

Abstract
Inactivation of the tumor suppressor Rb in the mouse induces cell death, which depends entirely (in lens, CNS) and only partly (PNS, skeletal muscles) on Apaf1/Ced4, an apoptosomal factor thought to be required for processing procaspase-9 following mitochondrial permeabilization. Here, we report that in response to cytotoxic drugs, Apaf1−/− primary myoblasts but not fibroblasts undergo bona fide apoptosis. Cell demise was associated with disruption of mitochondria but not endoplasmic reticulum. Processing of procaspase-9 occurred in Apaf1−/− myoblasts but not fibroblasts, and ablation of Casp9 prevented drug-induced apoptosis in both cell types. Deregulation of the Rb pathway by overexpression of E2F1 also induced caspase-9-dependent, Apaf1-independent apoptosis in myoblasts. Despite its requirement for apoptosis in vitro, mutation in Casp9 abrogated cell death in the nervous system and lens but only partly in skeletal muscles of Rb-deficient embryos. In addition, developmental cell death in fetal liver and PNS was not inhibited in Casp9−/− embryos. Therefore, loss of pRb elicits apoptosome-dependent and apoptosome-independent cell death, and the requirement and coupling of caspase-9 to Apaf1 are both context-dependent.
Keywords: Apaf-1/Ced-4, apoptosis, caspase-9, E2F1, myogenesis, neurogenesis, retinoblastoma
Introduction
Inactivation of the Rb tumor suppressor pathway represents one of the hallmarks of cancer progression (Classon and Harlow, 2002; Zacksenhaus, 2003). In different tumors, Rb is often mutated or its protein product (pRb) is functionally inactivated primarily by phosphorylation (Sherr, 2000). pRb binds certain transcription factors such as E2F1 and modulates the expression of genes involved in S phase progression and DNA synthesis as well as cell death (Nahle et al, 2002; Chau and Wang, 2003). In the course of cancer progression, loss of pRb coincides with inactivation of proapoptotic factors, such as p53 (Classon and Harlow, 2002). However, the exact apoptotic pathways that are induced in response to Rb inactivation during development and cancer progression are not fully understood.
Rb knockout mice provide an excellent model to study survival pathways downstream of pRb. Mutation in Rb leads to mid-gestational death with massive apoptosis, ectopic DNA synthesis and incomplete differentiation in tissues where Rb is highly expressed (Lipinski and Jacks, 1999; Vooijs and Berns, 1999). This includes the developing nervous system (both PNS and CNS), liver, lens and muscles (Jiang et al, 1997). Rb−/− embryos expressing an Rb mini-transgene, mgRb, in the nervous system survive to term and reveal a required role for pRb in terminal myogenesis (Zacksenhaus et al, 1996; Jiang et al, 2001). Developing muscles in mgRb:Rb−/− fetuses exhibit cell death, reduced muscle mass, endoreduplication in residual myotubes and decreased expression of certain muscle markers (Zacksenhaus et al, 1996; Jiang et al, 2000). Placental rescue or conditional inactivation of Rb in the E6.5 embryo proper also extends the life span of Rb-null embryos to birth and reveals similar muscle defects (MacPherson et al, 2003; Wu et al, 2003). Hence, apoptosis in certain tissues (CNS) appears to result from the combined effects of pRb loss and external stress, whereas apoptosis in other tissues (lens, skeletal muscles) seems to be cell autonomous.
Two major apoptotic pathways have been described: the intrinsic mitochondrial pathway and the extrinsic death ligand pathway. In the latter, death ligands such as TNFα induce trimerization of death receptors, activation of apical caspase-8 and subsequently executioner caspases, such as caspase-3 (Ricci et al, 2003). In the intrinsic pathway, DNA damage, hypoxia, cytotoxic drugs or oncogene activation leads to mitochondrial outer membrane permeabilization (MOMP) and release of apoptogenic factors. One such factor, cytochrome c, binds Apaf1 (apoptosis protease activating factor-1) and promotes oligomerization and recruitment of apical procaspase-9 into a high-molecular-weight complex termed the apoptosome (reviewed in Ricci et al, 2003). Assembly of the apoptosome induces autoprocessing of caspase-9 and subsequent activation of caspase-3. Null mutations in Apaf1 or Casp9 lead to similar phenotypes (Cecconi et al, 1998; Hakem et al, 1998; Kuida et al, 1998; Yoshida et al, 1998). Most Apaf1−/− and Casp9−/− mice survive to embryonic day (E) 16.5 and exhibit severe brain abnormalities due to suppression of apoptosis and accumulation of neurons. Several other organs are also affected (e.g. lens, interdigital webs). In addition, both Apaf1−/− and Casp9−/− primary fibroblasts resist apoptosis and cooperate with Myc in promoting neoplastic transformation (Soengas et al, 1999).
Genetic analysis of Rb−/− embryos indicates that in the CNS and lens, cell death can be mitigated by concurrent inactivation of E2F1 or p53 (Morgenbesser et al, 1994; Macleod et al, 1996; Tsai et al, 1998; Jiang et al, 2000). In contrast, apoptosis in PNS and skeletal muscles is independent of these factors. Intriguingly, inactivation of Apaf1 completely suppresses cell death in the CNS and lens of Rb mutant embryos, but only partly rescues the PNS and skeletal muscles (Guo et al, 2001). To elucidate this Apaf1-independent cell death, we herein investigated mechanisms of apoptosis in Apaf1−/− primary myoblasts. We found that Apaf1 is dispensable for apoptosis induced by cytotoxic drugs or overexpression of E2F1 in primary myoblasts but not fibroblasts, whereas caspase-9 is required in both cell types. Despite its critical role in apoptosis in vitro, caspase-9 mediated cell death in only certain cell types in Rb mutant embryos, excluding skeletal muscles. Thus, the coupling of caspase-9 activation to Apaf1 in response to identical apoptotic stimuli and the requirement for the apoptosome as a whole are both cell-type-specific.
Results
Primary myoblasts lacking Apaf1 retain sensitivity to cytotoxic drugs
Primary myoblasts were isolated from limbs of four independent Apaf1−/− mutant embryos exhibiting obvious brain protrusion and used within passage 2–4 (Supplementary Figure S1A and B). The percentage of myoblasts was estimated to be over 90% based on immunostaining for Desmin, a myoblast marker (Supplementary Figure S1D). Both wild-type and Apaf1−/− primary myoblasts were able to differentiate into multinucleated myotubes and express myosin heavy chain (MHC) (Figure 1A), myogenin and Troponin T (not shown).
Figure 1.
