. (C) Phosphatase inhibitors did not mimic or cause additional up-shifting of KSR mobility. PD, PD98059. OA, Okadatic acid. (D) Addition of 1 mM CoCl2, CuSO4 or MgCl2 does not cause the up-shift of KSR mobility.
We also observed that Zn2+-induced KSR phosphorylation was not obviously affected by the addition of EGF to the medium (Figure 4A). Nor was this phosphorylation altered by addition of a MEK inhibitor (PD98059) (Figure 4C). These results suggest that the Zn2+-induced phosphorylation of KSR is neither inhibited by activation of Ras nor mediated by an ERK feedback activity. Furthermore, phosphatase inhibitors (okadaic acid or calyculin A) did not mimic or cause an additional up-shift of KSR mobility (Figure 4C; data not shown), suggesting that it is unlikely that Zn2+ acts to inhibit a serine/threonine phosphatase like PP2A. In addition, we have shown that the observed effect on KSR phosphorylation by heavy metal ions is likely Zn2+ specific, because the addition of high concentrations of Co2+, Cu2+, Mg2+ or Ca2+ did not lead to an up-shift of KSR mobility (Figure 4D, data not shown).
Discussion
Our observations lead us to propose an updated model for the functions of a number of regulators in Ras signaling during vulva induction in C. elegans (Figure 5). SUR-8 functions to facilitate the activation of Raf by Ras, a role that is consistent with the direct interactions between SUR-8 and Ras and SUR-8 and Raf (Sieburth et al, 1998; Li et al, 2000), as well as our new result that an activated lin-45/Raf is epistatic to loss of sur-8. Conflicting genetic and biochemical data concerning ksr-1 and sur-6 are resolved, as mutations in both have now been shown to be epistatic to activated lin-45/Raf. These genetic observations for KSR are consistent with biochemical studies indicating that KSR acts as a scaffolding protein to facilitate activation of MEK by Raf (Roy et al, 2002). The genetic data suggesting that ksr-1 and sur-6 function in a common regulatory event (Sieburth et al, 1999) are also supported by our findings. The fact that sur-6(lf) suppresses a lin-45(gf) mutant that mimics dephosphorylation of a potential PP2A target residue suggests that SUR-6/PR55 does not mediate dephosphorylation of this LIN-45/Raf residue alone. Thus, our results argue for another role for PP2A, mediated by the SUR-6/PR55 subunit. One attractive model would predict that SUR-6 mediates dephosphorylation of the inhibitory effect by PAR-1/C-TAK1 on KSR-1. Indeed, the mammalian homolog of SUR-6 was recently shown to associate with KSR-1 in a PDGF-stimulated-dependent manner, and dephosphorylation of the C-TAK1 target residue as well as KSR1 membrane targeting were shown to require PP2A activity (Ory et al, 2003). Finally, the genetic and biochemical data are also consistent with the suggestion that Zn2+ ions also act downstream of lin-45/Raf but either upstream or parallel and antagonistic to par-1. We thus suggest that increased intracellular Zn2+, a likely result of loss of sur-7 function, inhibits KSR activity by either promoting phosphorylation of the protein by another kinase or by C-TAK1 at sites other than S297 and S392.
Figure 5.
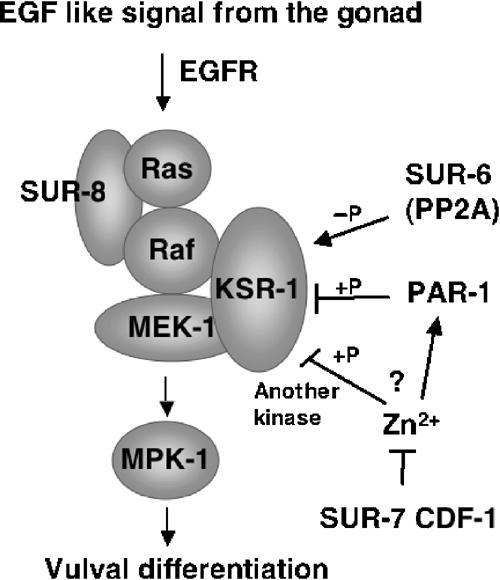
A model for the roles of several regulators of Ras-mediated vulval induction in C. elegans. See text for description.
