

Abstract
ATR/Rad3-like kinases promote the DNA damage checkpoint through regulating Chk1 that restrains the activation of cyclin-dependent kinases. In fission yeast, Crb2, a BRCT-domain protein that is similar to vertebrate 53BP1, plays a crucial role in establishing this checkpoint. We report here that Crb2 regulates DNA damage checkpoint through temporal and dynamic interactions with Rad3, Chk1 and replication factor Cut5. The active complex formation between Chk1 and Crb2 is regulated by Rad3 and became maximal during the checkpoint arrest. Chk1 activation seems to need two steps of interaction changes: the loss of Rad3–Chk1 and Rad3–Crb2 interactions, and the association between hyperphosphorylated forms of Chk1 and Crb2. Chk1 is the major checkpoint kinase for the arrest of DNA polymerase mutants. The in vitro assay of Chk1 showed that its activation requires the presence of Crb2 BRCT. Hyperphosphorylation of Crb2 is also dependent on its intact BRCT. Finally, we show direct interaction between Rad3 and Crb2, which is inhibitory to Rad3 activity. Hence, Crb2 is the first to interact with both Rad3 and Chk1 kinases.
Keywords: 53BP1, BRCT, Chk1, Rad3, S. pombe
Introduction
DNA damage checkpoint pathways ensure that the progression of the cell cycle is restrained while repair is carried out (Hartwell and Weinert, 1989). DNA damage is promoted by both extrinsic sources such as UV and ionizing radiation, and intrinsic sources from metabolism and events associated with DNA replication. Many gene products, including human ATR/ATM, fission yeast Rad3 and budding yeast Mec1, which constitute a conserved kinase family, are required for promoting the DNA damage checkpoint (Melo and Toczyski, 2002; Rouse and Jackson, 2002). DNA replication checkpoints that ensure the further progression of the cell cycle are activated when replication is impaired (Kolodner et al, 2002). The entry into M-phase is blocked by the S/M checkpoint when replication forks are stalled by an inhibitor of ribonucleotide reductase (RNR), hydroxyurea (HU). The intra S-phase checkpoint ensures the block of late replication origin firing when replication is inhibited in early S-phase (Shirahige et al, 1998). For ensuring the cell cycle arrest, the DNA damage checkpoint promotes the activation of the Chk1 (Walworth and Bernards, 1996) that inhibits Cdc25 phosphatase, an activator of mitotic cyclin-dependent kinase (CDK). For the checkpoint induced by HU, Cds1 is hyperactivated (Boddy et al, 1998). ATR/Rad3/Mec1 is required for all the known DNA integrity checkpoints.
Three types of components, a sensor, an adaptor and an effector, are thought to be required for checkpoint signalling (Melo and Toczyski, 2002; Rouse and Jackson, 2002). Chk1 and Cds1 are effector kinases. Different complexes, including Rad proteins in fission yeast, have been suggested to play roles in sensing DNA damage. Rad3 forms a complex with Rad26 equivalent to human ATR–ATRIP and budding yeast Mec1–Lcd1 (Edwards et al, 1999; Paciotti et al, 2000; Rouse and Jackson, 2000; Cortez et al, 2001).
Several adaptor proteins provide specificity to the checkpoint responses initiated by a common set of sensors and transducers (Melo and Toczyski, 2002; Rouse and Jackson, 2002). Candidates of adaptor proteins specific to damage checkpoint signalling are fission yeast Crb2, budding yeast Rad9 and vertebrate 53BP1 (Saka et al, 1997; Vialard et al, 1998; DiTullio et al, 2002; Schwartz et al, 2002; Wang et al, 2002). Crb2 is specifically involved in the DNA damage checkpoint and is required for the phosphorylation of Chk1 by Rad3 (Saka et al, 1997; Capasso et al, 2002). Crb2 is hyperphosphorylated upon DNA damage, and this hyperphosphorylation requires the checkpoint Rad proteins, but not Chk1. These data suggest that Crb2 acts downstream of the checkpoint Rad proteins, but upstream of Chk1. In budding yeast, Rad9 is phosphorylated following checkpoint activation, and only the phosphorylated Rad9 interacts with the effector kinase Rad53 (Sun et al, 1998).
We wished to understand the mechanistic role of Crb2 in establishing the DNA damage checkpoint. Crb2 contains two C-terminal BRCT motifs (Bork et al, 1997; Saka et al, 1997). Crb2 is required for the checkpoint induced by UV, methylmethane sulfonate (MMS) and ionizing radiation, and also by DNA polymerase mutations (Saka et al, 1997; Willson et al, 1997). Conversely, like Chk1, Crb2 is not required for a replication block resulting from nucleotide depletion, such as treatment with HU. Its hyperphosphorylation after DNA damage requires Rad3 kinase. Crb2 was originally identified by a two-hybrid interaction with Cut5/Rad4 (Saka and Yanagida, 1993), and as a novel checkpoint mutant (Willson et al, 1997). Cut5 (Saka and Yanagida, 1993) is known to interact with the N-terminal domain of Crb2. Temperature-sensitive (ts) cut5 mutations that occur within the BRCT domains of Cut5 abolish the interaction with Crb2. In addition to the shared phenotypes of Δchk1 and Δcrb2 strains, Crb2 and Chk1 can also interact in a yeast two-hybrid assay, and Crb2 is required for the formation of the Chk1 upper band in response to DNA damage. Thus Crb2 may serve as an adaptor protein for the DNA damage checkpoint ensuring signalling through Chk1.
In this study, we investigated the roles of the Crb2 BRCT domains. BRCT domains are present among a large number of proteins involved in DNA damage and DNA repair (Bork et al, 1997; Callebaut and Mornon, 1997). However, it is not known how Crb2 BRCT functions in the establishment of the DNA damage checkpoint. Structural studies (Joo et al, 2002) indicated that the two BRCT domains of 53BP1 are tandemly arranged and bind to p53 in vitro. We show that Crb2 BRCT domains are crucial for establishing the DNA damage checkpoint that senses both lesions made by UV irradiation and those caused by mutations in DNA polymerase subunits. Furthermore, dynamic interactions between Crb2 and Rad3 and between Crb2 and Chk1 are dramatically altered upon damage, and these alterations appeared to be correlated with arresting cell cycle progression upon DNA damage.
Results
BRCT domains are essential for UV-induced damage checkpoint
To examine whether the C-terminal BRCT domains of Crb2 have any role in vivo in establishing the DNA damage checkpoint, a C-terminal truncated crb2 gene (hereafter designated as crb2ΔC) was constructed and integrated with expression from the native promoter. Crb2ΔC (546 aa) was detected by Western blot (Figure 1A, right lane). The full-length (FL) Crb2 (calculated MW 87.4 kDa) produced diffuse phosphorylated bands (Saka et al, 1997) migrating at positions higher (apparent MW 100–110 kDa) than the expected MW (left lane).
Figure 1.