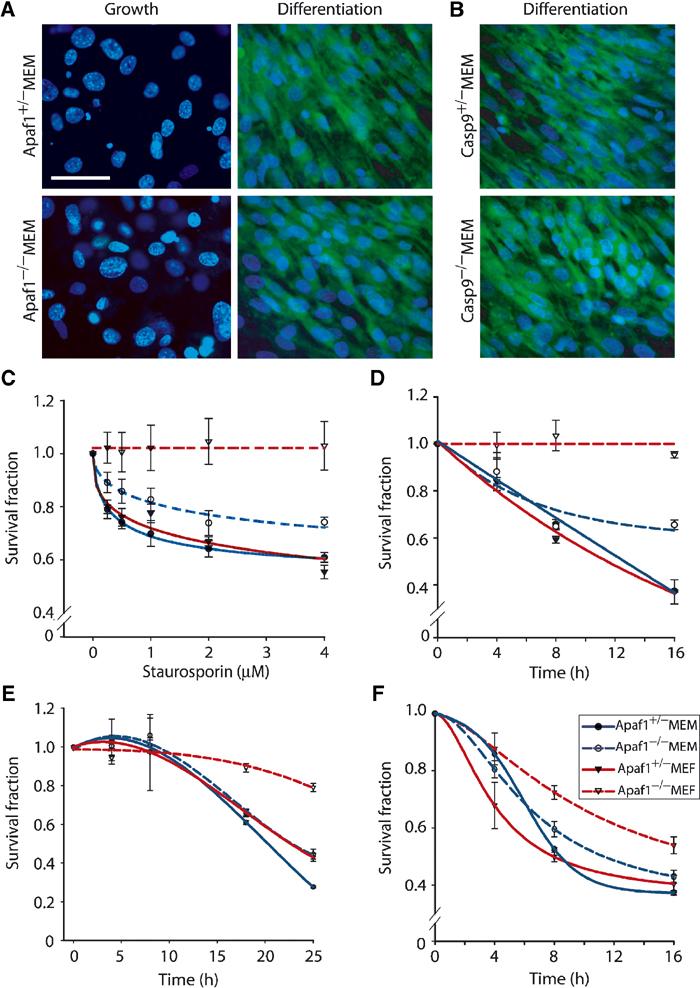
Cytotoxic drugs induce cell death in Apaf1−/− primary myoblasts but not fibroblasts. (A, B) Confocal microscopy images of Apaf1−/−, Casp9−/− and control MEMs cultured in ‘Growth' or ‘Differentiation' conditions, stained for MHC (cytoplasm—green) and counterstained with Hoechst (nucleus—blue). Bar=50 μm. (C–F) Representative MTT assays performed in duplicate on primary cultures treated with increasing concentrations of STS (C), using 2 μM STS for different duration (D); 50 μM CP (E); or 2 μM TNFα (F).
The response of Apaf1−/− primary myoblasts to cytotoxic drugs was determined by the MTT assay, which detects mitochondrial activity, and hence cell proliferation and viability. Treatment of normal mouse embryonic fibroblasts (MEFs) or myoblasts (MEMs) with staurosporin (STS) reduced cell viability in a concentration- and time-dependent manner (Figure 1C and D). Whereas Apaf1−/− MEFs were highly resistant, Apaf1−/− MEMs were almost as sensitive to STS as control cells. The residual resistance to STS in Apaf1−/− mutant MEMs might reflect low contamination with primary fibroblasts or some nonredundant requirement for Apaf1. The kinetics of mitochondrial inhibition with other drugs such as cisplatin (CP) was much slower; substantial reduction in MTT levels was observed only after 16–24 h (Figure 1E). Apaf1−/− MEFs were consistently more resistant to doxorubicin and etoposide relative to control MEF; whereas both wild-type and Apaf1−/− MEMs were sensitive. However, with the latter drugs, the levels of cell viability in both myoblast cultures fluctuated in different experiments (data not shown). We therefore used STS in most subsequent experiments. TNFα, which initiates apoptosis via the extrinsic pathway, suppressed cell viability in all cell types irrespective of Apaf1 (Figure 1F). Thus, the persistent cell death previously observed in skeletal muscles of Rb-Apaf1 double mutant embryos can be recapitulated in primary Apaf1−/− myoblast cultures treated with cytotoxic drugs.
Apaf1−/− myoblasts undergo bona fide apoptosis
To determine the nature of cell death observed in Apaf1−/− myoblasts, we treated the cultures with cytotoxic drugs and tested several parameters that represent hallmarks of apoptosis. Externalization of phosphatidylserine on cell membrane, an early event in apoptosis, was detected with fluorochrome-conjugated AnnexinV, a Ca2+-dependent phospholipid-binding protein, and quantified by flow cytometry. Loss of Apaf1 efficiently protected MEFs from apoptosis, as evident from the reduced percentage of AnnexinV-labeled cells in the right and upper-right quadrants (Figure 2A). Strikingly, Apaf1−/− MEMs apoptosed at a similar rate as control MEMs (Figure 2B). On average, the levels of apoptosis in four different sets of Apaf1−/− MEMs and control myoblasts were 29.6% and 24.9%, respectively (Figure 2C).
Figure 2.

Cytotoxic drugs induce bona fide apoptosis in Apaf1−/− myoblasts. (A, B) MEFs (A) or MEMs (B) were treated with vehicle alone (control) or 2 μM STS for 4 h, stained with FITC-conjugated AnnexinV and PI and quantified by flow cytometry. The percentage of cells in each quadrant is indicated. (C) Average level of apoptosis calculated from five different experiments using control and Apaf1−/− MEMs isolated from four independent litters. (D) TUNEL analysis of indicated cultures treated with buffer (−), CP or STS. (E) Western blot analysis of PARP cleavage in STS-treated cells. (F) Apoptotic morphology in Apaf1−/− myoblasts revealed by electron microscopy. Nuclear condensation (black arrowhead) and ruptured mitochondria (white arrowheads) are indicated. Additional EM micrographs are shown in Supplementary Figure S3.
To test whether cell death in Apaf1−/− myoblasts involved DNA fragmentation, we employed the Terminal-deoxynucleotidyl-transferase (TdT)-mediated dUTP Nick End-Labeling (TUNEL) assay (Gavrieli et al, 1992). Wild-type MEFs were sensitive to CP or STS treatments, whereas Apaf1−/− MEFs were resistant (Figure 2D). Remarkably, Apaf1−/− MEMs exhibited similar levels of TUNEL-positive cells as control MEMs. Thus, cytotoxic drugs can induce DNA degradation in myoblasts in the absence of Apaf1.
We next investigated whether cell death in Apaf1−/− MEMs was accompanied by caspase-dependent protein cleavage. Intriguingly, poly(ADP-ribose) polymerase (PARP), a substrate of caspase-3, was processed in control MEFs and MEMs as well as Apaf1−/− MEMs but not in Apaf1−/− MEFs (Figure 2E). Caspase-3 itself was processed in both controls and Apaf1−/− MEMs (Supplementary Figure S2A).
Ultrastructural analysis revealed that STS treatment induced typical chromosomal condensation, seen as large dense material in the nucleus in both control and Apaf1−/− MEMs (Figure 2F, black arrowhead; Supplementary Figure S3). Apaf1−/− MEFs did not exhibit similar chromosomal condensation in response to STS (Supplementary Figure S3). Mitochondrial swelling and loss of cristae were also detected in Apaf1−/− myoblasts and control cells but not in Apaf1−/− fibroblasts (Figure 2F, white arrowheads; Supplementary Figure S3). Taken together, cytotoxic drugs can induce classical caspase-dependent apoptosis in Apaf1−/− myoblasts in a manner that is indistinguishable from control myoblasts or fibroblasts.
Mitochondrion but not endoplasmic reticulum is implicated in apoptosis induced in Apaf1−/− myoblasts
In addition to the mitochondria, apoptosis can be induced by other organelles such as the endoplasmic reticulum (ER) (Ferri and Kroemer, 2001). ER stress leads to Ca+2-dependent induction of MOMP as well as direct activation of procaspase-9 by ER-bound caspase-12 (Rao et al, 2002). To test whether STS induces apoptosis in myoblasts via the ER, we examined the expression of chaperone Bip/Grp78, which is transcriptionally activated upon ER stress. Thapsigargin (TG), a known inducer of ER stress via specific inhibition of sarcoplasmic/ER Ca+2-ATPase (SERCA) (Lytton et al, 1991), stimulated Bip expression 2–4-fold in wild-type MEFs, MEMs and Apaf1−/− MEMs (Figure 3A and B). In contrast, STS treatment moderately inhibited Bip expression relative to untreated cells, indicating that this drug does not induce ER stress in primary myoblasts.
