

Abstract
In Drosophila, the Toll pathway establishes the embryonic dorsoventral axis and triggers innate immune responses to infection. The transmembrane receptor Toll acts through three death domain-containing proteins, the kinase Pelle and the adapters Tube and MyD88, in signaling to downstream NF-κB-like transcription factors. Here, we delineate the critical events in the earliest stages of Toll signaling. Mutational studies based on structural modeling reveal that the direct interaction of the bivalent Tube death domain with MyD88 is critical for signaling in vivo. The complex of MyD88 and Tube forms prior to signaling and is localized to the embryonic plasma membrane by MyD88. Upon Toll homodimerization, this complex is rapidly recruited to Toll. Binding of Pelle to the MyD88–Tube complex promotes Pelle activation, leading to degradation of the IκB-like inhibitor, Cactus. Together, these experiments convert a linear picture of gene function into a dynamic mechanistic and structural understanding of signaling complex assembly and function.
Keywords: death domain, dorsoventral patterning, IRAK, MyD88, NF-κB, Toll, Pelle, Tube
Introduction
In Drosophila, regulation of the Toll signal transduction pathway defines the embryonic dorsoventral axis (Belvin and Anderson, 1996; Drier and Steward, 1997; Wasserman, 2000). Localized activation of Toll, a transmembrane receptor, leads to the graded nuclear translocation of an NF-κB-related transcription factor, Dorsal. Homodimers of Dorsal are initially present throughout the embryonic cytoplasm, where they are bound to an IκB-like inhibitor, Cactus, that blocks nuclear translocation. Following fertilization, localized cleavage of the ligand Spätzle activates Toll in a graded ventral to dorsal pattern over the embryo surface. Signaling by activated Toll, via the adapter proteins MyD88 and Tube and the protein kinase Pelle, triggers degradation of Cactus, leading to the formation of a Dorsal protein nuclear concentration gradient. Through activation of ventral-specific loci and repression of dorsal-specific loci, the Dorsal gradient establishes embryonic dorsoventral polarity.
The Toll pathway functions again in both larvae and adults as part of the innate immune response (Lemaitre et al, 1996; Hoffmann and Reichhart, 2002). Upon fungal challenge, wild-type flies express an array of genes encoding antimicrobial peptides, including Drosomycin, a potent antifungal agent. This immune response is protective, since flies lacking Spätzle, Toll, MyD88, Tube, or Pelle fail to induce the Drosomycin gene and succumb to fungal infection much more readily than the wild type.
The general outline of the Toll pathway is well conserved across animal species ranging from insects to humans. Drosophila Toll and mammalian Toll-like receptors direct signaling through Pelle in Drosophila or the orthologous interleukin-1 receptor associated kinases (IRAKs) in mammals (Hecht and Anderson, 1993; Shelton and Wasserman, 1993; Janssens and Beyaert, 2003). Adapter proteins, such as MyD88 and Tube, link Toll receptors to these downstream kinases (Letsou et al, 1991; Horng and Medzhitov, 2001; Janssens and Beyaert, 2002; Tauszig-Delamasure et al, 2002). Toll receptors and MyD88 share a conserved intracellular signaling motif, the TIR (Toll, IL-1 receptor, and Resistance genes) domain (Hultmark, 1994; Jebanathirajah et al, 2002). MyD88, Tube, Pelle, and IRAK all contain a distinct homotypic interaction motif, the death domain (Feinstein et al, 1995).
In Drosophila embryos, Toll pathway activity must be precisely regulated in time and space to direct formation of the axis determining Dorsal gradient. However, insight into this regulation is limited by the numerous questions that remain unanswered about the basic signaling mechanism in flies. For example, evidence exists that aggregation of Tube or Pelle can activate signaling, but the nature of protein complexes functioning in vivo remains for the most part obscure. Moreover, there are contradictory findings about which proteins in the pathway directly interact to mediate signal transduction (Edwards et al, 1997; Shen and Manley, 1998; Horng and Medzhitov, 2001; Tauszig-Delamasure et al, 2002; Charatsi et al, 2003). Lastly, there has been little information about the biochemical state of the Toll pathway prior to or immediately after Toll activation.
Here we use molecular, biochemical, cell biological, and genetic techniques to dissect early events in Toll signaling. We find that specific interactions of the bivalent Tube death domain drive association of MyD88, Tube, and Pelle into a heterotrimer. Using site-directed mutations, we identify residues critical for the two underlying dimeric interactions and demonstrate that these associations are critical for signaling in both cultured cells and embryos. We show that prior to signaling, MyD88 and Tube interact and that MyD88 functions to localize Tube to the plasma membrane. We further demonstrate that Toll can be activated by homodimerization, leading to rapid recruitment of the signaling complex via death domain-independent binding of Toll to MyD88.
Results
Heterotrimer formation activates Pelle
We have previously shown that the death domain of Tube can bind to the death domain of MyD88 and to that of Pelle (Xiao et al, 1999; Sun et al, 2002). Moreover, we have demonstrated that these two interactions provide the basis for a stable death domain heterotrimer. To investigate how such interactions function in Toll signaling, we transfected constructs encoding epitope-tagged full-length or truncated MyD88, Tube, and Pelle into S2 cells, carried out anti-MyD88 immunoprecipitation, and then detected MyD88-associated proteins by immunoblotting. As with the death domain alone (Sun et al, 2002), Tube and Pelle stably associated with MyD88 in this assay (Figure 1, lane 3). Efficient recruitment of Pelle into the complex required exogenous Tube expression (Figure 1, compare lanes 2 and 3), consistent with the Tube death domain functioning as an adapter for the association of the MyD88 and Pelle death domain. Furthermore, full-length Tube and the N-terminal Tube death domain (DD) recruited Pelle equally well (Figure 1, compare lanes 3 and 4), indicating that the C-terminal Tube repeats are not essential for heterotrimer formation.
Figure 1.
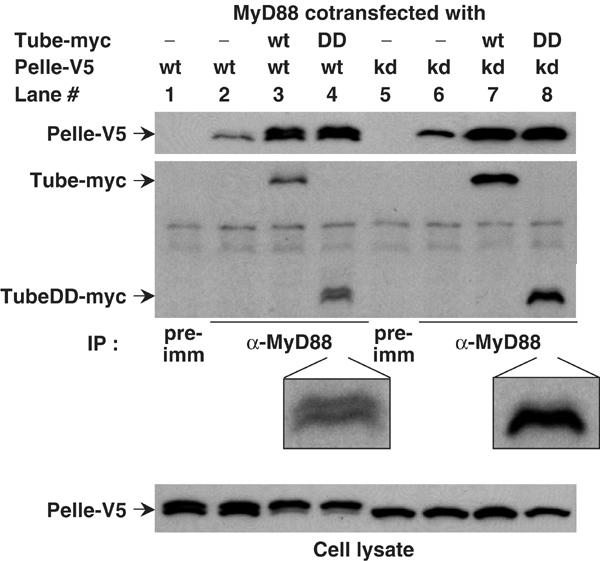
Tube-mediated interaction between Pelle and MyD88. S2 cells expressing MyD88 were transfected with Pelle or the kinase-dead (KD) PelleK240R, in combination with either full-length Tube or the Tube death domain (DD). Immunoprecipitates were prepared with preimmune (pre-imm) serum or anti-MyD88 antiserum as indicated. Pelle proteins were detected by anti-V5 antibody; Tube proteins were detected with anti-myc antibody. The TubeDD bands in lanes 4 and 8 are enlarged to facilitate viewing. The association of a small amount of Pelle with MyD88 in the absence of Tube transfection reflects the activity of endogenous Tube protein. MyD88 consistently appears as a doublet in these assays; the nature of the difference between the two species is not known.