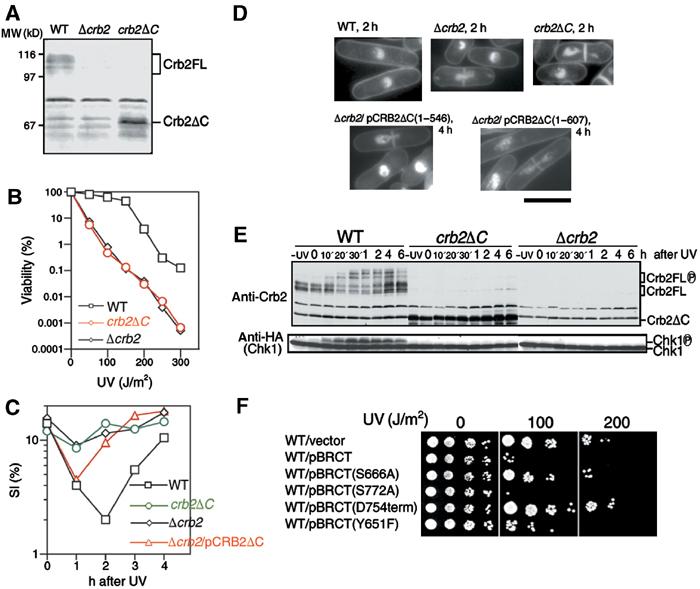
The BRCT domains of Crb2 are required for establishing the DNA damage checkpoint. (A) Identification of the Crb2ΔC protein (right lane) by immunoblot. Polyclonal antibodies against Crb2 revealed several contaminating bands. Full-length (FL) Crb2 is shown at the left (bands are multiple due to phosphorylation). The control Δcrb2 lacks the protein band (middle lane). (B) Viability of wild-type (WT), Δcrb2 null and C-terminal deletion crb2ΔC after UV irradiation (0–300 J/m2) was measured. (C) The SI% after UV irradiation was measured for wild-type, Δcrb2, crb2ΔC and Δcrb2 cells carrying plasmid pCRB2ΔC. (D) Light micrographs of UV-irradiated Δcrb2 carrying vector, or plasmid with the full-length crb2+ gene (FL) or C-terminal truncated plasmid pCRB2ΔC. Cells were stained by DAPI. Scale bar, 10 μm. (E) Immunoblot of cell extracts after UV irradiation (100 J/m2) using antibodies against Crb2 and Chk1-HA. Strains used carry the integrated Chk1-HA with the wild-type crb2+ gene, crb2ΔC or Δcrb2 null. (F) S. pombe strains, which can ectopically overproduce wild-type or mutant BRCT fragments, were made using REP41-based plasmid (Maundrell, 1990), and their UV hypersensitivities were examined in the absence of thiamine (the nmt1+ promoter on).
The crb2ΔC strain grew normally but was hypersensitive to UV irradiation (Figure 1B), responding in a manner that was indistinguishable from that of Δcrb2 null. To test their checkpoint proficiency, crb2ΔC was irradiated with UV (100 J/m2), and cell cycle progression was followed by measurement of the septation index (SI%; percent of cells with a medial septum). The reduction of SI% in the culture of the wild-type (WT) strain after UV irradiation indicated a temporary arrest in the cell cycle (Figure 1C). The SI% did not decrease significantly in Δcrb2 and crb2ΔC, however, indicating that the checkpoint arrest was abolished in these strains. The SI% in Δcrb2 null that carried plasmid pCRB2ΔC showed a small and transient decrease that resumed to unirradiated levels 1 h earlier than that of wild-type cells. This arrest was brief but reproducible, suggesting that the overproduced 67 kDa Crb2ΔC retained some function of Crb2 in response to DNA damage.
We examined the nuclear phenotypes of Δcrb2 and crb2ΔC cells stained with DAPI (Figure 1D). While Δcrb2 null or crb2ΔC containing the vector plasmid showed, upon UV irradiation (100 J/m2), the typical cut phenotype due to the lack of checkpoint arrest, Δcrb2 null cells that carried multicopy plasmid pCRB2ΔC(1–546) or pCRB2ΔC(1–607) were significantly elongated, and in addition showed the cut phenotype, suggesting a transient delay prior to mitotic entry. This phenotype was consistent with the assay of SI%. In wild type irradiated with the same UV dose, most cells showed a cell cycle delay with significant cell elongation, followed by a recovery to normal growth. We concluded that the BRCT domains of Crb2 were necessary to establish the damage checkpoint, although at least in the context of overexpression, the truncated proteins possessed a residual ability to signal a DNA damage response.
To confirm the lack of checkpoint activation, Western blot was performed for the hyperphosphorylated form of activated Chk1, which has reduced migration in SDS–PAGE. Extracts were prepared from wild-type, Δcrb2 and crb2ΔC cells (all containing an integrated Chk1-HA gene) after UV irradiation. The hyperphosphorylated upper band of Chk1 (Walworth and Bernards, 1996) appeared in wild-type extracts 10 min after irradiation and formed the peak of the band intensity around 30–60 min (Figure 1E). However, the upper band of Chk1 was absent in crb2ΔC and Δcrb2. The use of antiphospho antibody against Ser345 confirmed the temporal phosphorylation of Chk1 by Rad3 only in wild-type cells (Supplementary Figure S1A; Lopez-Girona et al, 2001). Moreover, there were no detectable slower migrating species for Crb2ΔC, suggesting that hyperphosphorylation did not occur for Crb2ΔC.
Overproduction of BRCT causes cells to be hypersensitive to UV
We examined whether overproduction of the Crb2 BRCT domains alone had any effect on the damage response. Wild-type cells overproducing BRCT (aa 535–778) became hypersensitive to UV (Figure 1F). In the absence of irradiation, BRCT-overproducing wild type grew normally, however. This dominant-negative effect was not observed if the 25 aa extreme C-terminus was deleted. Hence, both of the BRCT domains are necessary for this phenotype, in keeping with the presence of these domains in tandem repeats in the majority of BRCT proteins. In addition, overexpression of the BRCT domains containing a substitution (S666A; mutation in the linker region between two BRCTs) and another substitution (Y651F; highly conserved among BRCTs) largely abolished the overproduction effect. Presumably, the overexpression of BRCT is titrating a protein required for the damage response.
Chk1 activation requires the BRCT domains of Crb2
The previous two-hybrid analysis indicated that Chk1 was bound to the central domain of Crb2 (Saka et al, 1997; Esashi et al, 2000). As the upper (phosphorylated) band of Chk1 was not produced in Δcrb2 after UV irradiation, we presumed that Crb2 was required for the activation of Chk1. As a more direct assessment of Chk1 activation, we immunoprecipitated HA-tagged Chk1 and assayed its activity in vitro using GST-Cdc25 (60–160 aa) as a substrate (Figure 2A). Basal Chk1 activity was increased approximately three-fold following 100 J/m2 of irradiation, and no background phosphorylation of GST-Cdc25 was detected in immunoprecipitates of a kinase dead Chk1 (D155A substitution in the catalytic site).
Figure 2.