Figure 3.
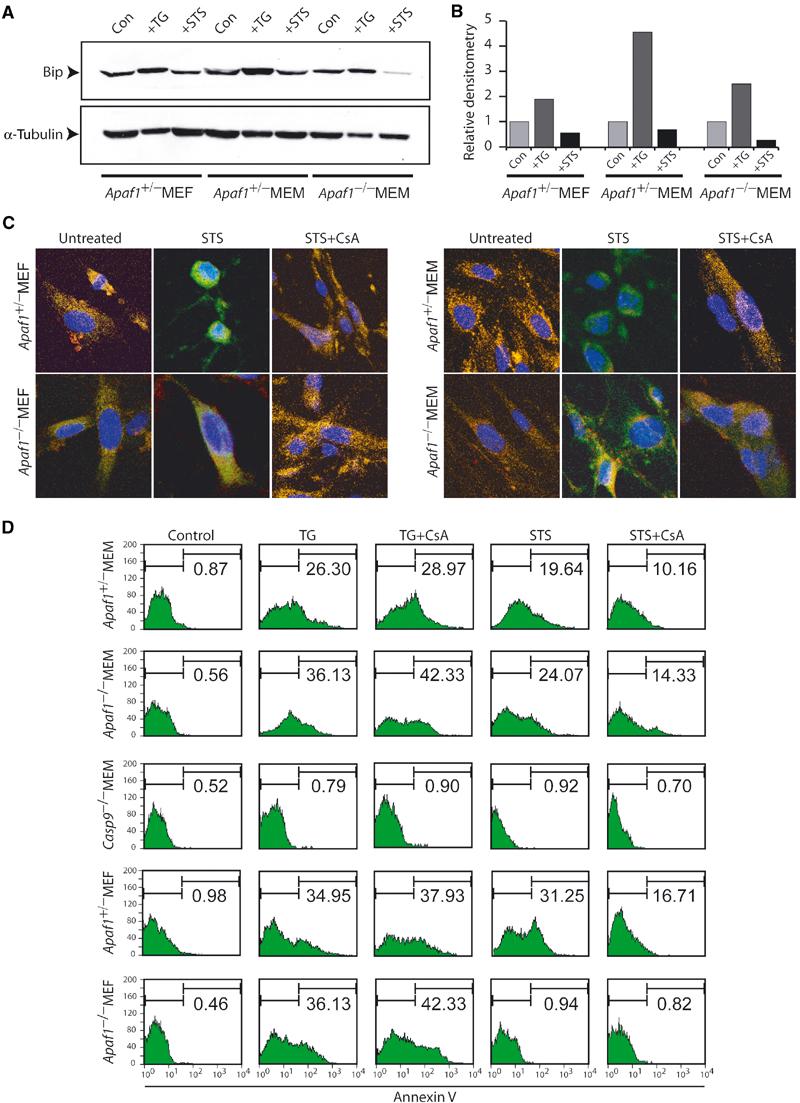
STS induces CsA-suppressible and ER-independent apoptosis in primary myoblasts. (A) Western blot analysis of control and Apaf1−/− myoblasts treated with vehicle (con), TG or STS with Bip antibodies. α-Tubulin was used as a loading control. (B) Levels of Bip relative to α-tubulin were determined by densitometry analysis of the Western blot in (A). TG but not STS induces Bip expression. (C) Confocal images of cytochrome c (green), mitotracker (red) and DAPI (blue) in untreated cultures or following 1 h treatment with STS alone or STS plus CsA. (D) AnnexinV analysis of indicated cultures treated with TG or STS either alone or together with CsA.
In untreated cells, cytochrome c was localized in mitochondria as evident from a characteristic filamentous and overlapping staining with a mitochondrial specific marker (mito-tracker) (Figure 3C). Following STS treatment, cytochrome c staining became more uniform and distinct from the mito-tracker marker, indicative of mitochondrial release. Cytochrome c release appeared in both fibroblasts and myoblasts, irrespective of the presence of Apaf1, and was unlikely the consequence of apoptosis as it was observed immediately 1 h post-STS treatment. Cyclosporine A (CsA), an inhibitor of MOMP (Ricci et al, 2003), suppressed cytochrome c release in all cultures (Figure 3C).
We then asked whether inhibition of cytochrome c release by CsA was associated with suppression of cell death in Apaf1−/− MEMs. Remarkably, cotreatment of control MEFs, MEMs and Apaf1−/− MEMs with STS and CsA reduced the level of apoptosis by approximately 50%, compared to drug treatment alone (Figure 3D). CsA did not attenuate TG-induced apoptosis (Figure 3D). Although Apaf1−/− MEFs were resistant to apoptosis initiated by STS, they readily apoptosed in response to TG. Collectively, these results implicate the mitochondrion or another CsA-sensitive organelle, but not the ER, in mediating Apaf1-independent cell death in skeletal myoblasts.
Cytotoxic drugs induce processing of caspase-9 in Apaf1−/− primary myoblasts
The suppression of apoptosis by CsA in Apaf1−/− mutant myoblasts inspired us to ask whether apoptosis was accompanied by caspase-9 activation, independently of Apaf1. Apoptosome assembly induces autocleavage of procaspase-9, a 49 kDa protein, into 37, 35, 12 and 11 kDa proteins (Li et al, 1997). Caspase-3 further amplifies caspase-9 activity by cleaving procaspase-9 into 37 and 12 kDa fragments (Fujita et al, 2001). In fibroblasts treated with doxorubicin or STS, processed forms of caspase-9 were observed only in the presence of Apaf1 (Figure 4A and B). By contrast, in myoblasts, procaspase-9 was efficiently processed in response to these drugs irrespective of Apaf1 (Figure 4A and B). A doublet of p37 and p35 kDa proteins corresponding to the cleaved forms of caspase-9 was clearly detected in drug-treated wild-type and Apaf1−/− myoblasts. As the p35 fragment is generated only by caspase-9 autocleavage, this observation suggests that autoprocessing of caspase-9 occurred independent of Apaf1 and caspase-3. Processing of procaspase-9 in Apaf1−/− MEMs was further confirmed using a caspase-9 antibody that recognizes only the p35 active form of caspase-9. Both STS and etoposide, but not TNFα, induced the formation of p35 caspase-9 (Supplementary Figure S2B). Since TNFα induced myoblasts cell death (Figure 1F) without activating caspase-9, the cleavage of procaspase-9 in Apaf1−/− myoblasts in response to cytotoxic drugs likely reflects a direct effect of STS rather than an indirect consequence of apoptosis.
Figure 4.

Activation of caspase-9 in Apaf1-deficient myoblasts. Western blot analysis of indicated cultures treated with Doxorubicin (DOX) (A) or STS (B) with antibodies that recognize both pro- and cleaved forms of caspase-9. (C) Bar graph representing relative enzymatic activity of caspase-9 in primary fibroblasts and myoblasts treated with 2 μM STS.
