

Abstract
The signaling pathway of insulin/insulin-like growth factor-1/phosphatidylinositol-3 kinase/Akt is known to regulate longevity as well as resistance to oxidative stress in the nematode Caenorhabditis elegans. This regulatory process involves the activity of DAF-16, a forkhead transcription factor. Although reduction-of-function mutations in components of this pathway have been shown to extend the lifespan in organisms ranging from yeast to mice, activation of Akt has been reported to promote proliferation and survival of mammalian cells. Here we show that Akt activity increases along with cellular senescence and that inhibition of Akt extends the lifespan of primary cultured human endothelial cells. Constitutive activation of Akt promotes senescence-like arrest of cell growth via a p53/p21-dependent pathway, and inhibition of forkhead transcription factor FOXO3a by Akt is essential for this growth arrest to occur. FOXO3a influences p53 activity by regulating the level of reactive oxygen species. These findings reveal a novel role of Akt in regulating the cellular lifespan and suggest that the mechanism of longevity is conserved in primary cultured human cells and that Akt-induced senescence may be involved in vascular pathophysiology.
Keywords: aging, Akt, endothelial cells, senescence
Introduction
Cellular senescence is the limited ability of primary human cells to divide when cultured in vitro and is accompanied by a specific set of changes in cell morphology, gene expression and function. These phenotypic changes have been implicated in human aging (Faragher and Kipling, 1998). This hypothesis, the cellular hypothesis of aging, was established by Hayflick (1975) and is supported by evidence that the replicative potential of primary cultured human cells is dependent on donor age and that the growth potential of cultured cells correlates well with the mean maximum lifespan of the species from which the cells are derived (Rohme, 1981), although some conflicting data have been reported (Cristofalo et al, 1998). Primary cultured cells obtained from patients with premature aging syndromes, such as Werner syndrome and Bloom syndrome, are known to have a shorter lifespan than the cells from age-matched healthy populations (Rohme, 1981; Thompson and Holliday, 1983), further supporting this hypothesis. Cell division is essential for the survival of multicellular organisms that contain renewable tissues, but also places the organism at the risk of developing cancer. It has been suggested that complex organisms have evolved at least two cellular mechanisms to prevent oncogenic transformation, which are apoptosis and cellular senescence (Campisi, 2001). Accordingly, age-associated diseases could be regarded as a by-product of the tumor suppressor mechanism, cellular senescence (Weinstein and Ciszek, 2002).
Many molecular mechanisms have been suggested to contribute to human aging and its associated diseases. Recent genetic analyses have demonstrated that reduction-of-function mutations in the signaling pathway of insulin/insulin-like growth factor-1 (IGF-1)/phosphatidylinositol-3 kinase (PI3K)/Akt (also known as protein kinase B) extend the longevity of the nematode Caenorhabditis elegans (Kenyon et al, 1993; Morris et al, 1996; Paradis and Ruvkun, 1998; Guarente and Kenyon, 2000; Kenyon, 2001; Lee et al, 2001; Lin et al, 2001; Longo and Finch, 2003). The forkhead transcription factor DAF-16, which is phosphorylated and thereby inactivated by Akt (Lee et al, 2001; Lin et al, 2001), plays an essential role in this longevity pathway (Lin et al, 1997; Ogg et al, 1997). More recently, it has been reported that the genes regulating longevity are conserved in organisms ranging from yeast to mice. Mutation of Sch9, which is homologous to Akt, extends the lifespan of yeast (Fabrizio et al, 2001), and mutations that decrease the activity of the insulin/IGF-1-like pathway increase the longevity of fruit flies (Tatar et al, 2001) and mice (Bluher et al, 2003; Holzenberger et al, 2003). These mutations that extend the lifespan are associated with increased resistance to oxidative stress, which is partly mediated by the increased expression of antioxidant genes (Honda and Honda, 1999; Fabrizio et al, 2003; Murphy et al, 2003).
In mammalian cells, activation of Akt has been reported to induce proliferation and survival, thereby promoting tumorigenesis (Datta et al, 1999; Blume-Jensen and Hunter, 2001; Testa and Bellacosa, 2001). Overexpression of Akt can transform NIH3T3 cells (Cheng et al, 1997), while introduction of Akt antisense RNA inhibits the tumorigenic phenotype of cancer cells expressing high levels of Akt (Cheng et al, 1996). The mechanisms by which Akt promotes cell proliferation and survival are likely to be multifactorial, because it has been reported to directly phosphorylate several components of the cell cycle machinery as well as the cell death machinery (Datta et al, 1999). Akt counteracts the effect of cyclin-dependent kinase inhibitors on cell cycle progression by modulating their intracellular localization and level of transcription (Medema et al, 2000; Shin et al, 2002; Viglietto et al, 2002; Zhou et al, 2001a). Akt also increases the cyclin D1 level by inhibiting its degradation, which is important in the G1/S phase transition (Diehl et al, 1998). Moreover, it is known that Akt phosphorylates and inactivates proapoptotic factors such as BAD (Datta et al, 1997; del Peso et al, 1997) and procaspase-9 (Cardone et al, 1998), thereby promoting cell survival. Although these reports have suggested an important role of Akt in human malignancy (Blume-Jensen and Hunter, 2001; Testa and Bellacosa, 2001), it has mainly been examined in immortal cell lines and the impact of Akt activation on the growth and lifespan of primary cultured human cells is unknown.
In the present study, we found that inhibition of Akt could prolong the lifespan of primary cultured human endothelial cells, whereas constitutive activation of Akt promoted senescence-like growth arrest via a p53/p21-dependent pathway. Akt-induced growth arrest was inhibited by a mutated forkhead transcription factor that was resistant to Akt phosphorylation. These findings disclose a novel role of Akt in regulating the lifespan of cells and suggest that the mechanism of longevity is conserved in primary cultured human cells.