At present, it is not totally clear why the expression of the previously used hsp-dRaf(gf) construct resulted in constitutive signaling activity that is independent of KSR function in both Drosophila and C. elegans. As mentioned in the text, this raf(gf) construct contains a Drosophila raf transcript fused to a transmembrane domain of the Torso receptor (Dickson et al, 1992). The prevailing model suggests that KSR facilitates interaction between Raf and Mek at the plasma membrane upon activation of Ras (Morrison, 2001). The Raf-Torso fusion protein, which is constitutively targeted to the plasma membrane due to the transmembrane domain of Torso, may alter the conformation of Raf in such a way that it drastically increases its affinity for Mek without a requirement fort KSR. Nevertheless, since dephosphorylation of the two 14-3-3 binding sites of B-raf was clearly shown to be critical for Raf activation (Chong et al, 2001), the alanine substitutions at these two sites in Raf provide a gain-of-function mutant gene that appears to simply render the protein, to a large extent, independent of upstream activation, making it ideal for genetic epistasis analysis.
Why should the cellular concentration of Zn2+ have such a specific effect on Ras signaling? While proper levels of Zn2+ may have an important influence on general developmental decisions, an attractive model is that the Ras-mediated cell-signaling pathway is used to sense and regulate the cellular levels of Zn2+ and/or other heavy metal ions. For example, a target of MAPK may be involved in regulating the activity or transcription of ion transporters to complete feedback regulation of cellular levels of these ions. This in turn would downregulate Ras-mediated signaling. At least one other heavy metal ion transporter, when its function is reduced, can affect signaling through the Ras/MAPK pathway. We blasted the Wormbase C. elegans protein database with the CDF-1 protein sequence. Numerous worm proteins were identified as potential members of this family, and we performed RNAi against the four most similar proteins (Y105E8A.3, T18D3.3, Y39E4A.2 and ZC395.3). Interestingly, only RNAi against the gene ZC395.3 could suppress the Muv phenotype of let-60/ras(gf) (27.9% Muv). This gene encodes the least similar of the four proteins to CDF-1, and has previously been given the name toc-1 (transporter of divalent cations) (Ewbank et al, 1997). It will be important to address if increased production of zinc transporters or activity of these proteins is modulated in response to increased or hyperactive signaling through the Ras/MAPK cascade.
Materials and methods
C. elegans strains and genetics
Mutants were derived from the wild-type Bristol N2 strain and cultured under standard conditions at 20°C unless otherwise noted (Brenner, 1974). Some strains used for mapping and genetic analysis were obtained from the Caenorhabditis Genetic Center in Minnesota.
sur-7 analysis and RNAi
sur-7(ku119) was isolated as a recessive suppressor of the let-60(n1046) Muv phenotype as previously described (Sundaram and Han, 1995). let-60(n1046); sur-7(ku119) was outcrossed five times, creating the strains MH664 [let-60(n1046);sur-7(ku119)] and MH801 [sur-7(ku119)] used for the majority of genetic analyses. sur-7 was genetically mapped to the right arm of LGX between lin-15 and sup-10 using standard three-factor mapping. For SNP mapping, the strain lin-15(n765ts) sur-7(ku119) sup-10(n983) was generated. This triple mutant strain was then mated to the Hawaiian wild-type isolate CB4856 to generate trans-heterozygous progeny. These animals were allowed to self and the resulting progeny were then scored for either lin-15-non-sup-10 or sup-10-non-lin-15 recombinants. Nine SNPs in the lin-15 sup-10 interval were analyzed to determine the positions of recombination events (Koch et al, 2000), and each recombinant was tested for its ability to suppress let-60(n1046) and therefore the presence of wild type or mutant sur-7. Three informative recombinants narrowed the sur-7 locus to a two-cosmid region (F01G12 and T24D11) between PK6169 and F38E9.1. The cosmids were injected into MH664 [let-60(n1046);sur-7(ku119)] animals at 15 μg/ml along with 100 μg/ml sur-5∷GFP, which served as a marker for transgenic progeny (Gu et al, 1998). F01G12 was found to rescue the suppression of the Muv phenotype to >50% Muv in two of six transgenic lines generated. pJHY21 is an XbaI/SalI digested F01G12 fragment cloned into pBluescript. This 6 kb fragment contains ∼3 kb of the 5′ region to the open reading frame F01G12.2 and the entire OFR followed by ∼750 kb of the 3′ sequence. Seven of seven transgenic lines established with this subclone rescued the sur-7(ku119) phenotype. pJHY21 was used to generate a full-length translation SUR-7∷GFP fusion construct (pJHY26) by introducing a unique restriction site just prior to the predicted stop codon (Quickchange site-direct mutagenesis kit from Stratagene) and ligating GFP coding DNA from the vector pPD118.85 (a gift from Dr Andrew Fire).