To ascertain that processing of procaspase-9 was accompanied by increased enzymatic activity, we assayed the cleavage of LEHD-AMC, a synthetic fluorogenic substrate of caspase-9 (Figure 4C). Lysates from STS-treated Apaf1−/− MEFs yielded a very low signal. In contrast, Apaf1−/− MEMs exhibited a high level of caspase-9 activity similar to control MEFs and MEMs. The caspase-9 inhibitor LEDH-CHO suppressed this activity. Thus, procaspase-9 can be processed and rendered biologically active in skeletal myoblasts in the absence of Apaf1.
Caspase-9 is required for apoptosis in primary myoblasts
We next investigated whether caspase-9 was not only processed but also required for apoptosis in Apaf1−/− myoblasts. First, we tested the effect of specific caspase inhibitors on apoptosis in the various cell types (Supplementary Figure S2C). The caspase-8 inhibitor (Z-IETD-FMK), which abrogates death ligand-mediated apoptosis, had a minimal to marginal effect on cell death in the different cell types. Conversely, caspase-3 (Z-VAD-fmk) and caspase-9 (AC-LEHD-CHO) inhibitors significantly suppressed apoptosis in both control and Apaf1−/− myoblasts.
To corroborate these results, we generated caspase-9-deficient myoblasts from five E16.5 Casp9−/− null embryos and control littermates (Hakem et al, 1998) (Figure 1B; Supplementary Figure 1C). While ∼40% of wild-type MEMs died by apoptosis, Casp9−/− myoblasts were dramatically resistant (∼0.15%) (Figure 5A and B). TUNEL analysis also revealed 15% positive nuclei in control MEMS but only 0.18% in Casp9−/− myoblasts (Figure 5C). Processing of caspase-3 (Supplementary Figure S2A) and PARP (Figure 5D) was completely inhibited in Casp9−/− myoblasts treated with STS, whereas TNFα-induced processing of PARP was unaffected. Induction of apoptosis by TG was also dependent on caspase-9, even though Apaf1 was dispensable (Figure 3D). Thus, caspase-9 is required for apoptosis induced by cytotoxic drugs in both myoblasts and fibroblasts. However, activation of capsase-9 can be uncoupled from Apaf1 in response to cytotoxic drugs in myoblasts or ER stress in both cell types.
Figure 5.
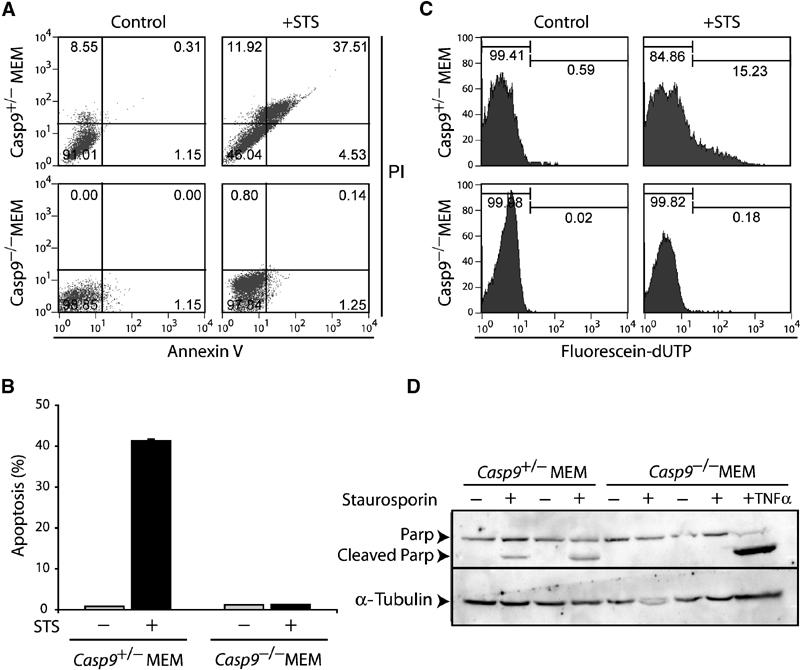
Caspase-9 is required for apoptosis induced by cytotoxic drugs in primary myoblasts. (A) AnnexinV analysis of control and Casp9−/− myoblasts treated with STS. (B) Average level of apoptosis (AnnexinV assay) in control and Casp9−/− MEMs isolated from four independent litters. (C) TUNEL analysis of control and Casp9−/− myoblasts treated with STS. (D) Western analysis of PARP in two independent control and Casp9−/− myoblast cultures treated with STS.
Uncoupling of caspase-9 activation from Apaf1 in myoblasts occurs as a result of deregulation of the pRb pathway
We next asked whether the differential roles of Apaf1 and caspase-9 in myoblasts and fibroblasts in response to cytotoxic drugs could be extended to apoptosis induced by the deregulation of Rb. In one approach, we attempted to induce myoblast cell death by serum starvation. However, in contrast to MEFs, serum deprivation in MEMs induced only a low level of apoptosis. On average, the percentage of apoptosing Rb mutant and control MEMs was ∼6.9 and ∼4%, respectively (measured by AnnexinV staining; data not shown). Rb−/−:Casp9−/− double mutant MEMs consistently exhibited about half the level of apoptosis compared to Rb−/− MEMs (∼3.1%), as did Casp9−/− MEMs (2.7%). While these results suggest that caspase-9 mediates apoptosis downstream of pRb, the low levels of apoptosis in myoblasts precluded conclusive interpretation.
To circumvent this problem, we induced cell death by overexpressing E2F1 using an adenovirus vector in which E2F1 is fused to green fluorescent protein (GFP; Ad.E2F1-GFP; Gill and Hamel, 2000). Previous analysis established that overexpression of E2F1 in primary myoblasts results in similar defects as loss of pRb, except that the level of apoptosis is markedly higher (Wang et al, 1995). Following Ad.E2F1-GFP infection, GFP-positive cells were gated and tested for apoptosis by PE-conjugated AnnexinV assay (red). In agreement with previous studies (Moroni et al, 2001), apoptosis induced by E2F1 was reduced from approximately 35% in control MEFs to ∼4.5% in Apaf1−/− fibroblasts (Figure 6B). However, in myoblasts, apoptosis was only reduced from 36 to 20% in the absence of Apaf1, but to ∼6% in the absence of caspase-9. Thus, both cytotoxic drugs and deregulation of the pRb pathway induce caspase-9 activation independently of Apaf1 in primary myoblasts in vitro.
Figure 6.