Results
Akt activation reduces the lifespan of human endothelial cells
We first investigated whether Akt activity was associated with cellular senescence of primary cultured human endothelial cells. Senescent endothelial cells had higher phospho-Akt levels than young endothelial cells (Figure 1A). To assess the actual role of Akt activity in regulating cellular lifespan, we infected primary cultured human endothelial cells with a retroviral vector encoding either constitutively activated myc-tagged Akt (AktCA) or dominant-negative myc-tagged Akt (AktDN). The empty retroviral vector pLNCX (Mock), encoding a neomycin resistance gene alone, was also transduced into endothelial cells as a control. Infected cells were purified using G418 for 7 days and then recultured until the cells underwent senescence. The 8th day after infection is designated as day 0 in all of the following experiments. Western blot analysis with anti-c-Myc antibody and anti-Akt antibody demonstrated that both AktCA and AktDN proteins were expressed by the endothelial cells, showing an approximately 4- to 8-fold increase of total Akt protein compared to endogenous Akt protein (Figure 1B). Long-term culture studies showed that constitutive activation of Akt significantly shortened the lifespan of the endothelial cells, whereas inhibition of Akt activity delayed senescence compared with mock-infected cells (Figure 1B). Introduction of AktDN influenced cellular lifespan in the late passages, but not in the early passages, suggesting that Akt activity increased with further cell division and thus promoted senescence. Expression of AktCA markedly reduced cell growth by day 7 (Figure 1C). AktCA-transduced endothelial cells were flattened and enlarged, while mock- or AktDN-infected endothelial cells exhibited normal morphology and growth (Figures 1C and D). Senescence-associated β-galactosidase activity was also increased in AktCA-transduced cells (Figure 1D). These changes of the phenotype, which were suggestive of senescence, were observed in various types of endothelial cells including microvascular endothelial cells (Supplementary Figure 1). The same senescence-like changes also occurred in confluent endothelial cells (Supplementary Figure 2). Thus, constitutive activation of Akt induced a senescence-like phenotype in human endothelial cells irrespective of the cell type and growth pattern. To further explore the relationship between Akt activity and cell growth, we isolated clones from AktCA-infected endothelial cells and determined the phospho-Akt level and the cell number on day 30. Clones obtained from mock-infected cell populations could be expanded up to 1–3 × 106 cells on average and revealed little Akt activity (Figure 1E, lane 1). In contrast, most of the AktCA-infected clones showed almost complete growth arrest and high levels of phospho-Akt expression (Figure 1E, lanes 5–7). However, some AktCA-infected clones showed low phospho-Akt levels and continued to proliferate (Figure 1E, lanes 2–4). Such proliferating populations may lead to underestimation of the growth inhibitory effect of AktCA in long-term culture experiments. The level of phospho-Akt was inversely correlated with the number of cells on day 30 (Figure 1E, right graph). Thus, we concluded that Akt is a negative regulator of the lifespan of primary cultured human endothelial cells.
Figure 1.
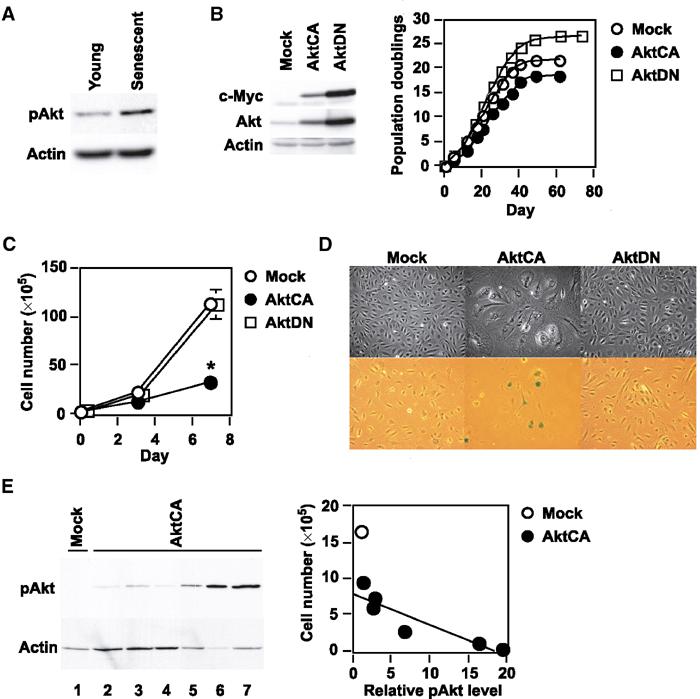
Akt negatively regulates the lifespan of primary cultured human endothelial cells. (A) Whole-cell lysates (30 μg) of young (passage 4) or senescent (passages 14–15) human endothelial cells were analyzed for the expression of phospho-Akt (pAkt, Ser473) and actin (loading control) by Western blotting. (B) Human endothelial cells were infected with pLNCX (Mock), AktCA or AktDN. After purification, infected cell populations were passaged until they underwent senescence, and the number of cumulative population doublings was determined. Similar results were obtained from three independent experiments. To validate the transduction of AktCA and AktDN, whole-cell lysates (30 μg) of each infected population were examined for the expression of exogenous myc-tagged Akt (c-Myc) and total Akt (Akt). (C) Human endothelial cells infected with pLNCX (Mock), AktCA or AktDN were purified with G418 for 7 days and seeded at a density of 3 × 105 cells per 100 mm plate on day 0. Cell number per 100 mm plate was then counted at indicated time points. *P<0.001 versus Mock, ANOVA, n=4. (D) Cell morphology (upper panel) and senescence-associated β-galactosidase staining (lower panel) in endothelial cells infected with pLNCX (Mock), AktCA or AktDN. (E) Independent clones were isolated from pLNCX (Mock)- or AktCA-infected endothelial cells. At 30 days after isolation, the cell number of each clone was counted. Whole-cell lysates (∼10 μg) of isolated clones were also prepared and analyzed for the expression of phospho-Akt by Western blotting (left panel, mock-infected clone for lane 1 and AktCA-infected clones for lanes 2–7). The cell number of each clone was as follows: 16.6 × 105 for lane 1; 6–10 × 105 for lanes 2–4; 0.1–2 × 105 for lanes 5–7. As the availability of samples was limited in the case of most AktCA-infected clones, the lysates used were less than 10 μg (lanes 5–7). Therefore, the levels of phospho-Akt were standardized on the basis of actin expression, and the relative level of phospho-Akt and the cell number of each clone were plotted in the graph (right panel, r=0.92, P<0.01). The corrected value of phospho-Akt in mock-infected clones (lane 1) is set at 1.