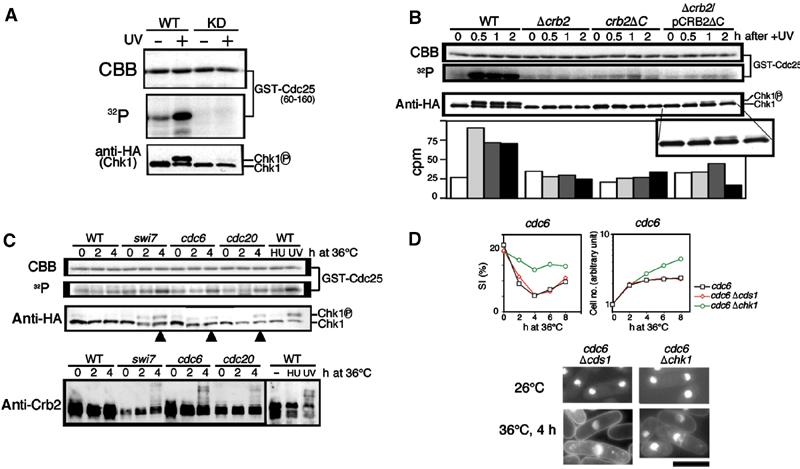
The activation of Chk1 activity requires functional Crb2 and occurs in UV-irradiated and polymerase-defective cells. (A) Extracts of S. pombe integrated with Chk1-HA were employed for immunoprecipitation. Chk1-HA immunoprecipitated by anti-HA antibodies was mixed with the substrate GST-Cdc25 fragment (60–160 aa) produced in E. coli and [γ-32P]ATP and incubated at 30°C for 30 min. The mixtures were run in SDS–PAGE, followed by autoradiography. The following extracts were made: wild type carrying Chk1-HA before (−) and after (+) UV irradiation; wild type carrying the kinase dead (KD) Chk1-HA before and after UV irradiation. CBB: Coomassie brilliant blue. (B) Wild type, Δcrb2, crb2ΔC and Δcrb2 carrying plasmid pCRB2ΔC were irradiated by UV. Extracts were prepared from cells before (0 h) or after (0.5, 1 and 2 h) irradiation. The Chk1 activity quantified using a liquid scintillation counter is shown at the bottom panel. The upper band of Chk1 was found in Δcrb2 cells carrying plasmid pCRB2ΔC. See the enlarged band patterns. (C) Cultures of wild type, swi7 (pol α), cdc6 (pol δ) and cdc20 (pol ɛ) were shifted from 26 to 36°C, and the extracts were made from cells prepared after 0, 2 and 4 h at 36°C. Extracts of wild type were also made in the presence of HU (15 mM, 1 h) and after UV irradiation (100 J/m2, 1 h). Extracts were used for the Chk1 activity and also for immunoblot to detect Chk1-HA and Crb2. (D) Polymerase-deficiency checkpoint requires Crb2 and Chk1 but not Cds1. Two double mutants Δchk1 cdc6 and Δchk1 cdc6 were cultured with single mutant at 36°C for 0–8 h. The SI% and the cell number were measured for the cultures shifted from 26 to 36°C. Cells stained by DAPI are shown. Scale bar, 10 μm.
We tested whether the activation of Chk1 was dependent on the presence of Crb2. Wild-type or Δcrb2 cells were UV irradiated, and were collected 0, 0.5, 1 and 2 h after irradiation. Neither the activation nor the phosphorylation of Chk1 was detected in Δcrb2, although the basal level of Chk1 activity was detected (Figure 2B). Two other strains, crb2ΔC and Δcrb2 carrying plasmid pCRB2ΔC, were examined. A small, but reproducible, increase of the Chk1 activity was observed in both crb2ΔC by 1.6-fold after 2 h and Δcrb2 containing pCRB2ΔC by 1.4-fold at 1 h after irradiation (Figure 2B). The tiny upper band of Chk1 was clearly seen for the latter strain (enlarged bands shown at the bottom). Crb2 was required for the activation of Chk1. In addition, as the C-deletion allele crb2ΔC could produce only a tiny activation and, if overproduced, resulted in a weak phosphorylation of Chk1 and Crb2ΔC (data not shown), the Crb2 BRCT domains seemed to be needed for the full activation of Chk1 after UV irradiation.
Chk1 is activated and Crb2 is hyperphosphorylated in polymerase mutants
Crb2 is required for the checkpoint induced in DNA polymerase mutants, but is not needed for checkpoint by 10 mM HU (Saka et al, 1997). A genetic requirement for Chk1 has also been shown for the arrest in polymerase mutants (Francesconi et al, 1997; Feng and D'Urso, 2001), but the increase of Chk1 activity has only been shown for DNA damage (Capasso et al, 2002). We hence examined the activation of Chk1 activity in polymerase mutant extracts. Schizosaccharomyces pombe has three classes of DNA polymerase, Swi7, Cdc6 and Cdc20, which encode DNA polymerase α, δ and ɛ, respectively. As shown in Figure 2C (third panel), the upper (phosphorylated) band of Chk1-HA was detected by Western blotting of swi7, cdc6 and cdc20 mutant extracts after 4 h at 36°C (the arrowheads), but not in HU-arrested cells. Consistently, the Chk1 activity (32P incorporation) significantly increased in these polymerase mutants (second panel; the control Coomassie brilliant blue (CBB)-stained GST-Cdc25 pattern shown in the top panel). There was no increase of Chk1 activity in HU-arrested cells (WT HU), however. The level of activation in polymerase ɛ (cdc20) mutant was slightly lower than that in UV-irradiated cells (UV).
Immunoblotting of Crb2 (Figure 2C, bottom panel) indicated that hyperphosphorylated bands of Crb2 were produced after 4 h at 36°C in the polymerase mutants. The hyperphosphorylated bands were hardly observed in HU-induced cells. These results showed that the activation of Chk1 in irradiated or polymerase mutant cells correlates with the appearance of the hyperphosphorylated bands of Crb2 and Chk1.
Checkpoint arrest induced by polymerase mutants require Crb2 BRCT domains and Chk1
We examined whether the BRCT domains of Crb2 were required for the checkpoint arrest induced in polymerase mutants. For this purpose, we used crb2ΔC, and assayed checkpoint kinetics in double mutants carrying ts mutations in swi7, cdc6 and cdc20 at 36°C. The SI% did not decrease in the double mutants, and the frequency of abnormal mitotic cells sharply increased, demonstrating that the checkpoint was not established in any of these three double mutants (Supplementary Figure 2). The presence of BRCT was thus essential for the checkpoint arrest induced in polymerase mutants at 36°C.
Two double mutants, cdc6 Δchk1 and cdc6 Δcds1, were used to observe whether checkpoint arrest could be maintained at 36°C. Cds1 is known to be hyperactivated in HU-arrested cells (Boddy et al, 1998; Lindsay et al, 1998). The checkpoint arrest was abolished in cdc6 Δchk1, but maintained in cdc6 Δcds1 (Figure 2D). The SI decreased in cdc6 Δcds1, but not in cdc6 Δchk1. Elongated arrested cells were observed in the double mutant with Δcds1. Similar results were obtained for cdc20 Δchk1, cdc20 Δcds1, swi7 Δchk1 and swi7 Δcds1 mutants (data not shown). Chk1 was thus not only activated in polymerase-deficient cells, but was also required for the checkpoint arrest at 36°C. These results are consistent with previous reports (Francesconi et al, 1997; Feng and D'Urso, 2001).
Overproduced Rad3 and Crb2 are in a complex in unirradiated cells
To obtain a mechanistic insight into how Crb2 can activate Chk1, we examined interactions among three proteins, between Crb2 and Rad3, between Chk1 and Rad3 and between Crb2 and Chk1, using an ectopic overproduction system in S. pombe (REP2 and REP41 contain the inducible nmt1 promoter; Maundrell, 1990). As described below, only the Crb2–Chk1 interaction has been detected without overproduction so far. One of the paired genes was expressed using strong REP2, while moderate REP41 was used for the other gene. Overproduction of Rad3 and Crb2, respectively, was done by REP2 and REP41 throughout the present study. For overexpression of Chk1, REP41 and REP2 were used depending on the paired gene employed.