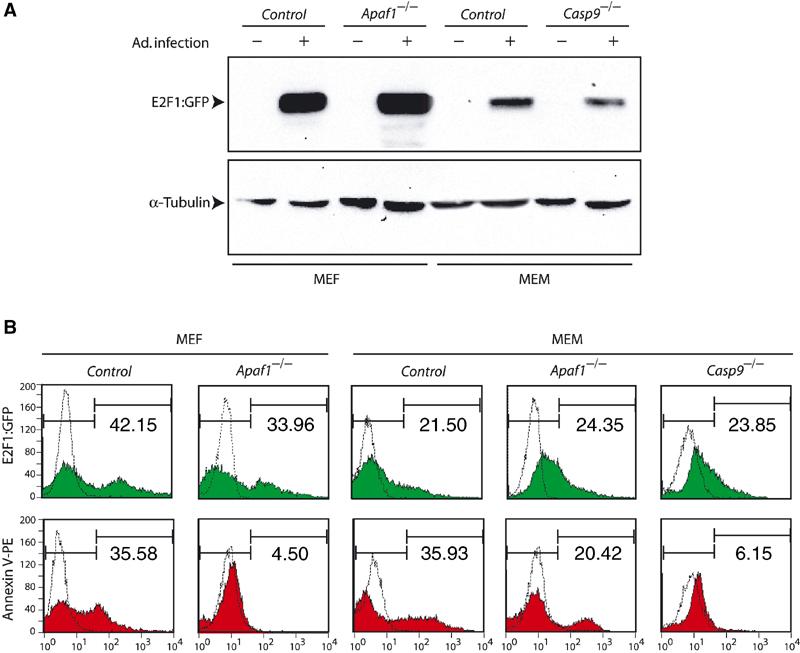
Caspase-9 but not Apaf1 is required for apoptosis induced by overexpression of E2F1 in primary myoblasts. (A) Representative Western blot analysis of primary cells infected with Ad.E2F1-GFP using anti-E2F1 antibodies. Protein loading was monitored with α-tubulin antibody. (B) Following infection with Ad.E2F1-GFP, GFP-positive cells were gated (top, green) and then analyzed by PE-conjugated AnnexinV binding assay (bottom, red) to quantify apoptosis. Numbers in each panel represent percentage of infection and apoptosis, respectively. Dotted curves represent uninfected negative controls.
Mutation in Casp9 only partly inhibits cell death in skeletal muscles of mgRb:Rb−/− mutant fetuses
The observation that caspase-9 was important in myoblasts in vitro even in the absence of Apaf1 prompted us to test whether this caspase was also required for the Apaf1-independent apoptosis that occurs during aberrant myogenesis in mgRb:Rb−/− mutant fetuses (Guo et al, 2001). To address this issue, we generated E16.5 mgRb:Rb−/−:casp9−/− composite mutant fetuses (Table I). As previously noted for mgRb:Rb−/−:Apaf1−/− mice (Guo et al, 2001), none of the mgRb:Rb−/−:Casp9−/− mutant fetuses displayed the characteristic brain protrusions seen in Casp9−/− embryos (not shown). In addition, while mgRb:Rb−/− mutant mice invariably exhibit a hunchback appearance due to lack of muscle toning (Zacksenhaus et al, 1996), three of eight mgRb:Rb−/−:Casp9−/− mutant mice did not show this phenotype, suggesting partial reversal of the muscle defect (not shown). A similar correction of posture was observed in some mgRb:Rb−/−:Apaf1−/− fetuses (Guo et al, 2001). However, histological examination revealed that like mgRb:Rb−/− and mgRb:Rb−/−:Apaf1−/− mutant mice, mgRb:Rb−/−:Casp9−/− skeletal myotubes contained giant nuclei indicative of endoreduplication and were shorter and less abundant relative to wild-type muscles, suggesting widespread cell death (not shown).
Table 1.
Frequency of progenya from Rb and Casp9 heterozygous intercrosses
| E13.5 | Rb+/±:Casp9+/± b | Rb−/−:Casp9+/± | Rb+/±:Casp9−/− | Rb−/−:Casp9−/− | Absorbed | Total |
|---|---|---|---|---|---|---|
| Observed | 81 | 18 | 27 | 5 | 10 | 131 |
| Expected | 73 (56.25%) | 25 (18.75%) | 25 (18.75%) | 8 (6.25%) | — | — |
| |
|
|
|
|
|
|
| E16.5 |
mgRb:Rb+/±:Casp9+/± |
mgRb:Rb−/−:Casp9+/± |
mgRb:Rb+/±:Casp9−/− |
mgRb:Rb−/−:Casp9−/− |
Absorbed |
Total |
| Observed | 102 | 20 | 19 | 8 | 6 | 149 |
| Expected |
84 (56.25%) |
28 (18.75%) |
28 (18.75%) |
9 (6.25%) |
— |
— |
| aGenotype was determined by PCR analysis of two independent biopsies from each embryo. | ||||||
| bEmbryos with at least one wild-type allele for Rb or Caspase9 were designated as +/±. | ||||||
To determine the effect of caspase-9 loss on apoptosis in mgRb:Rb−/− embryos, mutant and control mice were subjected to in situ TUNEL analysis (Figure 7). The level of cell death detected by the TUNEL assay varied substantially in different areas and different sections from the same tissue (i.e. DRG, muscles). These variations were observed in multiple experiments performed in parallel and probably reflected genuine fluctuations in apoptosis rather than technical inconsistencies. To overcome this problem, we performed TUNEL assays on serial sections across different areas from six sets of double mutants, single mutants and control littermates. A total of 86 and 174 sections of E13.5 and E16.5 embryos, respectively, were analyzed in the experiments described below.
Figure 7.
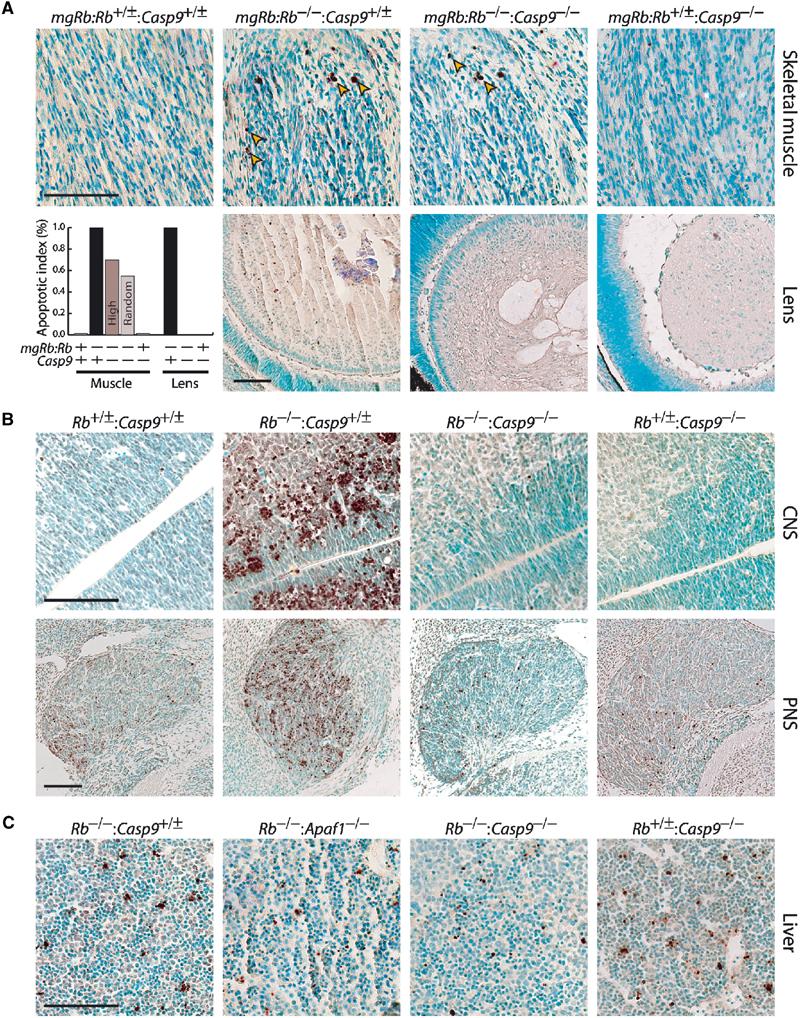
Loss of pRb elicits caspase-9-dependent and -independent cell death. (A) Sections through skeletal muscles and lens of representative E16.5 fetuses, analyzed by in situ TUNEL assay and counterstained with methyl green. Arrowheads point to TUNEL-positive nuclei (stained brown). The diagram shows average levels of cell death in multiple sections from six different litters plotted relative to mgRb:Rb−/− single mutant, which was assigned the value of 100%. For muscles, TUNEL-positive nuclei were scored in areas with a high level of cell death (high) or in multiple random areas of skeletal muscles under × 400 magnification (random). (B) Representative TUNEL assays of E13.5 embryos sectioned through the CNS (spinal cord) or PNS (trigeminal ganglia). (C) Widespread cell death (TUNEL-positive nuclei) in the fetal livers from E13.5 Casp9−/− and Apaf1−/− embryos in the presence or absence of Rb. Scale bar=100 μm.