Upregulation of p21 is essential for Akt-induced growth arrest
To clarify the mechanism of cell growth arrest induced by activation of Akt, we examined the expression of cell cycle regulatory proteins. Expression of p53 and p21Waf1/Cip1, but not p16Ink4a, was elevated, while the level of phosphorylated Rb was decreased in AktCA-infected cells compared with mock-infected cells (Figure 2A), suggesting that Akt may induce growth arrest by upregulating p53 and p21. To determine the role of p21 in Akt-induced cell growth arrest, we infected primary cultured mouse embryonic fibroblasts (MEF) derived from p21-deficient or wild-type mice with AktCA. Similar to endothelial cells, the growth of wild-type MEF was markedly reduced by activation of Akt compared with mock infection (Figure 2B, p21+/+). In contrast, Akt-induced cell growth arrest was restored in p21-deficient MEF (Figure 2B, p21−/−), suggesting that p21 is essential for Akt-induced growth arrest of these cells. It has been reported that expression of p21 is regulated by p53-dependent or -independent transcriptional mechanisms (el-Deiry et al, 1993) as well as protein degradation (Maki and Howley, 1997). To investigate the mechanism by which Akt activation increases p21 expression, we assessed the stability of p21 protein and the extent of p21 transcription. The half-life of p21 protein did not differ between mock- and AktCA-infected endothelial cells (Figure 2C). Northern blot analysis revealed that the level of p21 mRNA was significantly increased in Akt-infected cells compared with mock-infected cells (Figure 2D). Activation of Akt enhanced transcription of the luciferase reporter gene controlled by the promoter fragment of the human p21 gene (Figure 2E), indicating that activation of Akt caused the transcriptional upregulation of p21 expression.
Figure 2.
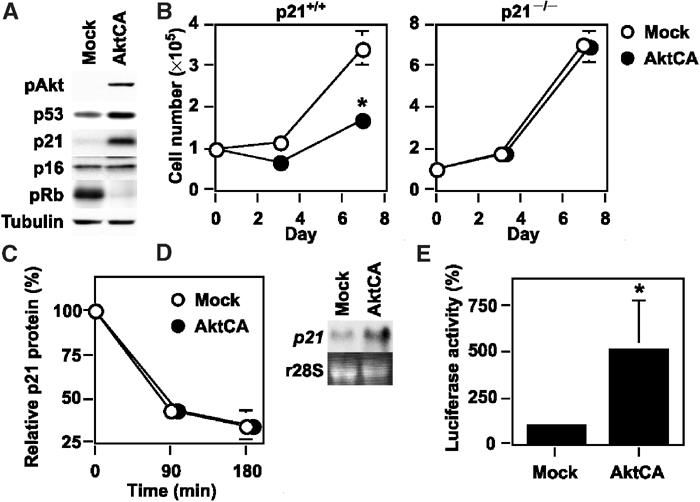
Upregulation of p21 is essential for Akt-induced growth arrest. (A) Whole-cell lysates (30 μg) of pLNCX (Mock)- or AktCA-infected endothelial cells on day 0 were examined for the expression of phospho-Akt (pAkt), cell cycle regulatory proteins and tubulin (loading control) by Western blotting. (B) MEF derived from wild-type (p21+/+) or p21-deficient mice (p21−/−) were infected with pLNCX (Mock) or AktCA, purified with G418 for 7 days and seeded at a density of 1 × 105 cells per 100 mm plate on day 0. Cell number per 100 mm plate was then counted at indicated time points. *P<0.001 versus Mock, ANOVA, n=4. (C) Human endothelial cells infected with pLNCX (Mock) or AktCA were treated with cycloheximide (10 μg/ml) for the indicated time interval. Whole-cell lysates (30 μg) were then prepared at each time point and assayed for the expression of p21 and actin (loading control) by Western blotting. The graph indicates the results of densitometric analysis for the levels of p21 protein relative to actin expression. The value at time 0 is set at 100%. (D) Total RNA (30 μg) was extracted from human endothelial cells infected with pLNCX (Mock) or AktCA and analyzed for p21 mRNA levels by Northern blotting (upper panel). Ribosomal RNA was used as an internal control (lower panel). (E) The luciferase reporter gene plasmid controlled by the promoter of the human p21 gene was transfected into endothelial cells infected with pLNCX (Mock) or AktCA 24 h before the luciferase activity was measured. The activity in mock-infected cells is set at 100%. *P<0.05 versus Mock, paired t-test, n=4.
Critical role of p53 transcriptional activity in Akt-induced growth arrest
To ascertain whether Akt activation induces the transcriptional activity of p53, we transfected Akt-infected endothelial cells with the luciferase reporter gene containing 13 copies of the p53-binding consensus sequence (PG13). Introduction of AktCA induced p53 promoter-driven luciferase activity compared with mock infection, but not luciferase activity driven by a promoter containing 15 copies of a similar sequence with mutation at critical positions (MG15) (Figure 3A). To further assess the relation between Akt and p53 transcription activity, we tested whether ablation of p53 could circumvent Akt-induced growth arrest. We infected human endothelial cells with a retroviral vector encoding the E6 oncoprotein of HPV16, which binds p53 and facilitates its destruction by ubiquitin-mediated proteolysis (pBabe E6). We also infected the same cells with the empty vector encoding resistance to puromycin alone (pBabe). Both cell populations were then subjected to infection with pLNCX or AktCA. Activation of Akt markedly inhibited the growth of pBabe-infected endothelial cells (Figure 3B, pBabe), while growth inhibition was completely abolished in E6-infected cells (Figure 3B, E6). Changes of cell morphology were also reversed to normal by introduction of E6 (Figure 3C). Ablation of p53 also lessened the decrease in the lifespan of AktCA-infected cells (Supplementary Figure 3). These results indicate a critical role of p53 in Akt-induced cell growth arrest. Introduction of AktCA did not induce p21 expression in E6-infected cells (Figure 3D), suggesting that constitutive activation of Akt increases induction of the transcription of p21 by a p53-dependent mechanism and thereby promotes cell growth arrest.
Figure 3.
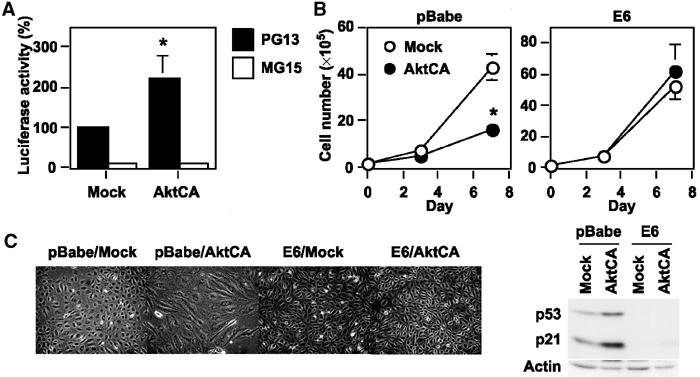
Critical role of p53 transcriptional activity in Akt-induced growth arrest. (A) The luciferase reporter gene plasmid pPG13-Luc containing the p53-binding sequence or pMG15-Luc containing the mutated p53-binding sequence was transfected into endothelial cells infected with pLNCX (Mock) or AktCA 24 h before the luciferase activity was measured. The activity of PG13-Luc in mock-infected cells is set at 100%. *P<0.005 versus Mock, ANOVA, n=4. (B) Human endothelial cells were infected with pBabe (empty vector) or pBabe E6 and purified with puromycin. Infected cells were then transduced with pLNCX or AktCA as described in Figure 1C and seeded at a density of 2 × 105 cells per 100 mm plate on day 0. Cell number was then counted at indicated time points. *P<0.05 versus Mock, ANOVA, n=4. (C) Morphology of cell populations prepared in (B). (D) Whole-cell lysates (30 μg) were extracted from cells prepared in (B) and examined for the expression of p53, p21 and actin (loading control).