We first examined whether Crb2 interacted with Rad3. The plasmid genes of GST-Rad3 and Crb2 were co-overexpressed after 16–18 h at 33°C in the EMM2 media in the absence of thiamine (promoter on). Glutathione beads were used to pull down GST-Rad3, and the level of co-precipitated Crb2, if any, was examined by immunoblot using anti-Crb2 antibodies (Figure 3A). GST-Rad3, but not GST only, efficiently co-purified with Crb2, indicating that they had formed the stable complex in S. pombe cells.
Figure 3.

Crb2 and Rad3 physically interact and alter their interaction after UV irradiation. (A) Physical interaction between Crb2 and Rad3 was examined in extracts of cells that co-overproduced GST-Rad3 (REP2) and Crb2 (REP41). (B) To determine which domain of Crb2 is responsible for this interaction, truncation series of Crb2 (a–e) were made, and co-overproduced with full-length GST-Rad3. (C) To determine which region of Rad3 is required for the interaction between Crb2 and Rad3, deletion series of Rad3 tagged with GST were made and co-overproduced with the BRCT domains of Crb2. (D) Cells overproducing checkpoint proteins (three panels from the right) showed cell cycle arrest phenotype in response to UV irradiation, indistinguishable from wild-type control (left panel). (E) UV sensitivities of cells overproducing checkpoint proteins were almost the same as wild-type control.
To determine which part of Crb2 was required for binding to GST-Rad3, NLS (nuclear localization signal of SV40 large T antigen) was fused to truncated fragments of Crb2 (a–e, Figure 3B). The full-length Crb2 is located in the nuclear chromatin (Saka et al, 1997). These truncated fragments were ectopically overexpressed, and we examined which could form the complex with GST-Rad3. The results showed that the C-terminal fragment (535–778) was bound to GST-Rad3. The BRCT domains were the site for interaction with Rad3.
To decide the region of Rad3 required for the interaction with Crb2, truncated GST fragments were expressed and examined for association with the BRCT domains of Crb2 (Figure 3C). The N-terminal fragment (1–439) of Rad3 efficiently bound to the HA-NLS-Crb2 BRCT. The C-terminal domain (1633–2386) of Rad3 containing the catalytic sequence failed to interact with BRCT of Crb2. The kinase domain of Rad3 was not essential for association with the BRCT fragment of Crb2.
Overproducing cells show normal checkpoint arrest and UV sensitivity
We examined whether checkpoint response was altered by overproducing checkpoint proteins. Wild-type cells ectopically co-overproducing Rad3 and Crb2, Rad3 and Chk1, and overproducing only Chk1 were UV irradiated. Overproduction of Rad3 and Crb2 showed no toxic effect. For the case of Chk1, special care was taken to reduce the overproduction effect (Al-khodairy et al, 1994). To maintain normal cell cycle progression, Chk1 was first mildly overproduced for 10 h in the absence of thiamine, and then thiamine (final 2 μM) was added to the culture medium to repress the nmt1 promoter. Under this Chk1 overproduction condition, the cell cycle continued normally. The time-course changes of the SI% were measured for three overproducing strains (Figure 3D). Their cell cycle arrest phenotype in response to UV irradiation was found to be indistinguishable from that of wild-type control. UV sensitivities of these cells were nearly identical to wild-type control (Figure 3E). Overproduced checkpoint proteins under the present conditions thus did not appear to impair the ability of checkpoint arrest and DNA repair functions.
Dissociation of Crb2 from Rad3 upon UV irradiation
Next, we examined whether the complex made between the overproduced Crb2 BRCT and Rad3 was modulated after irradiation. To this end, GST-Rad3 (REP2) and HA-NLS-Crb2BRCT (REP41) were ectopically co-overproduced in wild-type cells. In the absence of irradiation, full-length GST-Rad3 or C-terminally truncated GST-Rad3 (1–1476) formed the complex with Crb2 BRCT (Figure 4A, left panel). At 1 h after UV irradiation (100 J/m2), the complex formation was indeed diminished (right panel). Crb2BRCT was no longer detected in the GST-Rad3 or GST-Rad3 (1–1476) pull-down fractions. To confirm dissociation of Crb2BRCT from GST-Rad3 after UV irradiation, a time-course analysis was performed (Figure 4B). After 60 min, the association between Rad3 and Crb2 BRCT was reduced to the minimum, as indicated by immunoblot and its quantitative data. Timing for the minimal interaction corresponds to the maximal cell cycle arrest.
Figure 4.
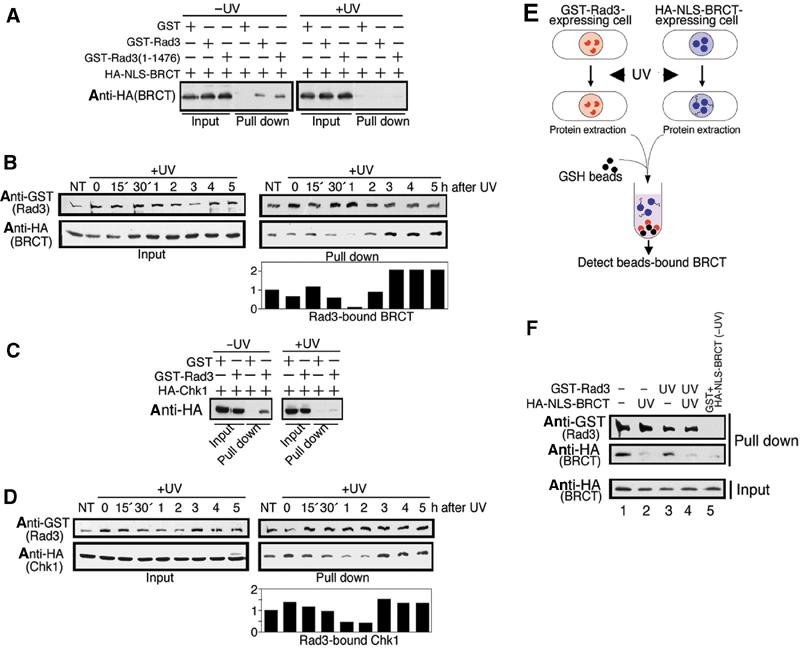
UV irradiation leads to transient dissociation of GST-Rad3 from Crb2 and Chk1. (A) Wild-type cells co-overproducing GST-Rad3 or C-truncated GST-Rad3 (1–1476) and Crb2 BRCT (535–778) tagged with HA and NLS were irradiated with UV (100 J/m2). At 1 h after UV irradiation, cells were collected and GST-Rad3 was pulled down. (B) Time-course analysis of the same cells above was performed 0–5 h after UV irradiation. Quantitative data are shown below. NT: no treatment. (C) Wild-type cells co-overproducing GST-Rad3 and HA-Chk1 were irradiated with UV. At 1 h after UV irradiation, cells were collected and GST-Rad3 was pulled down. (D) Time-course analysis of the same cells described in (C) was performed after UV irradiation. Quantitative data are shown below. (E) Strategy to determine whether BRCT or Rad3 was responsible for transient dissociation. (F) Individual cultures ectopically overproducing either HA-NLS-BRCT or GST-Rad3 under the nmt1+ promoter were first UV irradiated (UV) or not (−), and then they were mixed. GST-Rad3 was pulled down and the resulting pull-down fractions were immunoblotted using antibodies against HA or GST.