As previously established, mgRb:Rb−/− fetuses exhibited widespread cell death during terminal myogenesis (Figure 7A) (Zacksenhaus et al, 1996). Strikingly, loss of caspase-9 only moderately reduced the level of cell death in mgRb:Rb−/−:Casp9−/− composite mutants. TUNEL-positive nuclei were scored in areas with a high level of cell death or randomly in multiple muscle areas. As previously found in mgRb:Rb−/−:Apaf1−/− mutant embryos (Guo et al, 2001), the average level of apoptosis in mgRb:Rb−/−:Casp9−/− fetuses was approximately 70% (high areas) and 60% (random areas) relative to mgRb:Rb−/− single mutant mice (Figure 7A, diagram). Thus, despite the strict requirement for caspase-9 in myoblasts in vitro (Figures 5 and 6), loss of pRb initiates a cell death pathway that bypasses this apical caspase during myogenesis in vivo.
Rb mutant embryos undergo massive cell death in the lens epithelium (Morgenbesser et al, 1994; Liu and Zacksenhaus, 2000) (Figure 7A, second row). Concurrent inactivation of caspase-9 completely alleviated cell death in mgRb:Rb−/−:Casp9−/− double mutant lenses (Figure 7A; diagram). The inhibition of cell death in the lens was in stark contrast to the persistent apoptosis in muscles, clearly distinguishing the mgRb:Rb−/−:Casp9−/− from mgRb:Rb−/− fetuses.
Caspase-9 is required for apoptosis in CNS, partly in PNS but not in liver
To determine the role of caspase-9 in cell death in the CNS and PNS of Rb null embryos, we generated E13.5 Rb−/−:Casp9−/− double mutant embryos lacking the mgRb mini-gene (Table I). In the CNS, very few TUNEL-positive nuclei were detected in control and Casp9−/− mutant embryos, whereas in Rb−/− embryos, extensive cell death was observed in the postmitotic zone throughout the spinal cord and brain ventricles (Figure 7B). Loss of caspase-9 dramatically inhibited cell death in the CNS of Rb−/−:Casp9−/− mutant embryos (Figure 7B).
Contrary to the CNS, moderate levels of cell death were observed in the PNS of control and Casp9−/− embryos (Figure 7B). This indicates that caspase-9 is not required for developmentally regulated cell death in the PNS. The low level of cell death in the PNS varied from section to section and was most evident in trigeminal ganglia (Figure 7B). In dorsal root ganglia, only 1–6 nuclei were typically observed in different sections (not shown). The level of apoptosis in the PNS increased dramatically in Rb−/− embryos (Figure 7B). Concomitant loss of pRb and caspase-9 reduced cell death to levels seen in control and Casp9−/− embryos, but was not completely inhibited as in the CNS (Figure 7B). Moderate levels of cell death were also found in the developing livers of Rb−/−, Rb−/−:Apaf1−/−, Rb−/−:Casp9−/− and Casp9−/− mutant embryos (Figure 7C) and control animals (not shown). We conclude that loss of caspase-9 inhibits aberrant cell death induced in Rb-mutant CNS and PNS, but not physiological cell death in the developing PNS and liver.
Discussion
Diverged apoptotic pathways downstream of pRb
We report that inactivation of the tumor suppressor Rb elicits apoptosome-dependent and apoptosome-independent apoptotic pathways. Data presented in Figure 7 and previously (Macleod et al, 1996; Jiang et al, 2000; Guo et al, 2001), indicate that mutation in Rb results in a p53-Apaf1-caspase-9-dependent cell death in the CNS and lens, but p53-Apaf1-caspase-9-independent death in skeletal muscles. The persistent cell death observed in skeletal muscles in the absence of Apaf1 (Guo et al, 2001) or caspase-9 (Figure 7) suggests the existence of alternative apoptotic pathways that can bypass the apoptosome and induce classical apoptotic death (TUNEL-positive). Mutation in Rb may render myoblasts insensitive to survival factors (Ostrovsky and Bengal, 2003) or hypersensitive to death ligands (e.g. TNFα) that can induce apoptosis independent of the mitochondria (Figures 1F and 5D) (Phillips et al, 1999; Chau et al, 2002).
In the PNS, we observed a moderate level of cell death both in wild-type and Casp9−/− embryos (Figure 7B). This indicates that the requirement for caspase-9 can be circumvented by death stimuli generated during normal development of peripheral neurons. However, the excessive apoptosis seen in Rb mutant PNS is suppressed in Rb−/−:Casp9−/− mutant embryos to levels comparable to control and Casp9−/− mutant mice. We previously showed that mutations in Apaf1 reduce but do not completely inhibit cell death in the PNS of Rb null embryos (Guo et al, 2001). Thus, activation of procaspase-9 in the PNS is mediated partly by Apaf1 and partly by an unknown factor/mechanism.
Apoptosis induced during neurogenesis but not hematopoiesis in Bcl-xL knockout embryos (Motoyama et al, 1995) can be mitigated by concurrent inactivation of Apaf1 (Yoshida et al, 2002). Mutation in Casp9 also inhibits apoptosis in Bcl-xL−/− neurons (Zaidi et al, 2001). Thus, inactivation of the apoptosome has similar effects on Rb−/− and Bcl-xL−/− embryos, with the exception that in our hand some apoptotic cells are observed in the Rb−/−:Apaf1−/− and Rb−/−:Casp9−/− PNS. In accordance with recent studies in vitro (Deverman et al, 2002), these observations indicate that pRb and Bcl-xL operate in a common survival pathway at least in certain tissues.
Uncoupling the apoptosome
Our results show for the first time that caspase-9 activation can be uncoupled from Apaf1 in response to classical inducers of MOMP, including STS, CP, doxorubicin and etoposide, as well as overexpression of E2F1, in a cell type-dependent manner (Figures 4 and 6; Supplementary Figure S2). The latter is consistent with previous findings that link DNA damage to induction of E2F1 via ATM (Lin et al, 2001). Uncoupling of the apoptosome has been observed in Sendai virus-infected cells (Bitzer et al, 2002) or following activation of other cellular organelles like the ER (Figure 3 and Rao et al, 2002), but not by cytotoxic drugs or deregulation of E2F1 as reported here.