Forkhead transcription factor mediates Akt-induced growth arrest
In C. elegans, a reduction-of-function mutation in the PI3K/Akt pathway leads to activation of the forkhead transcription factor DAF-16, resulting in extension of the lifespan, and this effect is inhibited by mutations of antioxidant genes (Murphy et al, 2003). Recent evidence indicates that the mammalian forkhead transcription factor FOXO3a (also known as FKHR-L1) upregulates radical scavenger genes that have a protective effect against oxidative damage in human cells (Kops et al, 2002; Nemoto and Finkel, 2002). To investigate the role of FOXO3a in Akt-induced growth arrest, we examined the expression of FOXO3a and antioxidant genes. Phosphorylated FOXO3a (the inactive form) was increased in AktCA-infected endothelial cells compared with mock-infected cells (Figure 4A). The level of manganese superoxide dismutase (MnSOD), but not catalase, was reduced in AktCA-infected endothelial cells (Figure 4A). Consistent with the decreased level of MnSOD, AktCA-infected endothelial cells exhibited an increase of reactive oxygen species (ROS), as assessed using the redox-sensitive fluorophore 2′,7′-dichlorofluorescein diacetate (DCF) (Figure 4B). Since oxidative stress is postulated to induce the activation of p53 (Finkel and Holbrook, 2000), we examined the effect of an ROS scavenger, N-acetyl cysteine (NAC), on p53 promoter activity (PG13) in AktCA-infected endothelial cells. The enhancement of p53 promoter-driven luciferase activity by AktCA was significantly lessened after treatment with NAC, suggesting that ROS are involved in Akt-induced senescence-like growth arrest (Figure 4C). To further determine the causal link between Akt-induced growth arrest and phosphorylation of FOXO3a, we tested a mutated FOXO3a that was resistant to phosphorylation by Akt. Introduction of this FOXO3a mutant prevented senescence-like growth arrest and cellular morphological changes induced by activation of Akt (Figures 4D and E). Moreover, induction of p21 expression by Akt activation was effectively inhibited by the mutant form of FOXO3a (Figure 4F). These results suggest that constitutive activation of Akt inhibits the transcriptional activity of FOXO3a and thereby downregulates MnSOD, leading to an increase of ROS that promotes senescence-like growth arrest via the p53/p21-dependent pathway (Figure 4G). This signaling pathway could be recaptured in endothelial cells undergoing replicative senescence (Supplementary Figure 4), which suggests that Akt-induced growth arrest is relevant to physiological senescence and may also be involved in human vasculopathy.
Figure 4.
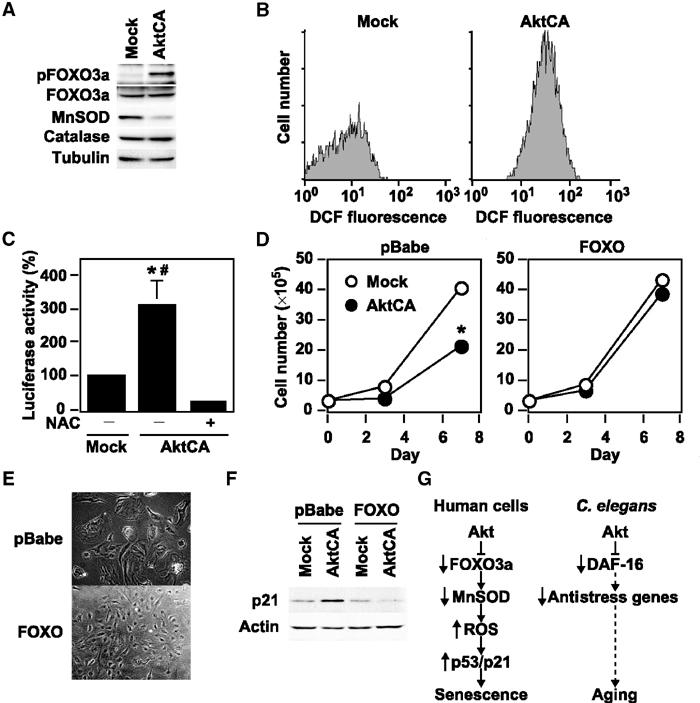
FOXO3a mediates Akt-induced growth arrest via the ROS/p53/p21-dependent mechanisms. (A) Whole-cell lysates (30 μg) of pLNCX (Mock)- or AktCA-infected endothelial cells were examined for expression of phospho-FOXO3a (pFOXO3a, Thr32), total FOXO3a (FOXO3a), MnSOD, catalase and tubulin (loading control) by Western blotting. (B) Human endothelial cells infected with pLNCX (Mock) or AktCA were loaded with DCF for 30 min and analyzed by FACS. Representative results from two independent experiments are shown. (C) The luciferase reporter gene plasmid PG13-Luc was transfected into endothelial cells infected with pLNCX (Mock) or AktCA and cultured in the absence or presence of NAC (0.5 mM). At 24 h after transfection, the luciferase activity was measured. The activity in mock-infected cells is set at 100%. *P<0.01 versus Mock, #P<0.001 versus AktCA+NAC, ANOVA, n=4. (D) Human endothelial cells were infected with pBabe (empty vector) or pBabe mutant FOXO3a (FOXO). Infected cell populations were then transduced with pLNCX (Mock) or AktCA and seeded at a density of 3 × 105 cells per 100 mm plate on day 0. Cell number was then counted at indicated time points. *P<0.05 versus Mock, ANOVA, n=3. (E) Morphology of Akt-infected cell populations prepared in (D). (F) Whole-cell lysates (30 μg) prepared in (D) were examined for the expression of p21 and actin (loading control) by Western blotting. Constitutive activation of Akt inhibits the transcriptional activity of FOXO3a and thereby downregulates MnSOD, leading to an increase of ROS that promotes senescence-like growth arrest via the p53/p21-dependent pathway. (G) Proposed signaling pathway of Akt-induced senescence in human endothelial cells compared with that in C. elegans. Akt inactivates FOXO3a and thereby downregulates its target antioxidant gene MnSOD, leading to an increase of ROS. ROS induces p53 activity, resulting in upregulation of p21 expression, which promotes cellular senescence in human endothelial cells. In C. elegans, the PI3K/Akt pathway also negatively regulates longevity by inactivating DAF-16 activity. This regulatory pathway partly involves the decreased expression of anti-stress genes including SOD.