Given that Pelle functions as a protein kinase both in signal transduction and in feedback regulation (Shelton and Wasserman, 1993; Towb et al, 2001), we were interested in the relationship between kinase activity and complex assembly. For these experiments, we took advantage of a site-directed, active-site Pelle mutation, PelleK240R (Shelton and Wasserman, 1993). Like wild-type Pelle, the catalytically inactive PelleK240R protein associated with MyD88 in a Tube-dependent manner (Figure 1, lanes 6 and 7), suggesting that Pelle kinase activity does not play a significant role in assembly of the trimeric death domain complex.
Although Pelle kinase activity is not required for complex formation, heterotrimer formation affects Pelle autophosphorylation. When expressed in Drosophila S2 cells, wild-type Pelle appears as two species with different electrophoretic mobility and with the faster migrating species corresponding to that seen with PelleK240R (Figure 1, compare cell lysates in lanes 1 and 5). The appearance of a slower migrating form of Pelle has been observed in a number of assays and has been found to represent the product of Pelle activation and autophosphorylation (Shen and Manley, 1998; Towb et al, 2001; Shen and Manley, 2002). When we cotransfected Pelle with both MyD88 and Tube, we saw a dramatic increase in the amount of autophosphorylated Pelle in the cell lysates (Figure 1, bottom panel, compare lanes 1 and 3). This shift did not occur when we coexpressed Pelle with either MyD88 or Tube alone (Figure 1, lane 2; data not shown), nor when we transfected cells with the catalytically inactive form, PelleK240R (Figure 1, bottom panel, lanes 7 and 8). Thus, under the conditions of this assay, recruitment of Pelle by Tube into a MyD88 complex triggers Pelle activation. This is the first demonstration of Pelle activation by interaction with upstream signaling components.
Recruitment of catalytically active Pelle into a death domain heterotrimer also results in the appearance of a Tube death domain doublet (Figure 1, compare lanes 4 and 8). The simplest interpretation of this finding is that activated Pelle phosphorylates the Tube death domain, as suggested by previous studies (Großhans et al, 1994, 1999; Edwards et al, 1997; Towb et al, 2001). We also find a reduction in the amount of Tube protein in cell lysate or in the anti-MyD88 immune complex when coexpressed with wild-type, but not kinase-dead Pelle (Figure 1, lanes 3 and 7; data not shown), indicating that active Pelle decreases Tube stability.
The Tube death domain is polar and bivalent
Our data indicate that the Tube death domain can simultaneously interact with the death domain of Pelle and MyD88. Based on the Pelle–Tube dimer structure, the Pelle binding surface consists of the turn between alpha helices 1 and 2, the turn between alpha helices 5 and 6, and the C-terminal tail of the Tube death domain (Xiao et al, 1999). This definition of the binding surface was confirmed by mutational analysis, demonstrating a critical role for a glutamate (E50) situated on the turn between helices 1 and 2 (Figure 2A, left). We therefore speculated that the death domain of MyD88 might have access to sites on the opposite surface of the Tube death domain.
Figure 2.
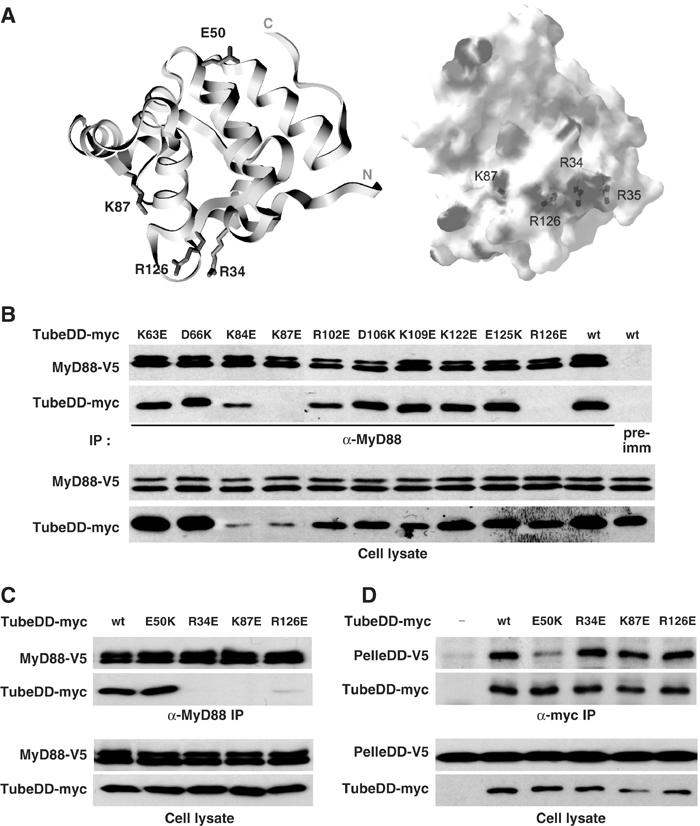
The Tube death domain is bivalent. (A) Left: A ribbon diagram of the crystal structure of Tube death domain with labeling of the side chains that are required for association with either the Pelle death domain or the MyD88 death domain. The diagram is oriented with the Pelle interaction surface at the top and the MyD88 interaction surface at the bottom. Right: An electrostatic diagram of the Tube surface with the positively charged residues in blue and negatively charged residues in red. This diagram is oriented with the MyD88 interaction surface facing the reader; the Pelle interaction surface is hidden. (B) Identification of Tube death domain residues involved in MyD88 association. Ten charged residues that are located between α-helices in the Tube death domain were mutated to ones with the opposite charge. The association of each mutant with MyD88 was assayed after anti-MyD88 immunoprecipitation from cells stably expressing a V5 epitope-tagged MyD88. Wild-type Tube death domain was used as control in immunoprecipitations with either anti-MyD88 or preimmune (pre-imm) serum. (The expression level differs among the Tube mutants due to variable transfection efficiency; see also panels C and D.) (C, D) The wild-type Tube death domain and its mutants (E50K, R343E, K87E, and R126E) were assayed for association with MyD88 (C) or the Pelle death domain (D) in coimmunoprecipitations with V5-tagged MyD88 or Pelle death domain (PelleDD), respectively.
To identify residues in Tube that are specifically required for interaction with MyD88, we carried out site-directed mutagenesis of the Tube death domain. We reasoned that mutating a critical acidic or basic residue would be likely to have a dramatic and readily observable effect, since electrostatic interactions are known to contribute to death domain interactions (Qin et al, 1999; Xiao et al, 1999; Sun et al, 2002). Focusing on charged residues in exposed turns that do not face the Pelle death domain, we converted side chains to ones of opposite charge and then assayed each mutant individually for coimmunoprecipitation with MyD88.
Of the 10 mutations in Tube generated in our initial screen, two, K87E and R126E, abolished MyD88 binding (Figure 2B). The two mutated residues, although well separated on the linear amino-acid sequence, lie in close proximity in the three-dimensional structure of the Tube death domain (Figure 2A, left). Moreover, each residue has a positively charged side chain. Examination of the surrounding surface region revealed two additional positively charged residues: R34 and R35. We mutated each and found that a charge-reversal mutation of R34, but not R35, blocked MyD88 binding (Figure 2C; see also below). We have thus identified a cluster of two arginines and one lysine (R34, R126, and K87), located in three distinct loop regions of the Tube death domain that fold together in the three-dimensional structure (Figure 2A) and are each required for binding to MyD88.