What then triggers the processing of caspase-9 in Apaf1−/− myoblasts? One possible mechanism, which we began to explore, is that in myoblasts, cytotoxic drugs exert their effect through other organelles, in addition to mitochondria. Although the ER is unlikely to be activated by STS (Figure 3), other organelles may engage procaspase-9 in response to cytotoxic drugs. Noncaspase related factors such as DCC may also target caspase-9 (Forcet et al, 2001). Our observation that CsA suppresses apoptosis in Apaf1−/− myoblasts (Figure 3) directly implicates the mitochondrion. Myoblasts and peripheral neurons may efficiently release mitochondrial apoptogenic factors (e.g. Smac/Diablo) that may facilitate the autoprocessing of caspase-9 even in the absence of Apaf1, as previously suggested (McNeish et al, 2003). A complication in testing this possibility is that smac/diablo null mice develop normally, indicating that this IAP inhibitor plays a redundant role, possibly with HtrA2/Omi (Okada et al, 2002). It is also possible that an Apaf1-like molecule compensates for the loss of Apaf1 in these tissues. Several Apaf1-like factors with CARD, NOD and a leucine-rich repeat in place of the WD domain have been identified (Inohara and Nunez, 2003). However, there is no evidence for the existence of a genuine Apaf1-like protein, comprising CARD, NOD and WD repeats, in the human genome (Aravind et al, 2001). Further research may elucidate the mechanism by which cytotoxic drugs and E2F1 induce procaspase-9 processing in Apaf1−/− primary myoblasts, and may unravel novel therapeutic targets for treating muscle atrophies and other degenerative diseases.
Context-specific requirement for the apoptosome
Mutations in Apaf1 and Casp9 clearly inhibit physiological as well as Rb−/−-induced cell death in the developing CNS (Figure 7) (Hakem et al, 1998; Yoshida et al, 2002). Inactivation of these apoptosomal factors also impedes apoptosis in reconstituted thymocytes and splenocytes in response to some but not all stimuli. Notably, Apaf1 is dispensable for the negative selection of thymocytes (Hara et al, 2002), indicating that in contrast to the response to cytotoxic drugs in vitro, alternative pathways mediate developmental cell death in vivo. This is in agreement with our observation of persistent apoptosis in fetal livers of Casp9−/− and Apaf1−/− mutant embryos (Figure 7C). Importantly, promoter silencing and mutations in Apaf1 were identified in melanoma (Soengas et al, 2001) and other types of tumors (reviewed in Yoshida, 2003), implicating this factor as a critical player downstream of MOMP in these cell types in the course of cancer progression. On the other hand, Apaf1−/− ES and neuronal cells treated with certain cytotoxic drugs exhibit protracted cell death but ultimately die by Bcl2-suppressible and AIF-mediated pathways in vitro (Haraguchi et al, 2000; Cregan et al, 2002). Here, we showed that the coupling of caspase-9 to Apaf1 and the role of the apoptosome as a whole are both context-dependent. Given this diversity, it is becoming apparent that distinct pathways are activated to execute cell demise depending on the tissue, developmental stage and apoptotic stimuli.
Implications for therapy
Previous analysis of Rb−/−:Casp3−/− mutant embryos revealed that caspase-3 is required for apoptosis in the PNS but not the CNS (Simpson et al, 2001). Our observation of caspase-9-dependent apoptosis in the CNS (Figure 7B) implicates other executioner caspases, such as 2, 6 and 7 expressed in the CNS, as mediators of apoptosis downstream of caspase-9 in these neurons. Taken together, caspase-9 may provide a target of choice for chemoprevention of injured neurons in the nervous system. Likewise, the complete inhibition of apoptosis in the lens of Rb−/−:Casp9−/− mutant embryos (Figure 7B) suggests that inhibitors to this apical caspase may impede lens degenerative diseases.
Materials and methods
Isolation, culturing and Adnoviral infection of primary myoblasts and fibroblasts
Limb muscles from E16.5 fetuses were incubated in 0.25% trypsin/EDTA and 300 units/ml collagenase II (Sigma) at 37°C for 30 min, dispersed by repeated tituration with Pasteur pipettes and cultured in growth media (GM; 1:9 DMEM:HAM-F10 media (Sigma), 20% FBS (Hyclone)). Myoblasts were further enriched by differential plating. Briefly, cells were seeded onto poly-L-lysine (Sigma)-coated plates to allow contaminating fibroblasts to adhere for 30 min at 37°C; myoblasts remaining in media suspension were collected and used by passage 4. Control fibroblasts were isolated from the skin of the same embryos. To induce differentiation, myoblasts were allowed to reach confluency on gelatin-coated plates in GM and then switched to differentiation media (DM; HAM-F10, 2% horse serum and 10 μg/ml insulin). Ad.E2F1-GFP was propagated in 293 cells as described (Gill and Hamel, 2000).
MTT assay
A total of 5000 cells/well were seeded into 96-well plates and grown overnight. After drug treatment, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide in PBS, Sigma) was added to 1 mg/ml for 2 h at 37°C, followed by 100 μl of extraction buffer (20% (w/v) SDS, 50% N,N-dimethylformamide, 20% acetic acid and 2.5% 1 N HCl, pH 4.7) overnight. Colorimetric reading at 570 nm was performed using a microplate reader (Molecular Devices).
Western blotting, immunohistochemistry, confocal and electron microscopy
Detailed procedures are available upon requests. For Western blots we used anti-mouse-specific caspase-9 polyclonal antibody (1:1000 dilution; New England Biolabs) and anti-p35 caspase-9 polyclonal antibody (1:200 dilution, SantaCruz) followed by anti-rabbit IgG-horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000 dilution; New England Biolabs). Similar results were obtained using anti-caspase-9 antibody provided by Dr X Wang (Li et al, 1997) (not shown). Additional antibodies included rabbit anti-parp (1:1000 dilution; New England Biolabs), rabbit anti-Bip/Grp75 (1:200 dilution; SantaCruz), goat anti-Apaf1 (1:1000 dilution; Upstate); goat anti-Fast skeletal Troponin T (1:100 dilution; SantaCruz), rabbit anti-myogenin (1:200 dilution; SantaCruz) and monoclonal anti-α-tubulin antibody (1:1500 dilution; Sigma). HRP activity was detected with SuperSignal West Dura chemiluminescence (Pierce) and captured by a Nikon CCD camera via BioRad Flour-S-Max Multimager.
For cytochrome c labeling, cells on coverslips were incubated with mitotracker Red 580 (200 nM, Molecular probes), fixed with 100% methanol at −20°C for 5 min and blocked for 5 min in 1% BSA. Monoclonal anti-cytochrome c antibody (1:50 dilution, SantaCruz) and Alexa 488 (1:100 dilution) goat anti-rabbit secondary antibody were added for 45 min each. Anti-myosin heavy chain (MHC; Sigma-Aldrich, MI) and anti-desmin (SantaCruz, CA) were used at 1:20 dilution. Confocal images were captured at 0.5 μm optical section using a 63 × c-apochromat objective lens and an LSM 510 Zeiss Axiovert 100M confocal microscope. Electron micrographs were taken with Hitachi H600 transmission electron microscope.
AnnexinV and TUNEL assays on single cells
AnnexinV and propidium iodide (PI) assays were performed using FITC-, GFP- or PE-AnnexinV kits (BD Biosciences, San Jose, CA) and quantified with a FACSCalibur flow cytometer (BD Biosciences). In all, 10 000 events were counted for each sample. For TUNEL, 2 × 106 STS-treated or control cells were fixed in 4% paraformaldehyde for 5 min followed by 1 min of 0.1% trypsin digestion. After two washes in PBS, cells were treated with terminal deoxynucleotidyl transferase (100 U/ml, Boeringer, Mannheim) and 10 μM fluorescein-dUTP (Boeringer, Mannheim) at 37°C for at least 1 h. Cells were washed twice in PBS and subjected to flow cytometry analysis.