Pathophysiological role of Akt-induced endothelial cell senescence
To investigate whether atherogenic stimuli could activate Akt in human atheroma tissues, we examined Akt activity in coronary arteries obtained at autopsy from patients who had ischemic heart disease. We detected Akt activity in endothelial cells on the surface of coronary atherosclerotic lesions, but not in those of the internal mammary arteries from the same patients, which showed minimal atherosclerotic changes (Figure 5A). To investigate the potential role of Akt-induced endothelial senescence in the pathogenesis of vasculopathy, we examined the effect of Akt on angiogenic activity and the expression of proinflammatory molecules. Tube formation by AktCA-infected endothelial cells was significantly reduced compared with that by mock-infected cells (Figure 5B). In addition, expression of intercellular adhesion molecule (ICAM)-1 was increased in AktCA-infected endothelial cells (Figure 5C). To further explore the role of Akt-induced senescence, we tested the influence of insulin on endothelial cell senescence. Treatment with insulin at a pathological dose caused increases in phospho-FOXO3a and p53 activity (Figures 6A and B), which were comparable to the changes seen in AktCA-infected cells. This increase was inhibited by introduction of AktDN (data not shown), indicating that it was dependent on Akt activity. Insulin induced p53 activity in a dose-dependent manner (Figure 6B). Moreover, continuous incubation with insulin was found to accelerate senescence of human endothelial cells, and this effect was also dose dependent (Figure 6C). Thus, it is conceivable that constitutive activation of Akt by growth factors may promote endothelial cell senescence and thereby contribute to vascular pathophysiology.
Figure 5.
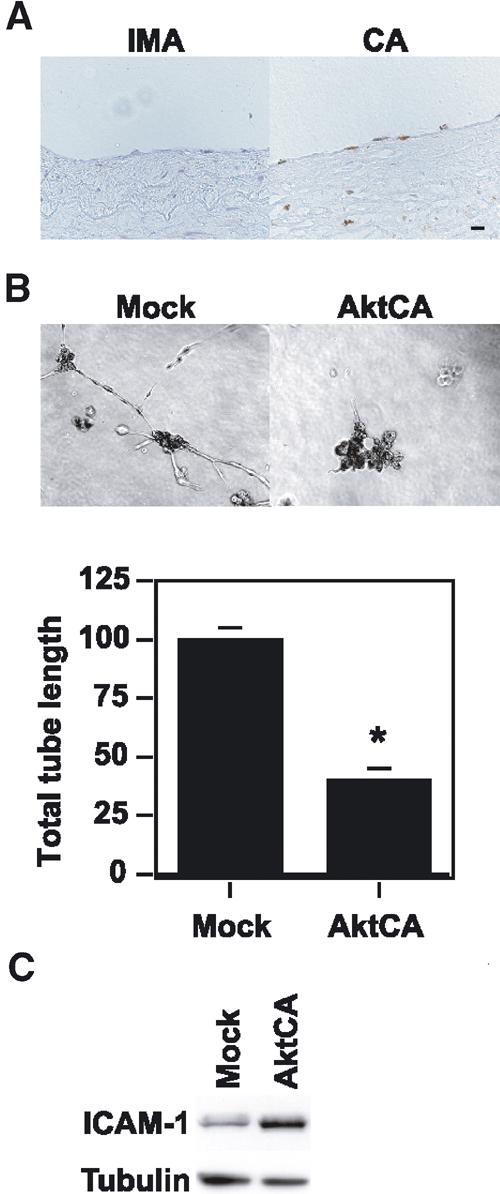
Pathophysiological role of Akt-induced endothelial cell senescence. (A) Immunohistochemistry for phospho-Akt (brown) in the coronary arteries (CA) and the internal mammary arteries (IMA) from the same patients. Scale bar: 10 μm. (B) Tube formation assay. Human endothelial cells infected with pLNCX (Mock) or AktCA were seeded onto Matrigel. After 48 h, the total tube length was estimated by an angiogenesis image analyzer (Kurabo, Osaka, Japan). The graph shows relative tube length in Mock- and AktCA-infected cells. The length in Mock-infected cells is set at 100%. *P<0.005 versus Mock, unpaired t-test, n=4. (C) Whole-cell lysates (30 μg) of pLNCX (Mock)- or AktCA-infected endothelial cells were examined for the expression of ICAM-1 and tubulin (loading control) by Western blotting.
Figure 6.
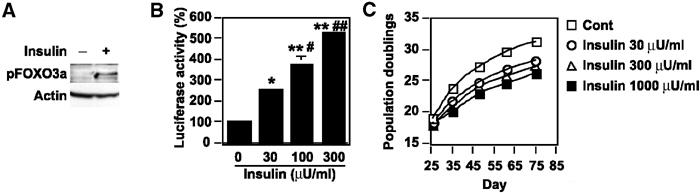
Insulin promotes endothelial cell senescence. (A) Whole-cell lysates (30 μg) of human endothelial cells treated with insulin (1000 μU/ml) for 30 min were analyzed for the levels of phosphorylated FOXO3a and actin (loading control) by Western blotting. (B) The luciferase reporter gene plasmid PG13-Luc was transfected into endothelial cells in the presence of insulin at the indicated dose. At 24 h after transfection, the luciferase activity was measured. The activity in controls is set at 100%. *P<0.05, **P<0.0001 versus control, #P<0.01, ##P<0.001 versus insulin 30 μU/ml, n=4. (C) Human endothelial cells were cultured in the presence of insulin at the indicated dose and passaged. The number of cumulative population doublings was determined (n=3).