To determine whether the mutated arginines and lysine are specifically required for MyD88 binding, we assayed the interaction of these Tube death domain mutants with MyD88 and with Pelle. As shown in Figure 2C and D, the R34E, K87E, and R126E mutations each block binding to MyD88, but not to the Pelle death domain. Mutation of E50 had the reciprocal effect, disrupting interaction with the Pelle DD but not MyD88. We conclude that the death domain of Tube is bivalent and that the mutations identified here (R34, R126, and K87) and previously (E50) define two distinct binding surfaces that interact independently with the death domain of MyD88 and Pelle.
Identification of MyD88 death domain residues required for Tube binding
Since a cluster of positively charged residues in the Tube death domain mediate binding to MyD88, we predicted that the corresponding surface on MyD88 would involve negatively charged residues. To test this hypothesis, we individually mutated each acidic residue in the MyD88 death domain from E80 to E187 into lysine (Figure 3A) and then assayed the mutant proteins in binding assays with the Tube death domain. In those instances where adjacent residues in the MyD88 death domain were acidic, they were mutated together.
Figure 3.
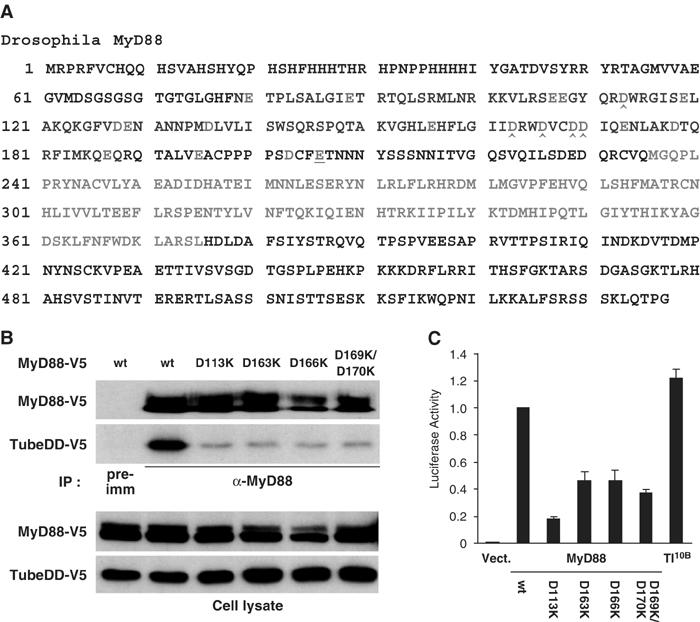
Identification of MyD88 Death Domain residues required for the MyD88–Tube Interaction. (A) The Drosophila MyD88 polypeptide sequence is shown with the death domain (79–188, in blue) and the TIR domain (236–375, in green). Negatively charged residues (red) were changed to Lys; consecutive acidic residues were changed together. The interaction between these MyD88 mutants and the Tube death domain was assayed with anti-MyD88 immunoprecipitation essentially as described in Figure 2. Residues with mutations abolishing Tube binding (D113K, D163K, D166K, and D169K/D170K) are marked (∧). (B) Anti-MyD88 immunoprecipitation was used to compare the interaction of the Tube death domain with either wild-type MyD88 or the four indicated MyD88 mutants. (C) MyD88 initiated signaling as assayed with a Drosomycin–luciferase reporter. Wild-type or mutant forms of MyD88 as indicated, or the constitutively active form of Toll, Toll10B, were transfected into S2 cells together with pGL3–Drosomycin. The reporter assay was carried out as described in Materials and methods.
Of the 14 charge-reversal mutations we generated in the MyD88 death domain, four dramatically reduced interaction with the Tube death domain. As shown in Figure 3B, these were D113K, D163K, D166K, and D169K/D170K. Furthermore, when assayed by transient transfection, these mutations also disrupted the ability of MyD88 to activate a Drosomycin–luciferase reporter (Figure 3C). Whereas either wild-type MyD88 or Toll10B induced a robust activation of the reporter, each of the four MyD88 mutants showed significantly reduced activity in this assay (Figure 3C). These studies indicate that the MyD88–Tube interaction is an essential component of MyD88-mediated signaling.
A surprising feature of the Tube–Pelle interaction, revealed by our earlier study, is the involvement of Tube sequences C-terminal to the death domain (Xiao et al, 1999). To determine whether the C-terminal extension of the MyD88 death domain has a similar role in binding to Tube, we individually mutated three acidic residues in this region (E195, D203, and E206). None affected the MyD88–Tube interaction (data not shown). The E206K mutation did, however, block the interaction of MyD88 with Toll and failed to function in embryo injection assays (Figure 3A, underlined; see below). Consistent with this finding, the linker region between the death domain and the TIR domain of mammalian MyD88 has been shown to be critical for its function (Burns et al, 2003).
The MyD88–Tube interaction is required for Toll signaling in embryos
Having identified mutations in both MyD88 and Tube that block their interaction, we used these mutations to probe the role of this interaction in embryonic dorsoventral patterning. Specifically, we assayed mutant MyD88 and Tube proteins for the ability to support Toll signaling in an embryo microinjection assay. Embryos lacking either MyD88 or Tube develop without ventral or lateral cuticle markers. Microinjection of RNA transcribed in vitro from the corresponding wild-type cDNA rescues the phenotype, restoring the missing cuticle features (Letsou et al, 1991; Charatsi et al, 2003). The extent to which mutant proteins restore the wild-type pattern provides a sensitive assay of their activity in Toll signaling in vivo.
As shown in Table I, the MyD88 mutations that block interaction with Tube in the S2 cell assay severely compromise Toll signaling in embryos. The D163K, D166K, and D169K/D170K mutations each prevent restoration of any patterning elements in myd88 null embryos and are thus indistinguishable from a complete loss-of-function mutation. A fourth interaction mutation, D113K, resulted in misregulated signaling (Table I); the observed phenotype resembled the effect of weakly lateralizing mutations in Toll (Anderson et al, 1985).
Table 1.
Critical role for the Tube–MyD88 interaction in Toll signaling
| RNA injected | No. of embryos scored | Partial rescue (FK only) (%) | Strong rescue (FK and VD) (%) |
|---|---|---|---|
| MyD88 alleles | |||
| Wild-type | 44 | 66 | 11 |
| E80K | 46 | 30 | 70 |
| E89K | 48 | 25 | 73 |
| E107K/E108K | 36 | 53 | 39 |
| D113Ka | 29 | (3)b | 0 |
| E119K | 64 | 67 | 14 |
| D128K/E129K | 34 | 6 | 0 |
| D136K | 39 | 72 | 8 |
| E156K | 42 | 24 | 62 |
| D163Ka | 36 | 0 | 0 |
| D166Ka | 24 | 0 | 0 |
| D169K/D170Ka | 28 | 0 | 0 |
| E173K | 31 | 58 | 26 |
| D178K | 24 | 0 | 0 |
| E187K | 21 | 5 | 90 |
| E195K | 35 | 11 | 54 |
| D203K | 55 | 33 | 51 |
| E206K | 33 | 6 | 0 |
| Tube alleles | |||
| Wild-type | 56 | 27 | 66 |
| R34Ea | 46 | 4 | 0 |
| R35E | 90 | 3 | 84 |
| K87Ea | 76 | 8 | 83 |
| K87Da | 44 | 20 | 0 |
| R126Ea |
27 |
0 |
0 |
| For each construct, the columns indicate the number of injected embryos for which cuticle pattern elements could be scored, the percentage of embryos scored that differentiated filzkörper (FK), a dorsal–lateral patterning element, and the percentage of embryos scored that differentiated both ventral denticle (VD) bands and FK. | |||
| aMutations that block the interaction of Tube and MyD88 in the coimmunoprecipitation assay. | |||
| bFor the D113K mutation, 31% (9/29) of injected embryos have fine lateral denticle bands extending around the embryonic circumference, but no FK. | |||
In most cases, death domain mutations that did not block interaction in S2 cells (e.g., E80K, E107K/E108K, E156K) provided rescue comparable to the wild type. The D128K/D129K DD mutation provided an exception, since it did not affect the MyD88–Tube interaction, but provided almost no rescuing activity. This mutation may hinder another, as yet uncharacterized, function of MyD88.