The molecular lesion associated with sur-7(ku119) was identified by sequencing PCR amplification of genomic DNA from let-60(n1046);sur-7(ku119) and let-60(n1046) lysates.
The full-length EST, yk462h8 (a gift from Yuji Kohara at the National Institute of Genetics), was used to generate double-stranded RNA for sur-7(RNAi) experiments following standard procedures (Fire et al, 1998). RNA was injected into the intestines of young adults and their progeny scored for phenotypes.
Constructing a lin-45(gf) allele and assaying vulval differentiation using a mek(gf)/mpk(gf) strain
A dpy-20(e1282) strain carrying an extrachromosomal array containing a lin-45(gf) gene (Chong et al, 2001) behind a hypodermal specific promoter from the col-10 gene, and the transgenic marker sur-5∷GFP was first constructed. This strain was subjected to γ-irradiation to induce germline integration following standard procedures (Epstein and Shakes, 1995). Two integrated lines were obtained and subjected to three rounds of backcrossing before use in epistatic analyses. The integrated line kuIs57 was found to possess a more penetrant Muv phenotype and was used for all experiments. PJHY26 (sur-7∷GFP) was similarly integrated into a wild-type N2 strain to create the line kuIs61. To make certain that the suppression of the Muv phenotype of the col-10:lin-45(gf) transgene by sur-7(ku119) is not due to a suppression of the col-10 promoter activity, we constructed a col-10:GFP and introduced it into C. elegans. The GFP expression of this fusion construct in the sur-7 mutant is no different from that in wild type.
A strain carrying an integrated DNA array that contains gf mutant copies of both the mpk-1 and the Drosophila mek genes was previously described and shown to effectively cause a highly penetrant Muv phenotype (Lackner and Kim, 1998). The Muv phenotype was shown to be epistatic to Vulvaless phenotype of genes acting upstream of mek but suppressed by mutations in downstream transcription factors. To determine if sur-7(ku119) is epistatic to this Muv phenotype, a strain containing both ku119 and this array were constructed. Adult animals growing at 20°C were allowed to lay eggs for 6 h and eggs were placed at 25°C for 48 h before scoring the Muv phenotype.
Heavy metal toxicity assays
For metal toxicity, the protocol in Bruinsma et al (2002) was followed. Molten nematode growth medium (NGM) was supplemented with varying concentrations of heavy metal prior to pouring on culture plates. OP50, the standard bacterial strain for culturing C. elegans, was then spotted on these plates and worms were cultured at 20°C according to standard protocols. L4-young adult worms were placed on metal-supplemented plates overnight and transferred to fresh plates the following day. These animals were allowed to lay eggs for 3 h and then removed. Four days later, the progeny were assayed for the percentage of egg-laying animals for Zn2+, Cd2+ and Co2+ sensitivity assays or the percentage of L1 arrested larvae for Cu2+ sensitivity assays. Zn2+ and Cu2+ assays were performed in triplicate and an average of 100 animals were scored for each data point.
Cell culture and transfection
HEK293 cells were cultured in DMEM (Invitrogen) containing 10% FBS. Cells were transfected with Lipofectamine (Invitrogen) according to the manufacturer's directions in serum-free DMEM. At 24 h after transfection, cells were treated with ZnSO4 for 3 h and/or 50 ng/ml EGF for 3 min. Cells were lysed in NP-40 lysis buffer and the mouse KSR protein was analyzed by SDS–PAGE and Western blot analysis using anti-M2 antibody (Sigma). For lambda phosphatase treatment, after cells were lysed, flag-tagged KSR proteins were immunoprecipitated using anti-M2 antibody and protein-G sepharose beads. Samples were washed three times with NP-40 lysis buffer, once with 20 mM tris buffer (pH 7.5), and then incubated with lambda phosphatase for 30 min at 30°C.