Discussion
Our results suggested a critical role of Akt activation in regulating the lifespan of primary cultured human cells in a manner similar to the control of longevity by the PI3K/Akt signaling pathway in C. elegans. We have previously demonstrated that senescent endothelial cells are present in human atherosclerotic plaques, but not nonatherosclerotic lesions, and express high levels of proinflammatory molecules that are known to promote atherogenesis (Minamino et al, 2002). Since Akt is known to be activated by various atherogenic stimuli (Cantley, 2002), our findings imply that constitutive activation of Akt by atherogenic stimuli may induce endothelial cell senescence in atheroma tissue and thereby contribute to atherogenesis. Consistent with this hypothesis, we observed that Akt was phosphorylated in human atheroma but not in normal arteries, and that expression of proinflammatory molecules was increased in AktCA-infected endothelial cells compared with mock-infected cells. Moreover, insulin increased p53 activity via an Akt-dependent mechanism and reduced the lifespan of endothelial cells. Thus, an Akt-induced senescence-like phenotype may be particularly involved in diabetic vasculopathy, since hyperinsulinemia could constitutively activate Akt in endothelial cells.
Although one supposes that Akt-induced senescence might be an artifact, the following points suggest that our findings are valid. First, we observed that Akt activity was increased in endothelial cells undergoing replicative senescence and that inhibition of this endogenous increase in Akt activity by AktDN led to prolongation of the cellular lifespan. This is compatible with many earlier studies demonstrating that reduction-of-function mutations in the insulin/PI3K/Akt pathway extend longevity in organisms ranging from yeast to mice (Longo and Finch, 2003). This signaling pathway (including phosphorylation of FOXO3a and downregulation of MnSOD induced by Akt) could be recaptured in endothelial cells undergoing replicative senescence (Supplementary Figure 4), which suggests that Akt-induced growth arrest may be relevant to physiological senescence. Second, we found that growth was significantly decreased in cloned populations obtained from cells infected with AktCA that exhibited a moderate increase in Akt activity (Figure 1, lanes 2–4). The level of Akt activity in these cells was similar to that in endothelial cells undergoing replicative senescence, suggesting that a physiological level of Akt activation may be able to promote cellular senescence. Third, we found that phospho-FOXO3a levels in AktCA-infected cells were comparable to those in endothelial cells stimulated by insulin at the level seen in patients with type II diabetes. The p53 transcriptional activity in AktCA-infected cells was also similar to that of cells treated with insulin. These results indicate that the pathophysiological activation of Akt was mimicked by infection with AktCA.
Gain-of-function mutations in the PI3K/Akt signaling pathway are frequently found in human cancers (Testa and Bellacosa, 2001). Thus, Akt-induced growth arrest may be another antitumorigenesis mechanism similar to Ras-induced senescence (Serrano et al, 1997; Campisi, 2001; Wright and Shay, 2001). We found that ablation of p53 prevented Akt-induced growth arrest, whereas both the p53- and p16-dependent pathways are reported to be essential for Ras-induced senescence (Serrano et al, 1997; Lin et al, 1998). Oncogenic Ras also induced premature senescence of primary cultured human vascular cells, which was suppressed by inhibition of mitogen-activated protein kinase kinase but not PI3K (Minamino et al, 2003), suggesting that Akt-induced growth arrest may be distinct from Ras-induced senescence. A recent study demonstrated that senescent cells could potentiate the oncogenic transformation of nearby normal cells (Krtolica et al, 2001), which suggests that induction of senescence by Akt as well as Ras may actually be pro-oncogenic.
We found that Akt increased the transcriptional activity of p53, resulting in upregulation of p21 in primary cultured human endothelial cells. Our results are consistent with previous reports that Akt mediates induction of p21 expression by various stimuli in myoblasts and vascular cells (Lawlor and Rotwein, 2000a, 2000b; Schonherr et al, 2001). However, Akt is also reported to induce cytoplasmic localization of p21 (Zhou et al, 2001a), thereby promoting cell proliferation, and to promote nuclear translocation of Mdm2 (a negative regulator of p53), leading to a reduction of both p53 levels and transactivation (Mayo and Donner, 2001; Zhou et al, 2001b). Such changes were not observed in human endothelial cells (H Miyauchi, unpublished data). It is noteworthy that most other studies have examined immortal cells, in which the normal cell cycle machinery might be impaired, and the effects of constitutive Akt activation have not been explored. Although the effects of tissue-specific transgenic expression of constitutively activated Akt alleles have been reported in several different murine models, most of these animals do not develop tumors (Vivanco and Sawyers, 2002), suggesting that activation of Akt is insufficient to cause cancer unless combined with other oncogenic stimuli. Thus, like Ras, Akt may promote cell proliferation and survival or senescence-like growth arrest, depending on various factors including the cellular context as well as the duration and extent of its activation.
In conclusion, we found that Akt negatively regulates the lifespan of primary cultured human endothelial cells via the p53/p21-dependent pathway, and this action is mediated at least partly by the forkhead transcription factor that regulates cellular ROS levels. Our data not only support the previous findings about the signaling pathway for longevity in C. elegans, but also provide a novel insight for research on the treatments of human vasculopathy and cancer.
Materials and methods
Cell culture
Human aortic endothelial cells, human dermal microvascular endothelial cells and human umbilical vein endothelial cells were purchased from Bio Whittaker (Walkersville, MD), and cultured according to the manufacturer's instructions. These cells gave similar results (data not shown). We defined senescent cells as the cultures that do not increase in the cell number and remain subconfluent for 2 weeks. We confirmed the senescent phenotype with SA-β-gal activity assay. Wild-type and p21-deficient MEFs were prepared from day 13.5 embryos derived from crosses between p21+/− mice (Jackson, Bar Harbor, ME) and cultured in DMEM plus 10% fetal bovine serum. Senescence-associated β-galactosidase staining was performed as described (Minamino et al, 2002). Tube formation assay was performed according to the manufacturer's instructions (BioCoat Angiogenesis System, Clontech, Palo Alto, CA).
Retroviral infection
The following plasmids were used for generating retroviruses: pLNCX (Clontech, Palo Alto, CA) and pBabe (a gift from Dr CW Lowe, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). We created the pLNCX-based vector expressing a constitutively active form of Akt (AktCA) or a dominant-negative form of Akt (AktDN) by using the fragment derived from the plasmid pBS-CA-Akt or pBS-DN-Akt (Fujishiro et al, 2001), respectively (a gift from Dr T Asano, Tokyo University, Tokyo, Japan). The pBabe-based vector expressing a mutant form of FOXO3a was constructed by using the fragment derived from pECE FOXO3aTM (Brunet et al, 1999) (a gift from Dr ME Greenberg, Harvard Medical School, Boston, MA). We also constructed the pBabe-based vector expressing E6 (pBabe E6). Details of the construct are available upon request. Retroviral stocks were generated by transient transfection of packaging cell line (PT67, Clontech) and stored at −80°C until use. Human endothelial cells (passage 4–6) were plated at 5 × 105 cells per 100-mm-diameter dish 24 h before infections. For infections, the culture medium was replaced by retroviral stocks supplemented with 8 μg/ml polybrene (Sigma, Tokyo, Japan). At 48 h after infections, the infected cell populations were selected by culture in 500 μg/ml G418 for 7 days (pLNCX-based vectors). After selection, 1–3 × 105 cells were seeded onto 100-mm-diameter dishes on the 8th day postinfection. The 8th day after infection is designated as day 0. For double infection, endothelial cells were infected with pBabe, pBabe FOXO3a or pBabe E6 purified with 0.8 μg/ml puromycin for 4 days and subjected to the second infection as described above.