In general, mutations in Tube that reduced interaction with MyD88 also blocked Tube activity in dorsoventral patterning. For example, both the R34E and R126E mutations disrupted binding in cells and blocked signaling in embryos (Table I). Furthermore, the assay exhibited considerable specificity, since the R35E mutation that alters a side chain next to the critical R34 residue behaved like the wild-type protein in both the embryo injection experiments and the immunoprecipitation assay (see below). We found only one case where the sensitivity of the two assays differed. Mutating K87 in Tube to either aspartate or glutamate greatly reduced the extent of coimmunoprecipitation for MyD88 and Tube (Figure 2 and data not shown), but only the K87D mutation eliminated activity in the embryo rescue assay (Table I; see also Xiao et al, 1999). Thus, the K87E protein has an activity that is sufficient to mediate signaling in embryos, but is insufficient to maintain an association of Tube and MyD88 under the conditions used for the immunoprecipitation.
Overall, the microinjection experiments demonstrate a critical role for the MyD88–Tube interaction in the Toll signaling process that establishes embryonic dorsoventral polarity.
MyD88 directly links Toll to Tube
In addition to its N-terminal DD, MyD88 has a C-terminal TIR domain that can mediate a homotypic association with the TIR domain of Toll. To determine if MyD88 simultaneously interacts with Toll and Tube, we expressed the constitutively active Toll10B protein and used coimmunoprecipitation as an assay for interactions with Tube and MyD88. When we expressed just Toll10B and Tube in S2 cells, we failed to detect significant association of the two proteins (Figure 4A, lane 2). In contrast, when we also expressed MyD88 we observed substantial amounts of Toll10B, as well as MyD88, in the anti-Tube immune complex (Figure 4A, compare lanes 3 and 2). MyD88 is thus required for the association of Tube and Toll.
Figure 4.
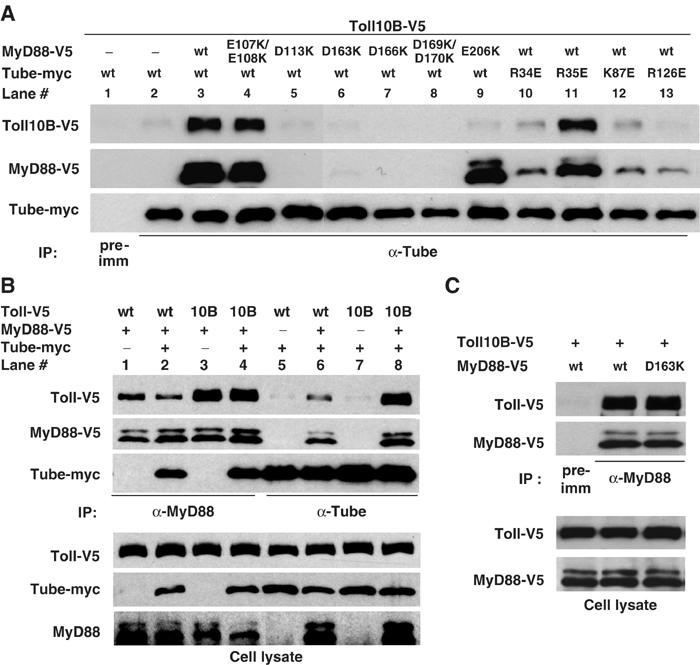
Association of Tube, MyD88, and Toll. (A) S2 cells were cotransfected with three different constructs: (1) V5-tagged Toll10B (Toll C781Y mutant); (2) vector DNA or V5-tagged MyD88; and (3) myc-tagged Tube. MyD88 and Tube mutants are as indicated. Immunoprecipitation was conducted with preimmune serum or anti-Tube serum. (Endogenous Tube may contribute to the residual coimmunoprecipitation of MyD88 and Toll10B in lanes 10, 12, and 13.) (B) Wild-type Toll or Toll10B was cotransfected into S2 cells with the listed combinations of empty vector, MyD88, and Tube. Anti-MyD88 or anti-Tube immunoprecipitation was then carried out as indicated. Toll and Tube protein in immunoprecipitates (upper) or cell lysates (bottom) were detected with anti-V5 and anti-myc antibodies, respectively. MyD88 protein was detected with anti-V5 antibody in immunoprecipitates and with anti-MyD88 antiserum in cell lysates. (C) Wild-type or a Tube binding-defective (D163K) mutant of MyD88 were assayed for their association with Toll10B as described in (B).
We next asked whether MyD88-mediated association of Tube and Toll is achieved through the interaction of the MyD88 and Tube death domain. We cotransfected Toll10B into S2 cells with either wild-type Tube and a MyD88 mutant defective in binding to Tube (Figure 4A, lanes 5–8) or wild-type MyD88 and a Tube mutant defective in binding to MyD88 (Figure 4A, lanes 10, 12, and 13). As controls, we also assayed mutations in each protein that do not block their interaction (MyD88 E107K/E108K and Tube R35E) (Figure 4A, lanes 4 and 11). Without exception, each mutation that blocks the Tube–MyD88 interaction markedly reduced the amount of Tube-associated Toll. In contrast, the two mutations that did not disrupt the Tube–MyD88 interaction also had little or no effect on the coimmunoprecipitation of Tube and Toll.
In the course of our mutagenesis, we identified a mutation, MyD88 E206K, that specifically blocks MyD88 binding to Toll. In the anti-Tube immunoprecipitation assay, MyD88 E206K was associated with Tube, but Toll10B was absent from the complex (Figure 4A, lane 9).
Overall, these studies demonstrate that the death domain mediated Tube–MyD88 interaction is essential for the formation of a Tube, MyD88, and Toll complex and is not dependent on the interaction of MyD88 and Toll.
Active Toll recruits the MyD88–Tube complex
To probe the role of Toll activation in the formation of a complex with MyD88 and Tube, we compared the activity of constitutively active (Toll10B) and wild-type Toll in the S2 cell interaction assay. As a first step, we immunoprecipitated MyD88 and determined the amount of Toll protein in the immune complex. We found significantly more Toll10B than wild-type Toll in such assays (Figure 4B, compare lanes 1 and 3), despite the fact that the epitope-tagged Toll and Toll10B were expressed at the same level (Figure 4B, cell lysate panel). This relationship held whether or not we also expressed Tube (Figure 4B, lanes 1–4). Furthermore, the association of MyD88 and Tube appeared unaffected by Toll activation (Figure 4B, compare lanes 2 and 4). These results show that MyD88 and Tube exist in a complex prior to signaling and that activated Toll recruits MyD88 via a Tube-independent mechanism.
In a parallel experiment, we used anti-Tube antiserum to isolate protein complexes (Figure 4B, lanes 5–8). Consistent with our previous experiments, MyD88 expression greatly enhances the Tube–Toll association (Figure 4B, compare lanes 5 and 6 or lanes 7 and 8). In addition, we again found that the MyD88–Tube complex preferentially interacts with the active form of Toll (Figure 4B, lanes 6 and 8), whereas Toll activation has no effect on the MyD88–Tube interaction.