Dissociation of Chk1 from Rad3 after UV irradiation
We found that HA-Chk1 (REP41) ectopically co-overexpressed with GST-Rad3 (REP2) formed a complex (Figure 4C). When cells that co-overproduced GST-Rad3 and HA-Chk1 in the EMM2 medium were irradiated by UV (100 J/m2), most GST-Rad3 obtained by pull-down no longer co-purified with Chk1-HA 1 h after irradiation. We then examined the time-course change of the interaction between Chk1 and Rad3. The result (Figure 4D) showed that the level of Chk1 bound to GST-Rad3 became transiently minimal approximately 1 h after UV irradiation, which was similar to the diminished interaction between GST-Rad3 and Crb2 BRCT domains. The upper phosphorylated bands were not clear when Chk1 was overproduced, an observation we have repeatedly made, presumably due to the difficulty to resolve the phosphorylated form from a more abundant level of protein. Although the in vivo complex formation between Rad3 and Chk1 after UV irradiation was previously described (Martinho et al, 1998), we show that the interaction is dynamic, becoming minimal 1 h after UV irradiation.
Irradiated BRCT is responsible for dissociation
The mechanism of dissociation upon damage is of interest. Even 30 min after irradiation, Crb2 BRCT domains were disassociated from Rad3. The N-terminal region of Rad3 or the BRCT domains of Crb2 may undergo modification or a conformational change, which would explain the loss of complex formation upon irradiation. We addressed the following question: which, Rad3 or Crb2 BRCT, is responsible for the loss of interaction in irradiated cells? To address this issue, cells singly overexpressing GST-Rad3 or HA-NLS-Crb2 BRCT were irradiated, followed by the culture for 1 h at 33°C (Figure 5E). Extracts were made, and then mixed with nonirradiated cell extracts of HA-Crb2 BRCT or GST-Rad3, respectively.
Figure 5.
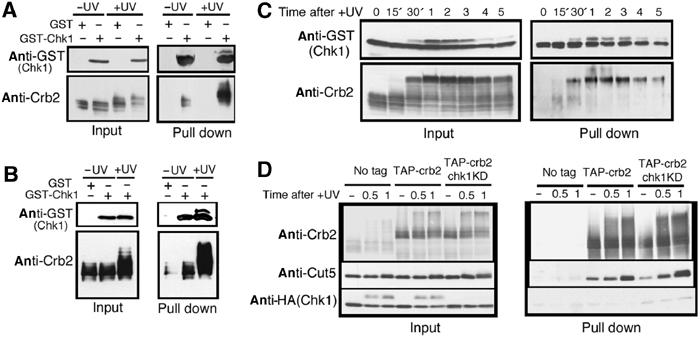
UV irradiation causes a transiently enhanced association between Crb2 and Chk1. (A) GST-Chk1 (REP2) and Crb2 (REP41) were co-overproduced under the inducible nmt1+ promoter in wild type. Cells overproducing both were collected before (−UV) or 1 h after (+UV) UV irradiation (100 J/m2). Crb2 protein in GST-Chk1 pull-down beads was analyzed by immunoblot using anti-Crb2 antibodies. (B) Same as (A) except that only GST-Chk1 was overproduced in wild-type cells. (C) Time-course analysis (0–5 h after +UV) of the same cells described in (B). (D) Association of Crb2 with Chk1, Chk1KD and Cut5 was examined at the single-copy level. Chk1, Chk1KD and Cut5 co-purified with TAP-Crb2 from cells before (−), and 0.5 and 1 h after UV irradiation are shown.
The complex formation was clearly restored for the irradiated Rad3 and nonirradiated Crb2 BRCT (Figure 4F, lane 3), but greatly diminished for the irradiated Crb2 BRCT and nonirradiated Rad3 (lane 2). If both were irradiated, the complex formation was abolished (lane 4). These results indicated that in response to irradiation, the alteration of Crb2 BRCT was responsible for the failure to form a complex.
Interaction between Crb2 and Chk1 is enhanced after UV irradiation
To determine whether the interaction between Crb2 and Chk1 is affected by UV irradiation, we first examined overproduction of GST-Chk1 (REP2) and Crb2 (REP41) as shown in Figure 5A. The relative amount of Crb2 bound to GST-Chk1 increased significantly following irradiation (+UV). Interestingly, the upper hyperphosphorylated form of Crb2 after irradiation was the predominant form associated with GST-Chk1.
We then examined whether the complex could be detected by overproduced GST-Chk1 and low-level native Crb2 produced by the single-copy wild-type gene. The amount of Crb2 bound to GST-Chk1 increased significantly following irradiation (Figure 5B). The upper hyperphosphorylated form of Crb2 after irradiation was again the predominant form associated with GST-Chk1. To test the hypothesis that the interaction between Crb2 and Chk1 was enhanced after UV irradiation, the time-course analysis was performed (Figure 5C). The level of Crb2 bound to GST-Chk1 (right bottom panel) peaked around 1–3 h after UV irradiation (100 J/m2). The interaction between GST-Chk1 and Crb2 was indeed enhanced during the period when Chk1 produced the hyperphosphorylated band and cells were checkpoint arrested. Note that in the time-course experiment, the native level of Crb2 was expressed, whereas Chk1 was overproduced.
We wanted to examine whether the complex could be detected in cells that expressed Chk1 and Crb2 by the native genes using the tandem affinity purification tag (TAP; Tasto et al, 2001) method. N-terminally TAP-tagged crb2+ integrant was constructed, and found to be normal in UV sensitivity. TAP-Crb2 protein was purified using IgG sepharose, and selectively eluted by TEV protease. The level of co-eluted Chk1-HA was then assayed by immunoblot. Two types of Chk1, wild-type Chk1-HA and kinase dead Chk1KD-HA, both of which were chromosomally integrated, were used. A small amount of Chk1KD was reproducibly obtained, dependent on the TAP tagging (Figure 5D; right, TAP-crb2 and chk1KD, bottom panel). The level of co-eluted wild-type Chk1 (TAP-crb2) was very low, comparable to the no-tag control. In this experiment, endogenous Cut5 was found to be co-purified with Crb2, and the level of Crb2-bound Cut5 increased in response to UV irradiation (right, middle panel). These results suggest that the direct interaction between Crb2 and Chk1 expressed at the single-copy gene level is less stable than the complex Crb2–Cut5.
Rad3 activity is reduced by interacting with Crb2 BRCT
Although Crb2 BRCT interacted with the nonkinase domain of Rad3, we tested whether the kinase activity of Rad3 might be altered by this interaction. PHAS-1 (phosphorylated heat- and acid-stable protein regulated by insulin) was used as the substrate (Chapman et al, 1999). GST-Rad3 was purified on glutathione sepharose. In all, 1 μg PHAS-1 and [γ-32P]ATP were added to the solution and incubated at 30°C for 30 min. Strong incorporation of 32P to PHAS-1 was observed (Figure 6, lane 2). No incorporation was found in PHAS-1 for the kinase dead Rad3 construct (KD; D2249E substitution). If Crb2 BRCT (0.2–2 μg) was added to the solution (lanes 2–6), the activity of Rad3 was greatly diminished. No incorporation of 32P was found in BRCT, indicating that this region of Crb2 is not a substrate for Rad3 in vitro. For a negative control of BRCT-mediated Rad3 inhibition, the mutant S666A in BRCT (lanes 7–10), which in the context of full-length Crb2 was nonfunctional (our unpublished result), was employed. In contrast to the wild-type BRCT, the increased level of BRCT S666A did not affect the activity of Rad3. Crb2 BRCT, in association with Rad3, might physically block the access of Rad3 to the substrate in vitro. Some other mechanisms to inhibit the activity are possible, however. We attempted to use the full-length Crb2, but it was very rapidly degraded if divalent cation was added, which is necessary for the kinase assay. An unidentified proteolytic activity seemed to be present in the full-length Crb2 preparations.