Western blotting and antibodies
Whole-cell lysates (30 μg) were resolved by SDS polyacrylamide gel electrophoresis (PAGE). Proteins were transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA) and incubated with the first antibody followed by an anti-rabbit immunoglobulin G–horseradish peroxidase antibody or anti-mouse immunoglobulin G–horseradish peroxidase antibody (Jackson, West Grove, PA). Specific proteins were detected using enhanced chemiluminescence (Amersham, Tokyo, Japan). The first antibodies used for Western blotting are as follows: antibodies to Akt, p53, ICAM-1, actin and tubulin (Santa Cruz, Santa Cruz, CA); antibodies to retinoblastoma protein and p16 (Pharmingen, Tokyo, Japan); anti-p21 antibody (Oncogene, Cambridge, MA); anti-phospho-Akt (Ser473) antibody (Cell Signaling, Beverly, MA); anti-catalase antibody (Sigma); antibodies to FOXO3a, phospho-FOXO3a (Thr32) and MnSOD (Upstate Biotechnology, Lake Placid, NY).
Northern blotting
Total RNA (30 μg) was extracted using RNA zol B (Tel Test, Friendswood, TX) according to the manufacturer's instructions, separated on a formaldehyde denaturing gel and transferred to a nylon membrane (Amersham). The blot was then hybridized with radiolabeled p21 cDNA probes using the Quickhyb hybridization solution (Stratagene, Tokyo, Japan) according to the manufacturer's instructions.
Luciferase assays
The reporter gene plasmid (1 μg) was transfected into endothelial cells infected with pLNCX (Mock) or AktCA 24 h before luciferase assay. The control vector encoding Renilla luciferase (0.1 μg) was co-transfected for an internal control. Luciferase assay was carried out using a dual-luciferase reporter assay system (Promega, Madison, WI) according to the manufacturer's instructions. The plasmids pPG13-Luc, pPG15-Luc and pWWP-LUC-1 (el-Deiry et al, 1993) were a gift from Dr B Vogelstein (Johns Hopkins University, Baltimore, MD).
Tissue specimens and histology
Human coronary arteries and internal mammary arteries were obtained from four autopsied individuals who had ischemic heart disease. For immunohistochemistry, the frozen sections (6 μm) were treated with 0.3% hydrogen peroxide in methanol for 20 min, preincubated with 5% goat serum and then treated with anti-phospho-Akt antibody (1:100; Santa Cruz, Santa Cruz, CA) for 1 h at 37°C. Next, the sections were incubated with a biotinylated goat secondary antibody, treated with the avidin–biotin complex (Elite ABC kit, Vector, Burlingame, CA) and stained with diaminobenzidine tetrahydrochloride and hydrogen peroxide. To verify the specificity of the first antibodies, we performed a control staining with nonimmune IgG and excluded the possibility of nonspecific signals. The studies on human samples were approved by our institutional review board.
Statistical analysis
All values were expressed as mean±s.e.m. Comparison of results between different groups was performed by one-way analysis of variance, paired t-test and unpaired t-test using StatView 4.5 (Abacus Concepts, Berkeley, CA).
Supplementary Material
Supplementary Figure 1 Akt induces cell growth arrest in human microvascular endothelial cells. (A) Human microvascular endothelial cells infected with pLNCX (Mock) or AktCA were purified with G418 for 7 days and seeded at the density of 3 × 105 cells per 100 mm plate on day 0. Cell number per 100 mm-plate was then counted on day 7. *P<0.0001 vs Mock, paired t-test, n=5. (B) Cell morphology of microvascular endothelial cells infected with pLNCX (Mock) or AktCA.
Supplementary Figure 2 Akt induces a senescence-like phenotype in confluent endothelial cells. (A) Human endothelial cells infected with pLNCX (Mock) or AktCA were purified with G418 for 7 days and were cultured at confluence. Whole-cell lysates (30 μg) were extracted and examined for p21 expression and actin (loading control) by Western blotting. (B) Cell morphology of confluent endothelial cells infected with pLNCX (Mock) or AktCA.
Supplementary Figure 3 Abrogation of p53 activity prolongs the lifespan of AktCA-infected endothelial cells. Human endothelial cells were infected with pBabe (empty vector) or pBabe E6 and purified with puromycin. Infected cells were then transduced with pLNCX or AktCA as described in Figure 1C. Double-infected cells were seeded at the density of 2 × 105 cells per 100 mm plate on day 0 and passaged. The number of cumulative population doublings was determined.
Supplementary Figure 4 Expression of phospho-FOXO3a, MnSOD, and p21 in endothelial cells undergoing replicative senescence. Whole-cell lysates (30 μg) of young (passage 4) or senescent (passage 14–15) human endothelial cells were analyzed for the expression of phospho-FOXO3a, MnSOD, p21, and actin (loading control) by Western blotting.
Acknowledgments
We thank Dr B Vogelstein, SW Lowe, ME Greenberg and T Asano for reagents. This work was supported by grants from Takeda Medical Research Foundation, Takeda Science Foundation, Japan Heart Foundation, Mochida Memorial Foundation, Uehara Memorial Foundation, Mitsubishi Pharma Research Foundation and the Ministry of Education, Science, Sports, and Culture of Japan (to TM and IK).
Competing interests statement The authors declare that they have no competing financial interests.