We confirmed by mutational analysis that the interaction of MyD88 with Toll is independent of the MyD88–Tube interaction. For these studies, we compared wild-type MyD88 with the D163K mutant that is deficient for Tube binding. We could detect no difference in the pairwise binding to Toll10B between the wild-type and D163K forms of MyD88 (Figure 4C). Thus, the death domain and the TIR domain of MyD88 independently associate with their binding partners.
MyD88 directs Tube membrane localization
Given our biochemical finding that a complex containing MyD88 and Tube is recruited by activated Toll, we predicted that MyD88 localization in embryos would resemble that of Tube. Using our anti-MyD88 antiserum, we examined MyD88 localization in syncytial wild-type embryos. A substantial fraction of the MyD88 was membrane associated, as is readily apparent by comparison with the membrane marker anti-PY20 (Figure 5A and E). Moreover, the pattern was indistinguishable from that seen for Tube (Figure 5C and G).
Figure 5.
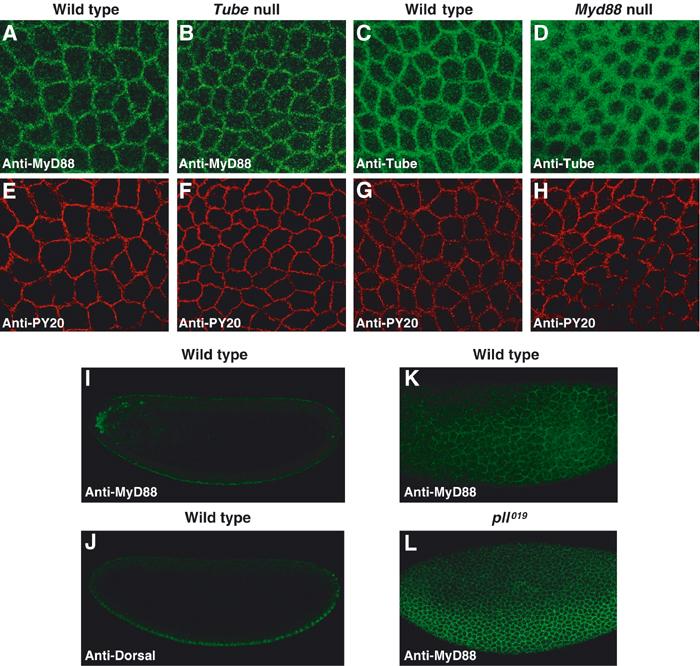
Distribution of MyD88 and Tube proteins during Drosophila dorsoventral patterning. (A–H) Lateral surface views of double-stained syncytial embryos. MyD88 in wild-type (A) and tube null (B) embryos, and Tube in wild-type (C) and myd88 null (D) embryos, were detected with anti-MyD88 and anti-Tube antibodies, respectively. The same embryos shown in panels A–D were stained for membrane antigen with antiphosphotyrosine antibody (anti-PY20) as shown in panels E–H. The level of Tube protein in wild-type and myd88 null embryos is indistinguishable, as assayed by immunoblotting (data not shown). (I, J) Confocal cross-sectional views of a wild-type embryo double-stained for MyD88 (I) and Dorsal (J). (K, L) Surface views of the lateral aspect of a wild-type embryo (K) and a catalytically inactive pelle mutant (pll019/Df(3R)IR16) embryo (L).
We have previously described a signal-dependent pattern of Tube localization, with highest Tube levels in the ventral regions where Toll activation is maximal (Towb et al, 1998). This gradient is very difficult to discern in surface views, but more apparent in confocal cross-sections oriented on the basis of the Dorsal nuclear concentration gradient. Furthermore, because Pelle negatively regulates Tube recruitment to signaling complexes (Towb et al, 2001), elimination of Pelle catalytic activity greatly enhances the gradient. We observed the same properties for MyD88 (Figure 5I–L), with the most dramatic gradient of localization seen in a background lacking Pelle kinase activity. The MyD88 staining pattern is in some cases clearly punctate (Figure 5A and B), as previously observed for Tube upon localized expression of activated Toll. Overall, MyD88 localization thus closely resembles that of Tube. The immunolocalization studies also reveal that the association with activated Toll of MyD88, like that of Tube, is subject to negative feedback regulation by the kinase activity of Pelle.
Since MyD88 and Tube interact, we next assayed whether the localization of either protein depended on the presence of the other. Whereas MyD88 localization appeared unaffected in Tube null embryos (Figure 5B and F), we observed two striking changes in Tube localization in embryos lacking MyD88. First, the Tube gradient disappeared, confirming that MyD88 is required to recruit Tube to sites of active signaling. Second, in place of the tight membrane localization seen in the wild type, the distribution of Tube in embryos lacking MyD88 was diffuse, comprising a broad region of the cytoplasm surrounding each syncytial nucleus (Figure 5D and H). Moreover, the alteration in localization occurred across the entire dorsoventral axis (data not shown), indicating that the absence of MyD88 perturbs Tube localization whether or not Toll is active.
These results indicate that MyD88 not only acts as a scaffold for Toll and Tube interaction, but also localizes the death domain complex to the plasma membrane prior to Toll activation. This membrane association of the presignaling complex is likely to play a critical role in the efficient recruitment of pathway components by activated Toll.
Inducible assembly of the Toll–MyD88–Tube complex
To explore the earliest events in Toll signaling, we developed a system for the rapid activation of the Drosophila pathway. We constructed an S2 cell expression plasmid encoding a chimeric protein (EGFR–Toll), consisting of the extracellular and transmembrane portions of the human epidermal growth factor (EGF) receptor and the intracellular region of Drosophila Toll. We reasoned that EGF-induced oligomerization of the Toll intracellular domain in the chimera would activate the pathway, as does oligomerization of downstream components (Galindo et al, 1995; Großhans et al, 1994, 1999).
We first tested whether the EGFR–Toll chimera functions as an EGF-inducible Toll molecule by stably transfecting S2 cells with an expression construct for the chimeric protein. EGF specifically induced a robust activation of the Drosomycin–luciferase reporter in S2 cells expressing EGFR–Toll (Figure 6A). Activation correlated with both the concentration of EGF and the duration of EGF treatment in the cell culture. A 4-h treatment with 0.5 μg/ml of EGF triggered a nearly 30-fold activation of the reporter (Figure 6A), more than four times the maximal induction of the Drosomycin gene seen with fungal infection of adult flies (De Gregorio et al, 2002).
Figure 6.