Acknowledgments
We sincerely thank D Sieburth, S Orita and D Green for initial genetic characterization of the ku119 allele; M Cui, F Wang, Q Crawford and B Kim for performing additional tests for the revision; J Lee for help on the lin-45(gf) allele and discussions; J Jakubowski-Bruinsma and K Kornfeld for helpful suggestions concerning the heavy metal tolerance assays; Y Kohara for cDNA clones; T Stiernagle of the Caenorhabditis Genetics Center for strains; and Dennis Eastburn for comments on the manuscript. This work was supported by grants from NIH and the Walther Cancer Institute to MH and K-LG. JHY and HC were supported by NIH predoctoral training grants and a fellowship from NIA (HC). K-LG is a John D and Katherine T MacArthur Fellow. MH is an Associate Investigator of HHMI.
References
- Abraham D, Podar K, Pacher M, Kubicek M, Welzel N, Hemmings BA, Dilworth SM, Mischak H, Kolch W, Baccarini M (2000) Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J Biol Chem 275: 22300–22304 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brtva TR, Drugan JK, Ghosh S, Terrell RS, Campbell-Burk S, Bell RM, Der CJ (1995) Two distinct Raf domains mediate interaction with Ras. J Biol Chem 270: 9809–9812 [DOI] [PubMed] [Google Scholar]
- Bruinsma JJ, Jirakulaporn T, Muslin AJ, Kornfeld K (2002) Zinc ions and cation diffusion facilitator proteins regulate Ras-mediated signaling. Dev Cell 2: 567–578 [DOI] [PubMed] [Google Scholar]
- Cacace AM, Michaud NR, Therrien M, Mathes K, Copeland T, Rubin GM, Morrison DK (1999) Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol Cell Biol 19: 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H, Lee J, Guan KL (2001) Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J 20: 3716–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens S, Bloss T, Vess C, Neumann D, Nies DH, Zur Nieden U (2002) A transporter in the endoplasmic reticulum of Schizosaccharomyces pombe cells mediates zinc storage and differentially affects transition metal tolerance. J Biol Chem 277: 18215–18221 [DOI] [PubMed] [Google Scholar]
- Cragg RA, Christie GR, Phillips SR, Russi RM, Kury S, Mathers JC, Taylor PM, Ford D (2002) A novel zinc-regulated human zinc transporter, hZTL1, is localized to the enterocyte apical membrane. J Biol Chem 277: 22789–22797 [DOI] [PubMed] [Google Scholar]
- Dickson B, Sprenger F, Morrison D, Hafen E (1992) Raf functions downstream of Ras1 in the Sevenless signal transduction pathway. Nature 360: 600–603 [DOI] [PubMed] [Google Scholar]
- Epstein HF, Shakes DC (1995) Caenorhabditis elegans: Modern Biological Analysis of an Organism. San Diego, CA: Academic Press [Google Scholar]
- Ewbank JJ, Barnes TM, Lakowski B, Lussier M, Bussey H, Hekimi S (1997) Structural and functional conservation of the Caenorhabditis elegans timing gene clk-1. Science 275: 980–983 [DOI] [PubMed] [Google Scholar]
- Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJ, Hofmann K, Bairoch A (2002) The PROSITE database, its status in 2002. Nucleic Acids Res 30: 235–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811 [DOI] [PubMed] [Google Scholar]
- Gaither LA, Eide DJ (2001) Eukaryotic zinc transporters and their regulation. Biometals 14: 251–270 [DOI] [PubMed] [Google Scholar]
- Gu T, Orita S, Han M (1998) Caenorhabditis elegans SUR-5, a novel but conserved protein, negatively regulates LET-60 Ras activity during vulval induction. Mol Cell Biol 18: 4556–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kemphues KJ (1995) par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell 81: 611–620 [DOI] [PubMed] [Google Scholar]
- Hu CD, Kariya K, Tamada M, Akasaka K, Shirouzu M, Yokoyama S, Kataoka T (1995) Cysteine-rich region of Raf-1 interacts with activator domain of post-translationally modified Ha-Ras. J Biol Chem 270: 30274–30277 [DOI] [PubMed] [Google Scholar]
- Huang L, Kirschke CP, Gitschier J (2002) Functional characterization of a novel mammalian zinc transporter, ZnT6. J Biol Chem 277: 26389–26395 [DOI] [PubMed] [Google Scholar]