Figure 6.
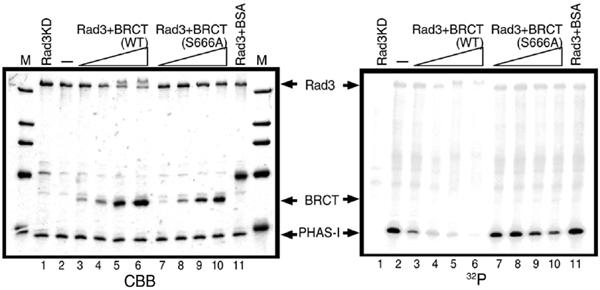
Rad3 activity is inhibited by the Crb2 BRCT domains in vitro. Rad3 purified from fission yeast cells that overproduced it was incubated with increasing amounts of purified BRCT of Crb2 (lanes 3–6). As a negative control, single amino-acid-substituted BRCT S666A was used (lanes 7–10). (Left panel) CBB-stained proteins. Rad3KD is the kinase dead mutant of Rad3 (lane 1). BSA: bovine serum albumin (lane 11). (Right panel) Autoradiography of the Rad3 assay using PHAS-1 as the substrate. M: molecular weight marker.
Discussion
Combining with the present and past results, we conclude that Crb2 is a multidomain interacting protein with three types of partners: Rad3, Chk1 and replication factor Cut5. Recently, many checkpoint adaptor/mediator proteins have been reported. Among them, Crb2 is the first that interacts with both upstream Rad3/ATM/ATR and downstream Chk1 kinases. Our results also suggest that the interactions between Crb2 and the partner proteins are dynamic during activation of the DNA damage checkpoint. In addition, complex formation of Crb2 with Cut5 is intensified after UV irradiation. As Crb2 is required for DNA repair as well as checkpoint arrest, it may mediate the damage signal to various molecules required for cellular responses. We show that the Crb2 BRCT domains are absolutely necessary for establishing the checkpoint. While our data show a dependency on the BRCT domains for Crb2 gross phosphorylation bands in vivo, they do not necessarily implicate the BRCT domains as the actual phosphorylation target. The BRCT domain of budding yeast Rad9 is required for checkpoint response and mediates the dimeric interaction after DNA damage, resulting in Rad53 activation (Soulier and Lowndes, 1999; Gilbert et al, 2001). We confirmed the dimeric interaction for Crb2 BRCT domain (our unpublished result).
Upon damage, an association between Crb2 and Chk1 could be detected. This association was dynamic, and became maximal when the checkpoint was firmly established. Conversely, the interactions between Rad3 and Chk1 and between Rad3 and Crb2 were diminished in overproducing cells when cells were UV irradiated. One has to be cautious for interpreting the results obtained by the overproducing systems, but we showed that checkpoint arrest and UV sensitivity were normal in cells overproducing the checkpoint proteins. Further, a dramatic alteration in the state of association upon DNA damage may at least reflect some aspects of checkpoint regulations. We tested whether the association was detectable for the expression by the single native genes. The dynamic association between Chk1 and Crb2 was clear when Crb2 was in its native level, indicating that the concentration of Chk1 was the limiting factor for association. If both Chk1 and Crb2 were expressed by the native single gene promoter, only weak association was observed particularly for Chk1KD. These results suggest that the interaction is transient and strengthened by high protein concentration.
Association between Rad3 and Crb2 and between Rad3 and Chk1 was not detected when any of their expression levels were reduced to that of the single native gene. We hence presume that the association is due to the high concentration effect by overexpression, and that association itself may not be physiologically relevant. Instead, the loss of interaction after UV irradiation may represent some important changes occurring to these molecules. The extract mixing experiment strongly suggests that the Crb2 BRCT domain rather than Rad3 after UV undergoes unidentified modification that is responsible for the loss of association. Hyperphosphorylated Chk1 and Crb2 no longer possessed the ability to interact with Rad3. A hypothesis is that the two steps are necessary for the activation of Chk1. The first is the loss of interaction between Rad3 and Crb2 and between Rad3 and Chk1, while the second is acquiring the interaction between hyperphosphorylated forms of Chk1 and Crb2.
The in vitro assay of Chk1 indicated that the activation of Chk1 requires the presence of Crb2. The N- and central domains of Crb2 could initiate a low-level activation of Chk1 after damage, but not sufficient for the maximum activation. The overproduced C-terminal BRCT of Crb2 was able to bind to the noncatalytic region of Rad3 in the undamaged cell, but no longer associated after irradiation. Rad3 is responsible for the upregulating phosphorylation of Chk1 (Walworth and Bernards, 1996; Lopez-Girona et al, 2001; Capasso et al, 2002), and capable of phosphorylating Chk1 in vitro (Martinho et al, 1998). Although the Crb2 BRCT domain is not the in vitro substrate of Rad3, the other domains of Crb2 might be directly phosphorylated by Rad3. 53BP1, a putative vertebrate homolog of Crb2, is phosphorylated in vitro by Rad3-like ATM (Xia et al, 2001). Both Chk1 and Crb2 might be the direct targets of Rad3. Phosphorylated Crb2 can act as an adapter to facilitate the phosphorylation of Chk1, bound to the central domain of Crb2 (Esashi et al, 2000). The Crb2–Chk1 complex phosphorylates its substrates to prevent Cdc2 activation and mitotic entry.
Crb2 is required not only for the DNA damage checkpoint but also for repair (Saka et al, 1997; Esashi and Yanagida, 1999; Caspari et al, 2002), so that the enzyme–substrate relationship between Crb2 and Rad3 may not be surprising. Rad3 may regulate Crb2, as hyperphosphrylation of Crb2 was not found in rad3 mutant (Saka et al, 1997). It is conceivable that Crb2 affects Rad3, as phosphorylation of Chk1 is scarce in Δcrb2 mutant cells. The kinase assay of Rad3 indicated that the association of BRCT with Rad3 was clearly an inhibitory event. An intact BRCT is necessary for this negative effect, as the single amino-acid substitution caused the loss of such in vitro inhibition. However, as the site of interaction in Rad3 is noncatalytic, and the interaction between Rad3 and BRCT is abolished in damaged cells, Crb2 should not be a simple inhibitor for Rad3. It is quite possible that regulation might be bidirectional between Rad3 and Crb2. Crb2 may be regulated by Rad3 and vice versa. Crb2 might be a substrate and also a regulatory cofactor for Rad3.