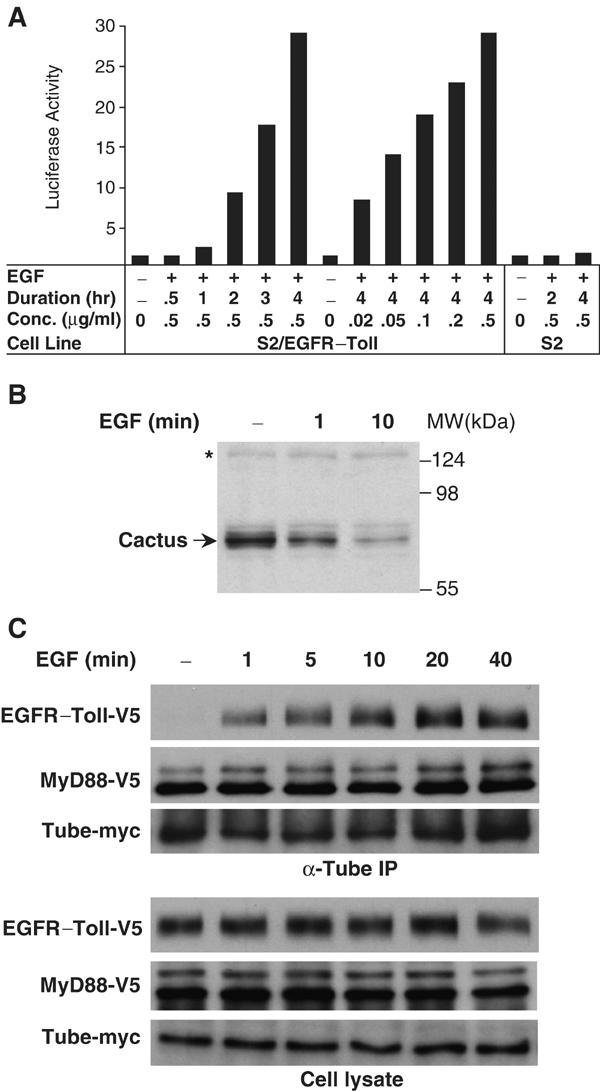
Inducible signaling complex assembly in the Drosophila Toll pathway. (A) Regulation of Toll signaling by a chimera of human EGF receptor and Drosophila Toll. S2 cells stably expressing this chimera (S2/EGFR–Toll) were transfected with Drosomycin–luciferase reporter and were treated either for various length of time (left), or with various EGF concentrations (right), as indicated; S2 cells with no EGFR–Toll expression were also tested for reporter activation after EGF treatment. Luciferase activity is reported as fold activation relative to untreated cells. (B) Inducible Cactus degradation in S2 cells expressing EGFR–Toll. Cell lysates were prepared from untreated cells or cells treated with 0.5 μg/ml EGF for 1 or 10 min. Arrow indicates Cactus protein in the cell lysates detected in an immunoblot using an anti-Cactus antiserum. Asterisk marks a nonspecific band indicating equivalent protein loading in each lane. (C) Inducible assembly of a protein complex in the Drosophila Toll pathway. V5 epitope-tagged EGFR–Toll was transfected into S2 cells together with myc-tagged Tube and V5-tagged MyD88. Transfected cells were treated with EGF (0.5 μg/ml) for the times indicated and then lysed. Proteins collected in an anti-Tube immunoprecipitation, as well as the expression of epitope-tagged proteins in S2 cells, were analyzed by immunoblotting with either anti-V5 or anti-myc antibody.
We next tested whether the effect of EGF in S2/EGFR–Toll cells mimics that of Spätzle in Drosophila embryos with respect to Cactus degradation. Using anti-Cactus antiserum, we assayed the degradation of endogenous Cactus in EGF-treated cells. As shown in Figure 6B, we observe degradation of Cactus within 1 min after EGF stimulation, with significant turnover by a 10-min time point. We conclude that EGF acts as an effective stimulant of the Toll pathway in S2 cells expressing EGFR–Toll.
To determine how protein complex assembly is regulated by Toll activation, we transfected S2 cells with EGFR–Toll and MyD88, both V5-tagged, as well as a myc-tagged Tube. Following treatment with EGF for various lengths of time, we prepared anti-Tube immunoprecipitates. The level of Tube-associated MyD88 protein was unaffected by EGF simulation. In contrast, we detected a significant level of the EGFR–Toll chimera in the anti-Tube immune complex within 1 min of EGF stimulation, with maximal association after 20 min (Figure 6C). We thus observe very similar kinetics for the assembly of the signaling complex and the proteolysis of the target, Cactus. We conclude that activated Toll initiates intracellular signal transduction by the rapid recruitment of a preformed complex containing MyD88 and Tube.
Discussion
We can now for the first time correlate the protein–protein interaction state of signaling components with that of the activity of Toll in the Drosophila system. Prior to signaling, we find that specific death domain interactions mediate the stable association of MyD88 and Tube in close proximity to the plasma membrane. Upon Toll dimerization, the Toll TIR domain becomes available for binding to the MyD88–Tube complex. Association of Pelle with this complex, through its direct interaction with Tube, promotes its activation. Phosphorylation by active Pelle then mediates signaling to Cactus as well as feeds back on the signaling complex.
Initiation of Toll signaling
Prior to signaling, MyD88 and Tube stably associate via death domain interactions. In contrast, MyD88 is not detectably bound to Toll prior to Toll activation. Thus, in the presignaling state the intracellular TIR domain of Toll must be inaccessible to MyD88 binding. It was therefore surprising to observe that MyD88 is membrane localized in the absence of Toll activation. The fact that the absence of MyD88 disrupts Tube membrane localization indicates that MyD88 mediates this membrane association of the presignaling complex. Although it is possible that MyD88 and Toll have a weak but significant association in the absence of Toll activation, it may also be that MyD88 binds other molecules at the plasma membrane, bringing the presignaling complex into proximity with the Toll intracellular domain. Consistent with this latter hypothesis, we have identified mutations in the MyD88 death domain that disrupt signaling, but do not block binding to Tube.
Results with the EGFR–Toll chimera clearly demonstrate that the TIR domain of Toll can be activated by ectodomain dimerization. Furthermore, they provide strong evidence that Drosophila Toll acts as a homodimer. For EGFR, it is known that the ectodomain is blocked from dimerization by interactions that are eliminated upon ligand binding (Schlessinger, 2002). By analogy, we argue that dimerization of the Toll ectodomain is also blocked in the absence of ligand. Support for this model comes from the observation of Winans and Hashimoto that deletion of the ectodomain, the ΔLRR form, constitutively activates Toll (Winans and Hashimoto, 1995). Furthermore, binding studies indicate that Spätzle can induce dimerization of the Toll extracellular domain (Weber et al, 2003). We imagine then that Spätzle-mediated dimerization of the Toll ectodomain dimerizes the intracellular TIR domain, thus triggering signaling. Dimerization is likely to be a general mechanism for Toll activation, since constitutive dimerization of a number of mammalian Toll-like receptors activates downstream pathways (Zhang et al, 2002).
Pelle activation and regulation
With Tube binding to both MyD88 and Pelle, signaling must involve, at least transiently, a tetrameric association of Toll, MyD88, Tube, and Pelle. However, evidence indicates that the tetrameric signaling complex is not long lived. First, the distribution of Pelle in the embryo is uniform, that is, Pelle, unlike Tube and MyD88, does not exhibit a steady-state gradient of localization in response to Toll signaling (Towb et al, 1998). Second, Pelle that is active as a protein kinase has a diminished association with Tube, relative to catalytically inactive Pelle, in yeast two-hybrid assays (Edwards et al, 1997). It is thus likely that activated Pelle directs its own dissociation from the signaling complex through phosphorylation of targets that include the Tube death domain. Both the dissociation and feedback likely have parallels in mammalian systems, in which IRAK-1 dissociates from the receptor and then associates with downstream proteins, such as TAK1, and in which IRAK family kinases are thought to negatively feed back on the receptor complex (Jiang et al, 2002).
Based on the chain of interactions we have identified and the apparent absence of a kinase acting upstream of Pelle, it appears likely that Toll dimerization drives activation of Pelle via autophosphorylation. Shen and Manley have found that Pelle undergoes autophosphorylation in a concentration-dependent manner (Shen and Manley, 2002) and there is evidence that oligomerization of the Pelle death domain can lead to kinase domain activation (Großhans et al, 1994, 1999). We report here that recruitment of Pelle into a complex with just MyD88 and Tube promotes efficient autophosphorylation of Pelle (Figure 1). We therefore speculate that Pelle is not part of the presignaling death domain complex, but is recruited to Toll subsequent to MyD88 and Tube.