References
- Bluher M, Kahn BB, Kahn CR (2003) Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299: 572–574 [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355–365 [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Campisi J (2001) Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 11: S27–S31 [DOI] [PubMed] [Google Scholar]
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655–1657 [DOI] [PubMed] [Google Scholar]
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282: 1318–1321 [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Altomare DA, Klein MA, Lee WC, Kruh GD, Lissy NA, Testa JR (1997) Transforming activity and mitosis-related expression of the AKT2 oncogene: evidence suggesting a link between cell cycle regulation and oncogenesis. Oncogene 14: 2793–2801 [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, Testa JR (1996) Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA 93: 3636–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofalo VJ, Allen RG, Pignolo RJ, Martin BG, Beck JC (1998) Relationship between donor age and the replicative lifespan of human cells in culture: a reevaluation. Proc Natl Acad Sci USA 95: 10614–10619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13: 2905–2927 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241 [DOI] [PubMed] [Google Scholar]
- del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278: 687–689 [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ (1998) Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12: 3499–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Liou LL, Moy VN, Diaspro A, SelverstoneValentine J, Gralla EB, Longo VD (2003) SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics 163: 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD (2001) Regulation of longevity and stress resistance by Sch9 in yeast. Science 292: 288–290 [DOI] [PubMed] [Google Scholar]
- Faragher RG, Kipling D (1998) How might replicative senescence contribute to human ageing? BioEssays 20: 985–991 [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247 [DOI] [PubMed] [Google Scholar]
- Fujishiro M, Gotoh Y, Katagiri H, Sakoda H, Ogihara T, Anai M, Onishi Y, Ono H, Funaki M, Inukai K, Fukushima Y, Kikuchi M, Oka Y, Asano T (2001) MKK6/3 and p38 MAPK pathway activation is not necessary for insulin-induced glucose uptake but regulates glucose transporter expression. J Biol Chem 276: 19800–19806 [DOI] [PubMed] [Google Scholar]
- Guarente L, Kenyon C (2000) Genetic pathways that regulate ageing in model organisms. Nature 408: 255–262 [DOI] [PubMed] [Google Scholar]
- Hayflick L (1975) Current theories of biological aging. Fed Proc 34: 9–13 [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421: 182–187 [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13: 1385–1393 [PubMed] [Google Scholar]
- Kenyon C (2001) A conserved regulatory system for aging. Cell 105: 165–168 [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366: 461–464 [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419: 316–321 [DOI] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA 98: 12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor MA, Rotwein P (2000a) Coordinate control of muscle cell survival by distinct insulin-like growth factor activated signaling pathways. J Cell Biol 151: 1131–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor MA, Rotwein P (2000b) Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21. Mol Cell Biol 20: 8983–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Hench J, Ruvkun G (2001) Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol 11: 1950–1957 [DOI] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW (1998) Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12: 3008–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C (1997) daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278: 1319–1322 [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C (2001) Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28: 139–145 [DOI] [PubMed] [Google Scholar]
- Longo VD, Finch CE (2003) Evolutionary medicine: from dwarf model systems to healthy centenarians? Science 299: 1342–1346 [DOI] [PubMed] [Google Scholar]
- Maki CG, Howley PM (1997) Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol Cell Biol 17: 355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo LD, Donner DB (2001) A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 98: 11598–11603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404: 782–787 [DOI] [PubMed] [Google Scholar]
- Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I (2002) Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105: 1541–1544 [DOI] [PubMed] [Google Scholar]
- Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, Toko H, Komuro I (2003) Ras-induced vascular smooth muscle cell senescence in human atherosclerosis. Circulation 108: 2264–2269 [DOI] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G (1996) A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382: 536–539 [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424: 277–283 [DOI] [PubMed] [Google Scholar]
- Nemoto S, Finkel T (2002) Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science 295: 2450–2452 [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G (1997) The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389: 994–999 [DOI] [PubMed] [Google Scholar]
- Paradis S, Ruvkun G (1998) Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev 12: 2488–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohme D (1981) Evidence for a relationship between longevity of mammalian species and life spans of normal fibroblasts in vitro and erythrocytes in vivo. Proc Natl Acad Sci USA 78: 5009–5013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonherr E, Levkau B, Schaefer L, Kresse H, Walsh K (2001) Decorin-mediated signal transduction in endothelial cells. Involvement of Akt/protein kinase B in up-regulation of p21(WAF1/CIP1) but not p27(KIP1). J Biol Chem 276: 40687–40692 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL (2002) PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 8: 1145–1152 [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS (2001) A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292: 107–110 [DOI] [PubMed] [Google Scholar]
- Testa JR, Bellacosa A (2001) AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA 98: 10983–10985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KV, Holliday R (1983) Genetic effects on the longevity of cultured human fibroblasts. II. DNA repair deficient syndromes. Gerontology 29: 83–88 [DOI] [PubMed] [Google Scholar]
- Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M (2002) Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med 8: 1136–1144 [DOI] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2: 489–501 [DOI] [PubMed] [Google Scholar]
- Weinstein BS, Ciszek D (2002) The reserve-capacity hypothesis: evolutionary origins and modern implications of the trade-off between tumor-suppression and tissue-repair. Exp Gerontol 37: 615–627 [DOI] [PubMed] [Google Scholar]
- Wright WE, Shay JW (2001) Cellular senescence as a tumor-protection mechanism: the essential role of counting. Curr Opin Genet Dev 11: 98–103 [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC (2001a) Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 3: 245–252 [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC (2001b) HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol 3: 973–982 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Akt induces cell growth arrest in human microvascular endothelial cells. (A) Human microvascular endothelial cells infected with pLNCX (Mock) or AktCA were purified with G418 for 7 days and seeded at the density of 3 × 105 cells per 100 mm plate on day 0. Cell number per 100 mm-plate was then counted on day 7. *P<0.0001 vs Mock, paired t-test, n=5. (B) Cell morphology of microvascular endothelial cells infected with pLNCX (Mock) or AktCA.
Supplementary Figure 2 Akt induces a senescence-like phenotype in confluent endothelial cells. (A) Human endothelial cells infected with pLNCX (Mock) or AktCA were purified with G418 for 7 days and were cultured at confluence. Whole-cell lysates (30 μg) were extracted and examined for p21 expression and actin (loading control) by Western blotting. (B) Cell morphology of confluent endothelial cells infected with pLNCX (Mock) or AktCA.
Supplementary Figure 3 Abrogation of p53 activity prolongs the lifespan of AktCA-infected endothelial cells. Human endothelial cells were infected with pBabe (empty vector) or pBabe E6 and purified with puromycin. Infected cells were then transduced with pLNCX or AktCA as described in Figure 1C. Double-infected cells were seeded at the density of 2 × 105 cells per 100 mm plate on day 0 and passaged. The number of cumulative population doublings was determined.
Supplementary Figure 4 Expression of phospho-FOXO3a, MnSOD, and p21 in endothelial cells undergoing replicative senescence. Whole-cell lysates (30 μg) of young (passage 4) or senescent (passage 14–15) human endothelial cells were analyzed for the expression of phospho-FOXO3a, MnSOD, p21, and actin (loading control) by Western blotting.