In this study, we showed that Crb2 and Chk1 are essential for the checkpoint arrest of DNA polymerase α, δ and ɛ mutants, consistent with previous genetical results (Francesconi et al, 1997; Feng and D'Urso, 2001). Note that Cds1 was hyperactivated in HU-arrested cells (Boddy et al, 1998; Lindsay et al, 1998) but not in polymerase-deficient cells (Aono et al, 2002). HU- and polymerase mutant-arrested cells may produce different types of aberrant replication forks. Alternatively, nucleotide depletion in HU-arrested cells might cause different defects. In polymerase-defective cells, like in UV-irradiated cells, both phosphorylation and activation of Chk1 were found. As hyperphosphorylation bands of Crb2 were also found, there was no gross difference between the checkpoint arrest due to UV irradiation and polymerase defects. These results, together with the increase of Chk1 activity in DNA polymerase mutant cells, firmly established that Chk1 is the major checkpoint kinase for the arrest of DNA polymerase mutants. Similar findings were made in higher eucaryotes (Guo et al, 2000; Feijoo et al, 2001). In budding yeast, Rad53 is implicated in the polymerase-deficient checkpoint (Wang and Elledge, 1999).
In short, Crb2 is a crucial player in damage checkpoint, regulating the cellular responses upon damage through temporal and dynamic interactions with Rad3 and Chk1. The active complex formation between Chk1 and Crb2 is regulated by Rad3 and became maximal during the checkpoint arrest. Previously, the N-terminus of Crb2 was shown to interact with Cut5 (Saka and Yanagida, 1993; Saka et al, 1994, 1997; Esashi et al, 2000). All the cut5 mutants so far identified failed to replicate DNA, and Cut5 mutant protein did not interact with Crb2 in the yeast two-hybrid system (Saka et al, 1997). In this study, we showed that Crb2 was physically bound to Cut5 in S. pombe cells at the single gene level. Moreover, the association increased in response to DNA damage. The implication of Cut5–Crb2 interaction in the DNA damage checkpoint remains to be understood.
Materials and methods
Strains, culture conditions and UV irradiation
S. pombe h− 972 and h+ 975 (Gutz et al, 1974) and their derivatives were used. For the construction of crb2ΔC single-copy mutant, crb2+ open reading frame up to the BsmI site was cloned into the integration vector pYC11 (Chikashige et al, 1989) with the native promoter of crb2+ and poly(A) signal sequences of nmt1+. The plasmid was transformed into the strain Δcrb2 leu1 mutant. Integration of the Leu+ plasmid at the crb2 locus was confirmed by PCR and Southern hybridization. The rich YPD and the minimal EMM2 (Mitchison, 1970) media were employed. For UV irradiation (UV STRATALINKER, Stratagene), early log phase cells were plated (∼1 × 108 cells/10 × 14 cm2). For light microscopy, cells were stained by calcoflour for SI% and by DAPI for the nuclear analysis after glutaraldehyde fixation.
Preparation of S. pombe cell extracts
Cells collected by centrifugation were disrupted by glass beads in the extraction buffer, and the cleared supernatants by 14 000 g centrifugation at 4°C for 10 min were used as extracts.
Chk1 kinase assay
The method described in Capasso et al (2002) was slightly modified. Chk1-HA was immunoprecipitated from extract (0.4 mg protein content) using 40 μl of protein A sepharose CL-4B (Amersham) conjugated with 2.5 μg of 12CA5 anti-HA antibody (Boehringer). The immunoprecipitate beads were washed three times with the extraction buffer (10 mM Na3PO4 at pH 7.0, containing 150 mM NaCl, 1% NP-40, 10 mM EDTA, 50 mM NaF, 2 mM DTT, 1 mM PMSF, 1/100 vol of protease inhibitor cocktail (Nacalai Tesque)) and three times with the Chk1 assay buffer (50 mM Tris–Cl at pH 7.0, containing 5 mM MgCl2, 0.4 mM MnCl2, 25% glycerol (molecular biology grade), 0.1% Triton X-100, 5 mM NaF, 5 mM β-glycerophosphate, 1 mM DTT) in the absence of ATP. An 8 μl aliquot of immunoprecipitate beads was used for the kinase reaction mixture containing 1 μg of the recombinant GST-Cdc25 (60–160 aa of S. pombe Cdc25) protein purified from Escherichia coli as a substrate, 10 μl of 2 × Chk1 assay buffer and 1 pmol [γ-32P]ATP (∼6000 Ci/mmol), resulting in a total volume of 28 μl. After incubation at 30°C for 30 min, samples were analyzed by SDS–PAGE and stained by CBB (R-250) followed by autoradiography.
GST pull-down assay for detecting protein interactions
Recombinant GST protein was overproduced using the inducible nmt1+ promoter. For simultaneous overproduction, two plasmids, REP2 (the strongest overproducer using the marker gene for Ura+) and REP41 (the moderate overproducer using the marker gene for Leu+), were transformed in fission yeast strain h− leu1 ura4 (Maundrell, 1990). Overproduction was carried out in cells at 33°C in the absence of thiamine for 16–18 h, and GST proteins were pulled down using the pull-down buffer (50 mM Tris–Cl at pH 8.0, containing 1 mM EDTA, 10% glycerol, 0.4 M NaCl, 0.1% NP-40, 1 mM Na3VO4, 50 mM NaF, 2 mM DTT, 1 mM PMSF and 1 mM Trasylol). The GST-fused proteins were pulled down using glutathione sepharose 4B beads (Amersham). Beads bound to proteins were gently washed five times with the pull-down buffer. The washed beads were analyzed in SDS–PAGE, and co-precipitated proteins were detected by immunoblotting.
Protein purification from E. coli
GST-fused recombinant proteins were purified using the pGEX expression system (Amersham). Cells treated with 0.5–1 mM IPTG for 3 h at 36°C were disrupted in extraction buffer (phosphate buffer saline at pH 8.5, containing 1% Triton X-100, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 1 mM Trasylol) by sonication. Purified proteins bound to glutathione beads were eluted in the buffer used in the following assay (containing 10 mM glutathione).
In vitro Rad3 assay
The wild-type and the kinase dead (D2249E) GST-Rad3 were overproduced under the nmt1+ promoter (REP1) in the absence of thiamine and purified as described in the GST pull-down assay section using 10 μl glutathione beads starting from the extracts containing 1.5 mg protein. Before the assay of kinase activity, the Rad3 beads were washed three times with the Rad3 assay buffer (10 mM HEPES pH 7.5, containing 50 mM NaCl, 10 mM MgCl2, 10 mM MnCl2, 1 mM DTT). In all, 10 μl of Rad3 beads was used for each reaction mixture containing 1 μg of PHAS-1 as a substrate, 2 μl of 10 × Rad3 assay buffer and 1 pmol [γ-32P]ATP, resulting in a total volume of 30 μl. After incubation at 30°C for 30 min, the samples were analyzed as the Chk1 assay described above.
Supplementary Material
Supplement Figures
Acknowledgments
We thank Drs Nancy Walworth, Paul Russell and Tony Carr for strains. This work was supported by the COE Scientific Research Grant from the Ministry of Education, Culture, Sports, Science and Technology. Matthew J O'Connell is a Scholar of the Leukemia and Lymphoma Society and work in his laboratory was supported by grants from the NHMRC (114229) and the NIH/NCI (CA100076-01).