Bivalent death domain as an adapter
Our data suggest that the Tube death domain serves as a docking site for simultaneous binding of two other death domain. While it is well known that many death domain form heterodimers, it remains an open question as to whether death domains other than Tube are bivalent. We note that the Tube death domain is polarized, in that positively charged residues form critical elements of the MyD88 binding site and negatively charged residues mediate interaction with Pelle (Xiao et al, 1999). Furthermore, these surfaces interact with residues of opposite charge in the death domain of MyD88 and Pelle. A second example of such an architecture may be the interaction of the caspase recruitment domains (CARDs) of Apaf-1 and procaspase-9, members of the death domain superfamily. Each of these domains has a polarized surface, with regions of opposite charge interacting and the remaining charged surfaces facing outward (Qin et al, 1999).
Evidence from mammalian systems indicates that signaling may involve not dimers and trimers, but rather larger complexes formed from multiple subunits of pathway components such as MyD88 and IRAK proteins (Jiang et al, 2002). Similarly, our data, while pointing out the architecture of dimeric protein interactions, are consistent with the formation of higher order assemblies (Towb et al, 1998). Indeed, the punctate immunolocalization pattern of MyD88 is suggestive of such aggregates.
Although mammals lack a Tube ortholog, particular IRAK isoforms might, like Tube, act as adapter/scaffolding molecules. In support of this suggestion, we note genetic and biochemical studies showing that the four IRAK isoforms are not functionally equivalent (Thomas et al, 1999; Kobayashi et al, 2002; Suzuki et al, 2002). Indeed, the kinase domains of both IRAK-2 and IRAK-M lack an aspartic acid residue required for catalytic activity (Janssens and Beyaert, 2003). Interaction between different IRAK isoforms has been observed in vitro (Wesche et al, 1999), and in vivo association of IRAK-1 or IRAK-4 with MyD88 requires the presence of IRAK-M (Kobayashi et al, 2002). It is thus possible that the death domain of one IRAK bridges the interaction of MyD88 with another IRAK, forming a heterotrimeric death domain complex analogous to that which we have characterized in flies.
Materials and methods
Reagents
S2 cell expression plasmids are all based on the vectors of the Drosophila Expression System (Invitrogen). Tube, MyD88, and Toll10B (C781Y) mutants were generated by PCR sewing (Ho et al, 1989). The chimeric EFGR–Toll protein was constructed by fusing the coding region of EGFR residues 1–669 with that of Toll residues 829–1097 using the same PCR-based method. The Drosomycin–luciferase construct was provided by J.-L. Imler (Tauszig et al, 2000). Anti-V5 and anti-myc (9E10) antibodies were purchased from Invitrogen and Santa Cruz Biotechnology, Inc., respectively. Recombinant human EGF was purchased from Calbiochem. Anti-Tube, anti-Cactus, anti-Dorsal, and anti-MyD88 antisera were described previously (Letsou et al, 1993; Gillespie and Wasserman, 1994; Reach et al, 1996; Sun et al, 2002). The antiphosphotyrosine antibody (monoclonal PY20) was purchased from Chemicon International, Inc.
S2 cell assays
S2 cells were maintained, transfected, and lysed as described previously (Sun et al, 2002). S2 cells transfected with pAc5.1/V5-His carrying EGFR–Toll were selected for stable expression as specified for the Drosophila Expression System (Invitrogen). For EGF treatment, the cell culture was fed with fresh serum-free medium 4 h before adding EGF. The EGF stimulation was terminated by resuspension of the treated cells in ice-cold PBS. Cells were pelleted by centrifugation for 5 min at 3000 rpm; cell pellets were resuspended in 300 μl (per 35 mm well of cells) of either ice-cold lysis buffer (50 mM Tris, pH 7.8, 150 mM NaCl, and 1% Nonidet P-40, for immunoprecipitation) supplemented with protease inhibitor cocktail (Roche Diagnostics) or Reporter Lysis Buffer (Promega, for reporter assay). After a freeze–thaw cycle to improve cell lysis, the cell lysates were cleared by centrifugation. An aliquot of lysate was mixed with SDS–PAGE sample buffer for immunoblot analysis. The reporter assay and immunoprecipitation assay was conducted essentially as described before (Sun et al, 2002). Each data point of the reporter assay shown in Results is the mean±s.d. of at least three independent assays.
RNA synthesis, embryo injection, and cuticle preparation
DNA templates of wild-type and mutant forms of MyD88 and Tube were PCR-amplified directly from the appropriate pMT/V5-His constructs, using primers that incorporated an Sp6 promoter sequence at the 5′ end. As in the cell culture experiments, each construct includes a V5 tag and a 6xHis tag (for MyD88) or a Myc tag (for Tube). RNA synthesis and sample preparation were as described previously (Xiao et al, 1999). The recipient myd88 null embryos were obtained from maternal flies of genotype kra1/kra1 for MyD88 construct injections (Charatsi et al, 2003); for Tube construct injections the maternal flies were genotype tubR5.6/Df(3R)XM3. Embryos were collected, microinjected, prepared, and scored as previously described (Wieschaus and Nüsslein-Volhard, 1986).
Immunofluorescence microscopy
Embryos were collected, fixed, and devitellinized essentially as described before (Galindo et al, 1995). The anti-MyD88 antibody was diluted (1:1000) in PBTA (1 × PBS, 1% BSA, 0.05% Triton X-100, 0.02% NaN3) and purified by preabsorption with fixed kra1 embryos at 4°C for about 2 days. For antiphosphotyrosine (PY20) and anti-MyD88 staining, embryos were incubated with PY20 (1:500) and anti-MyD88 overnight at 4°C and then washed five times in PBTA over a 1.5-h period. The secondary antibodies, Cy3-conjugated goat anti-mouse IgG and Cy5-conjugated goat anti-rabbit IgG (Jackson Immunoresearch), were diluted (1:1000) in PBTA and applied for 1 h at room temperature. Embryos were then washed six times over 1.5 h with PBTA and mounted on slides in Fluoromount G (Southern Biotechnology Associates). The anti-Tube staining and double labeling experiments with anti-Dorsal serum were carried out as described previously (Reach et al, 1996; Towb et al, 2001). Laser scanning confocal microscopy was used to analyze embryos labeled with fluorescent antibodies. Images were collected with a Leica SP2 AOBS Vis–UV microscope and processed with Leica TCS Confocal Software.
Acknowledgments
We thank Laura Hull for the construction of the EGFR-Toll chimera, Bernard Moussian for MyD88 mutant flies, Jean-Luc Imler for the luciferase reporter construct, Zhimin Lu and Tony Hunter for human EGFR cDNA, Ethan Bier and William McGinnis for use of their confocal microscope, James Chen and Stephen Sprang for assistance on the protein structure illustration, Kristin Benjamin for technical assistance, and Ethan Bier, Anthea Letsou, and Richard Firtel for helpful comments on the manuscript. The research described here was supported by National Institutes of Health Grant 5R01-GM50545 to SAW and ACS Grant PF-01-097-01-DDC to PT DNC was supported by a Julia Brown Undergraduate Scholarship.