- Hurd DD, Kemphues KJ (2003) PAR-1 is required for morphogenesis of the Caenorhabditis elegans vulva. Dev Biol 253: 54–65 [DOI] [PubMed] [Google Scholar]
- Jaumot M, Hancock JF (2001) Protein phosphatases 1 and 2A promote Raf-1 activation by regulating 14-3-3 interactions. Oncogene 20: 3949–3958 [DOI] [PubMed] [Google Scholar]
- Koch R, van Luenen HG, van der Horst M, Thijssen KL, Plasterk RH (2000) Single nucleotide polymorphisms in wild isolates of Caenorhabditis elegans. Genome Res 10: 1690–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld K (1997) Vulval development in Caenorhabditis elegans. Trends Genet 13: 55–61 [DOI] [PubMed] [Google Scholar]
- Kornfeld K, Hom DB, Horvitz HR (1995) The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in C. elegans. Cell 83: 903–913 [DOI] [PubMed] [Google Scholar]
- Lackner MR, Kim SK (1998) Genetic analysis of the Caenorhabditis elegans MAP kinase gene mpk-1. Genetics 150: 103–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Kaplan J (2001) The yeast gene MSC2, a member of the cation diffusion facilitator family, affects the cellular distribution of zinc. J Biol Chem 276: 5036–5043 [DOI] [PubMed] [Google Scholar]
- Li W, Han M, Guan KL (2000) The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev 14: 895–900 [PMC free article] [PubMed] [Google Scholar]
- Morrison DK (2001) KSR: a MAPK scaffold of the Ras pathway? J Cell Sci 114: 1609–1612 [DOI] [PubMed] [Google Scholar]
- Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK (2001) C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell 8: 983–993 [DOI] [PubMed] [Google Scholar]
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK (2003) Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol 13: 1356–1364 [DOI] [PubMed] [Google Scholar]
- Palmiter RD, Findley SD (1995) Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J 14: 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen IT, Saier MH Jr (1997) A novel family of ubiquitous heavy metal ion transport proteins. J Membr Biol 156: 99–103 [DOI] [PubMed] [Google Scholar]
- Raabe T, Rapp UR (2002) KSR—a regulator and scaffold protein of the MAPK pathway. Science's Stke [Electronic Resource]: Signal Transduction Knowledge Environ 2002: E28. [DOI] [PubMed] [Google Scholar]
- Rolls MM, Hall DH, Victor M, Stelzer EH, Rapoport TA (2002) Targeting of rough endoplasmic reticulum membrane proteins and ribosomes in invertebrate neurons. Mol Biol Cell 13: 1778–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M (2002) KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev 16: 427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieburth DS, Sun Q, Han M (1998) SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94: 119–130 [DOI] [PubMed] [Google Scholar]
- Sieburth DS, Sundaram M, Howard RM, Han M (1999) A PP2A regulatory subunit positively regulates Ras-mediated signaling during Caenorhabditis elegans vulval induction. Genes Dev 13: 2562–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg PW, Han M (1998) Genetics of RAS signaling in C. elegans. Trends Genet 14: 466–472 [DOI] [PubMed] [Google Scholar]
- Sundaram M, Han M (1995) The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83: 889–901 [DOI] [PubMed] [Google Scholar]
- Therrien M, Chang HC, Solomon NM, Karim FD, Wassarman DA, Rubin GM (1995) KSR, a novel protein kinase required for RAS signal transduction. Cell 83: 879–888 [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Der CJ (1998) Increasing complexity of the Ras signaling pathway. J Biol Chem 273: 19925–19928 [DOI] [PubMed] [Google Scholar]
- Volle DJ, Fulton JA, Chaika OV, McDermott K, Huang H, Steinke LA, Lewis RE (1999) Phosphorylation of the kinase suppressor of ras by associated kinases. Biochemistry 38: 5130–5137 [DOI] [PubMed] [Google Scholar]
- Wu Y, Han M (1994) Suppression of activated Let-60 ras protein defines a role of Caenorhabditis elegans Sur-1 MAP kinase in vulval differentiation. Genes Dev 8: 147–159 [DOI] [PubMed] [Google Scholar]