References
- Al-Khodairy F, Fotou E, Sheldrick KS, Griffiths DJ, Lehmann AR, Carr AM (1994) Identification and characterization of new elements involved in checkpoint and feedback controls in fission yeast. Mol Biol Cell 5: 147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aono N, Sutani T, Tomonaga T, Mochida S, Yanagida M (2002) Cnd2 has dual roles in mitotic condensation and interphase. Nature 417: 197–202 [DOI] [PubMed] [Google Scholar]
- Boddy MN, Furnari B, Mondesert O, Russell P (1998) Replication checkpoint enforced by kinases Cds1 and Chk1. Science 280: 909–912 [DOI] [PubMed] [Google Scholar]
- Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV (1997) A superfamily of conserved domains in DNA damage-responsive cell cycle checkpoint proteins. FASEB J 11: 68–76 [PubMed] [Google Scholar]
- Callebaut I, Mornon JP (1997) From BRCA1 to RAP1: a widespread BRCT module closely associated with DNA repair. FEBS Lett 400: 25–30 [DOI] [PubMed] [Google Scholar]
- Capasso H, Palermo C, Wan S, Rao H, John UP, O'Connell MJ, Walworth NC (2002) Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J Cell Sci 115: 4555–4564 [DOI] [PubMed] [Google Scholar]
- Caspari T, Murray JM, Carr AM (2002) Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev 16: 1195–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman CR, Evans ST, Carr AM, Enoch T (1999) Requirement of sequences outside the conserved kinase domain of fission yeast Rad3p for checkpoint control. Mol Biol Cell 10: 3223–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikashige Y, Kinoshita N, Nakaseko Y, Matsumoto T, Murakami S, Niwa O, Yanagida M (1989) Composite motifs and repeat symmetry in S. pombe centromeres: direct analysis by integration of NotI restriction sites. Cell 57: 739–751 [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- DiTullio RA Jr, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD (2002) 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat Cell Biol 4: 998–1002 [DOI] [PubMed] [Google Scholar]
- Edwards RJ, Bentley NJ, Carr AM (1999) A Rad3–Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nat Cell Biol 1: 393–398 [DOI] [PubMed] [Google Scholar]
- Esashi F, Mochida S, Matsusaka T, Obara T, Ogawa A, Tamai K, Yanagida M (2000) Establishment of and recovery from damage checkpoint requires sequential interactions of Crb2 with protein kinases Rad3, Chk1, and Cdc2. Cold Spring Harbor Symp Quant Biol 65: 443–449 [DOI] [PubMed] [Google Scholar]
- Esashi F, Yanagida M (1999) Cdc2 phosphorylation of Crb2 is required for reestablishing cell cycle progression after the damage checkpoint. Mol Cell 4: 167–174 [DOI] [PubMed] [Google Scholar]
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C (2001) Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol 154: 913–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, D'Urso G (2001) Schizosaccharomyces pombe cells lacking the amino-terminal catalytic domains of DNA polymerase epsilon are viable but require the DNA damage checkpoint control. Mol Cell Biol 21: 4495–4504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi S, Grenon M, Bouvier D, Baldacci G (1997) p56(chk1) protein kinase is required for the DNA replication checkpoint at 37 degrees C in fission yeast. EMBO J 16: 1332–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert CS, Green CM, Lowndes NF (2001) Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol Cell 8: 129–136 [DOI] [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG (2000) Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 14: 2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutz H, Heslot H, Leupold U, Loprieno N (1974) Schizosaccharomyces pombe. In Handbook of Genetics, King RC (ed) pp 395–446. New York: Plenum Press [Google Scholar]
- Hartwell LH, Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246: 629–634 [DOI] [PubMed] [Google Scholar]
- Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP (2002) Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes Dev 16: 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner RD, Putnam CD, Myung K (2002) Maintenance of genome stability in Saccharomyces cerevisiae. Science 297: 552–557 [DOI] [PubMed] [Google Scholar]
- Lindsay HD, Griffiths DJ, Edwards RJ, Christensen PU, Murray JM, Osman F, Walworth N, Carr AM (1998) S-phase-specific activation of Cds1 kinase defines a subpathway of the checkpoint response in Schizosaccharomyces pombe. Genes Dev 12: 382–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A, Tanaka K, Chen XB, Baber BA, McGowan CH, Russell P (2001) Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci USA 98: 11289–11294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinho RG, Lindsay HD, Flaggs G, DeMaggio AJ, Hoekstra MF, Carr AM, Bentley NJ (1998) Analysis of Rad3 and Chk1 protein kinases defines different checkpoint responses. EMBO J 17: 7239–7249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maundrell K (1990) nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J Biol Chem 265: 10857–10864 [PubMed] [Google Scholar]
- Melo J, Toczyski D (2002) A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 14: 237–245 [DOI] [PubMed] [Google Scholar]
- Mitchison JM (1970) Physiological and cytological methods for Schizosaccharomyces pombe. Methods Cell Physiol 4: 131–165 [Google Scholar]
- Paciotti V, Clerici M, Lucchini G, Longhese MP (2000) The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev 14: 2046–2059 [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2000) LCD1: an essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. EMBO J 19: 5801–5812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297: 547–551 [DOI] [PubMed] [Google Scholar]
- Saka Y, Esashi F, Matsusaka T, Mochida S, Yanagida M (1997) Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev 11: 3387–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka Y, Fantes P, Sutani T, McInerny C, Creanor J, Yanagida M (1994) Fission yeast cut5 links nuclear chromatin and M phase regulator in the replication checkpoint control. EMBO J 13: 5319–5329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka Y, Yanagida M (1993) Fission yeast cut5+, required for S phase onset and M phase restraint, is identical to the radiation-damage repair gene rad4+. Cell 74: 383–393 [DOI] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF (2002) Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell 9: 1055–1065 [DOI] [PubMed] [Google Scholar]
- Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H (1998) Regulation of DNA-replication origins during cell-cycle progression. Nature 395: 618–621 [DOI] [PubMed] [Google Scholar]
- Soulier J, Lowndes NF (1999) The BRCT domain of the S. cerevisiae checkpoint protein Rad9 mediates a Rad9–Rad9 interaction after DNA damage. Curr Biol 9: 551–554 [DOI] [PubMed] [Google Scholar]
- Sun Z, Hsiao JFay DS, Stern DF (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281: 272–274 [DOI] [PubMed] [Google Scholar]
- Tasto JJ, Carnahan RH, McDonald WH, Gould KL (2001) Vectors and gene targeting modules for tandem affinity purification in Schizosaccharomyces pombe. Yeast 18: 657–662 [DOI] [PubMed] [Google Scholar]
- Vialard JE, Gilbert CS, Green CM, Lowndes NF (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J 17: 5679–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walworth NC, Bernards R (1996) rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science 271: 353–356 [DOI] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Carpenter PB, Elledge SJ (2002) 53BP1, a mediator of the DNA damage checkpoint. Science 298: 1435–1438 [DOI] [PubMed] [Google Scholar]
- Wang H, Elledge SJ (1999) DRC1, DNA replication and checkpoint protein 1, functions with DPB11 to control DNA replication and the S-phase checkpoint in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 96: 3824–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson J, Wilson S, Warr N, Watts FZ (1997) Isolation and characterization of the Schizosaccharomyces pombe rhp9 gene: a gene required for the DNA damage checkpoint but not the replication checkpoint. Nucleic Acids Res 25: 2138–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Morales JC, Dunphy WG, Carpenter PB (2001) Negative cell cycle regulation and DNA damage-inducible phosphorylation of the BRCT protein 53BP1. J Biol Chem 276: 2708–2718 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figures