References
- Anderson KV, Jurgens G, Nüsslein-Volhard C (1985) Establishment of dorsal–ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell 42: 779–789 [DOI] [PubMed] [Google Scholar]
- Belvin MP, Anderson KV (1996) A conserved signaling pathway: the Drosophila Toll–Dorsal pathway. Annu Rev Cell Dev Biol 12: 393–416 [DOI] [PubMed] [Google Scholar]
- Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J (2003) Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med 197: 263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charatsi I, Luschnig S, Bartoszewski S, Nüsslein-Volhard C, Moussian B (2003) Krapfen/dMyd88 is required for the establishment of dorsoventral pattern in the Drosophila embryo. Mech Dev 120: 219–226 [DOI] [PubMed] [Google Scholar]
- De Gregorio E, Spellman PT, Tzou P, Rubin GM, Lemaitre B (2002) The Toll and Imd pathways are the major regulators of the immune response in Drosophila. EMBO J 21: 2568–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drier EA, Steward R (1997) The dorsoventral signal transduction pathway and the Rel-like transcription factors in Drosophila. Semin Cancer Biol 8: 83–92 [DOI] [PubMed] [Google Scholar]
- Edwards DN, Towb P, Wasserman SA (1997) An activity-dependent network of interactions links the Rel protein Dorsal with its cytoplasmic regulators. Development 124: 3855–3864 [DOI] [PubMed] [Google Scholar]
- Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E (1995) The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci 20: 342–344 [DOI] [PubMed] [Google Scholar]
- Galindo RL, Edwards DN, Gillespie SK, Wasserman SA (1995) Interaction of the pelle kinase with the membrane-associated protein tube is required for transduction of the dorsoventral signal in Drosophila embryos. Development 121: 2209–2218 [DOI] [PubMed] [Google Scholar]
- Gillespie SK, Wasserman SA (1994) Dorsal, a Drosophila Rel-like protein, is phosphorylated upon activation of the transmembrane protein Toll. Mol Cell Biol 14: 3559–3568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Großhans J, Bergmann A, Haffter P, Nüsslein-Volhard C (1994) Activation of the kinase Pelle by Tube in the dorsoventral signal transduction pathway of Drosophila embryo. Nature 372: 563–566 [DOI] [PubMed] [Google Scholar]
- Großhans J, Schnorrer F, Nüsslein-Volhard C (1999) Oligomerisation of Tube and Pelle leads to nuclear localisation of dorsal. Mech Dev 81: 127–138 [DOI] [PubMed] [Google Scholar]
- Hecht PM, Anderson KV (1993) Genetic characterization of tube and pelle, genes required for signaling between Toll and dorsal in the specification of the dorsal–ventral pattern of the Drosophila embryo. Genetics 135: 405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59 [DOI] [PubMed] [Google Scholar]
- Hoffmann JA, Reichhart JM (2002) Drosophila innate immunity: an evolutionary perspective. Nat Immunol 3: 121–126 [DOI] [PubMed] [Google Scholar]
- Horng T, Medzhitov R (2001) Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc Natl Acad Sci USA 98: 12654–12658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultmark D (1994) Macrophage differentiation marker MyD88 is a member of the Toll/IL-1 receptor family. Biochem Biophys Res Commun 199: 144–146 [DOI] [PubMed] [Google Scholar]
- Janssens S, Beyaert R (2002) A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci 27: 474–482 [DOI] [PubMed] [Google Scholar]
- Janssens S, Beyaert R (2003) Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell 11: 293–302 [DOI] [PubMed] [Google Scholar]
- Jebanathirajah JA, Peri S, Pandey A (2002) Toll and interleukin-1 receptor (TIR) domain-containing proteins in plants: a genomic perspective. Trends Plant Sci 7: 388–391 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Ninomiya-Tsuji J, Qian Y, Matsumoto K, Li X (2002) Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol Cell Biol 22: 7158–7167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110: 191–202 [DOI] [PubMed] [Google Scholar]
- Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA (1996) The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86: 973–983 [DOI] [PubMed] [Google Scholar]
- Letsou A, Alexander S, Orth K, Wasserman SA (1991) Genetic and molecular characterization of tube, a Drosophila gene maternally required for embryonic dorsoventral polarity. Proc Natl Acad Sci USA 88: 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsou A, Alexander S, Wasserman SA (1993) Domain mapping of tube, a protein essential for dorsoventral patterning of the Drosophila embryo. EMBO J 12: 3449–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y (1999) Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399: 549–557 [DOI] [PubMed] [Google Scholar]
- Reach M, Galindo RL, Towb P, Allen JL, Karin M, Wasserman SA (1996) A gradient of cactus protein degradation establishes dorsoventral polarity in the Drosophila embryo. Dev Biol 180: 353–364 [DOI] [PubMed] [Google Scholar]
- Schlessinger J (2002) Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110: 669–672 [DOI] [PubMed] [Google Scholar]
- Shelton CA, Wasserman SA (1993) Pelle encodes a protein kinase required to establish dorsoventral polarity in the Drosophila embryo. Cell 72: 515–525 [DOI] [PubMed] [Google Scholar]
- Shen B, Manley JL (1998) Phosphorylation modulates direct interactions between the Toll receptor, Pelle kinase and Tube. Development 125: 4719–4728 [DOI] [PubMed] [Google Scholar]
- Shen B, Manley JL (2002) Pelle kinase is activated by autophosphorylation during Toll signaling in Drosophila. Development 129: 1925–1933 [DOI] [PubMed] [Google Scholar]
- Sun H, Bristow BN, Qu G, Wasserman SA (2002) A heterotrimeric death domain complex in Toll signaling. Proc Natl Acad Sci USA 99: 12871–12876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC (2002) Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416: 750–756 [DOI] [PubMed] [Google Scholar]
- Tauszig S, Jouanguy E, Hoffmann JA, Imler JL (2000) Toll-related receptors and the control of antimicrobial peptide expression in Drosophila. Proc Natl Acad Sci USA 97: 10520–10525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauszig-Delamasure S, Bilak H, Capovilla M, Hoffmann JA, Imler JL (2002) Drosophila MyD88 is required for the response to fungal and Gram-positive bacterial infections. Nat Immunol 3: 91–97 [DOI] [PubMed] [Google Scholar]
- Thomas JA, Allen JL, Tsen M, Dubnicoff T, Danao J, Liao XC, Cao Z, Wasserman SA (1999) Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol 163: 978–984 [PubMed] [Google Scholar]
- Towb P, Bergmann A, Wasserman SA (2001) The protein kinase Pelle mediates feedback regulation in the Drosophila Toll signaling pathway. Development 128: 4729–4736 [DOI] [PubMed] [Google Scholar]
- Towb P, Galindo RL, Wasserman SA (1998) Recruitment of Tube and Pelle to signaling sites at the surface of the Drosophila embryo. Development 125: 2443–2450 [DOI] [PubMed] [Google Scholar]
- Wasserman SA (2000) Toll signaling: the enigma variations. Curr Opin Genet Dev 10: 497–502 [DOI] [PubMed] [Google Scholar]
- Weber AN, Tauszig-Delamasure S, Hoffmann JA, Lelievre E, Gascan H, Ray KP, Morse MA, Imler JL, Gay NJ (2003) Binding of the Drosophila cytokine Spatzle to Toll is direct and establishes signaling. Nat Immunol 4: 794–800 [DOI] [PubMed] [Google Scholar]
- Wesche H, Gao X, Li X, Kirschning CJ, Stark GR, Cao Z (1999) IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J Biol Chem 274: 19403–19410 [DOI] [PubMed] [Google Scholar]
- Wieschaus E, Nüsslein-Volhard C (1986) Looking at embryos. In Drosophila: A Practical Approach, Roberts DB (ed) pp 199–227. Oxford: IRL Press [Google Scholar]
- Winans KA, Hashimoto C (1995) Ventralization of the Drosophila embryo by deletion of extracellular leucine-rich repeats in the Toll protein. Mol Biol Cell 6: 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Towb P, Wasserman SA, Sprang SR (1999) Three-dimensional structure of a complex between the death domains of Pelle and Tube. Cell 99: 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Tay PN, Cao W, Li W, Lu J (2002) Integrin-nucleated Toll-like receptor (TLR) dimerization reveals subcellular targeting of TLRs and distinct mechanisms of TLR4 activation and signaling. FEBS Lett 532: 171–176 [DOI] [PubMed] [Google Scholar]