Caspase-9 activity and caspase inhibition analyses
In vitro caspase-9 activity was assayed according to the ApoAlert Caspase-9 kit (Clontech) using a GENios fluorometer (Tecan) with 380 nm excitation and 460 nm emission. Caspase inhibitors (100 μM, SantaCruz) were incubated with 2 μM STS for 4 h and analyzed by AnnexinV/PI assay.
Breeding and genotyping
Apaf1−/−, Casp9−/−, mgRb:Rb−/− and mgRb:Rb−/−:Casp9−/− embryos were obtained from timed pregnancies of heterozygous intercrosses. Duplicate tissues were isolated from each embryo for genotyping. PCR conditions have been described previously (Zacksenhaus et al, 1996; Hakem et al, 1998; Yoshida et al, 1998).
Histology and TUNEL analysis on sections
Briefly, embryos were fixed in 4% paraformaldehyde/PBS at 4°C overnight, embedded in paraffin and sectioned (8 μm). H&E and TUNEL assays, using biotin-dUTP, followed by DAB staining were performed as described (Jiang and Zacksenhaus, 2002).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Acknowledgments
We thank Paul Hamel for provision of Ad.E2F1-GFP, Xiaodong Wang for anti-caspase-9 antibody and Zhe Jiang for advice on TUNEL analysis. ATH has been supported by fellowships from the Frank Fletcher Memorial Fund, and Heart & Stroke Foundation of Canada. EZ holds a Cancer Research Society/Canadian Institute for Health Research (CIHR) Scientist Scholarship. This work was supported by grants to EZ from the Canadian National Institute for the Blindness (CNIB) and CIHR.
References
- Aravind L, Dixit VM, Koonin EV (2001) Apoptotic molecular machinery: vastly increased complexity in vertebrates revealed by genome comparisons. Science 291: 1279–1284 [DOI] [PubMed] [Google Scholar]
- Bitzer M, Armeanu S, Prinz F, Ungerechts G, Wybranietz W, Spiegel M, Bernlohr C, Cecconi F, Gregor M, Neubert WJ, Schulze-Osthoff K, Lauer UM (2002) Caspase-8 and Apaf-1-independent caspase-9 activation in Sendai virus-infected cells. J Biol Chem 277: 29817–29824 [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94: 727–737 [DOI] [PubMed] [Google Scholar]
- Chau BN, Borges HL, Chen TT, Masselli A, Hunton IC, Wang JY (2002) Signal-dependent protection from apoptosis in mice expressing caspase- resistant Rb. Nat Cell Biol 4: 757–765 [DOI] [PubMed] [Google Scholar]
- Chau BN, Wang JY (2003) Coordinated regulation of life and death by RB. Nat Rev Cancer 3: 130–138 [DOI] [PubMed] [Google Scholar]
- Classon M, Harlow E (2002) The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer 2: 910–917 [DOI] [PubMed] [Google Scholar]
- Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 158: 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Cook BL, Manson SR, Niederhoff RA, Langer EM, Rosova I, Kulans LA, Fu X, Weinberg JS, Heinecke JW, Roth KA, Weintraub SJ (2002) Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell 111: 51–62 [DOI] [PubMed] [Google Scholar]
- Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3: E255–263 [DOI] [PubMed] [Google Scholar]
- Forcet C, Ye X, Granger L, Corset V, Shin H, Bredesen DE, Mehlen P (2001) The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc Natl Acad Sci USA 98: 3416–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita E, Egashira J, Urase K, Kuida K, Momoi T (2001) Caspase-9 processing by caspase-3 via a feedback amplification loop in vivo. Cell Death Differ 8: 335–344 [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill RM, Hamel PA (2000) Subcellular compartmentalization of E2F family members is required for maintenance of the postmitotic state in terminally differentiated muscle. J Cell Biol 148: 1187–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Shi Y, Hiroki Y, Mak T, Zacksenhaus E (2001) Inactivation of the retinoblastoma tumor suppressor induces Apaf-1 dependent and independent apoptotic pathways during embryogenesis. Cancer Res 61: 8395–8400 [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW (1998) Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell 94: 339–352 [DOI] [PubMed] [Google Scholar]
- Hara H, Takeda A, Takeuchi M, Wakeham AC, Itie A, Sasaki M, Mak TW, Yoshimura A, Nomoto K, Yoshida H (2002) The apoptotic protease-activating factor 1-mediated pathway of apoptosis is dispensable for negative selection of thymocytes. J Immunol 168: 2288–2295 [DOI] [PubMed] [Google Scholar]
- Haraguchi M, Torii S, Matsuzawa S, Xie Z, Kitada S, Krajewski S, Yoshida H, Mak TW, Reed JC (2000) Apoptotic protease activating factor 1 (Apaf-1)-independent cell death suppression by Bcl-2. J Exp Med 191: 1709–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inohara N, Nunez G (2003) NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol 3: 371–382 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Gou Z, Saad F, Ellis J, Zacksenhaus E (2001) Retinoblastoma gene promoter directs transgene expression exclusively to the nervous system. J Biol Chem 276: 593–600 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Liang P, Leng R, Guo Z, Liu Y, Liu X, Bubnic S, Keating A, Murray D, Goss PE, Zacksenhaus E (2000) E2F1 and p53 are dispensable whereas p21Waf1/Cip1 cooperates with Rb to restrict endoreduplication and apoptosis during skeletal myogenesis. Dev Biol 227: 28–41 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Zacksenhaus E (2002) Activation of retinoblastoma protein in mammary gland leads to ductal growth suppression, precocious differentiation, and adenocarcinoma. J Cell Biol 156: 185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Zacksenhaus E, Gallie BL, Phillips RA (1997) The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene 14: 1789–1797 [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94: 325–337 [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489 [DOI] [PubMed] [Google Scholar]
- Lin WC, Lin FT, Nevins JR (2001) Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev 15: 1833–1844 [PMC free article] [PubMed] [Google Scholar]
- Lipinski MM, Jacks T (1999) The retinoblastoma gene family in differentiation and development. Oncogene 18: 7873–7882 [DOI] [PubMed] [Google Scholar]
- Liu Y, Zacksenhaus E (2000) E2F1 mediates ectopic proliferation and stage-specific p53-dependent apoptosis but not aberrant differentiation in the ocular lens of Rb deficient fetuses. Oncogene 19: 6065–6073 [DOI] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR (1991) Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem 266: 17067–17071 [PubMed] [Google Scholar]
- Macleod KF, Hu Y, Jacks T (1996) Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J 15: 6178–6188 [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T (2003) Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol 23: 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish IA, Bell S, McKay T, Tenev T, Marani M, Lemoine NR (2003) Expression of Smac/DIABLO in ovarian carcinoma cells induces apoptosis via a caspase-9-mediated pathway. Exp Cell Res 286: 186–198 [DOI] [PubMed] [Google Scholar]
- Morgenbesser SD, Williams BO, Jacks T, DePinho RA (1994) p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature 371: 72–74 [DOI] [PubMed] [Google Scholar]
- Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H, Helin K (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3: 552–558 [DOI] [PubMed] [Google Scholar]
- Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY (1995) Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science 267: 1506–1510 [DOI] [PubMed] [Google Scholar]
- Nahle Z, Polakoff J, Davuluri RV, McCurrach ME, Jacobson MD, Narita M, Zhang MQ, Lazebnik Y, Bar-Sagi D, Lowe SW (2002) Direct coupling of the cell cycle and cell death machinery by E2F. Nat Cell Biol 4: 859–864 [DOI] [PubMed] [Google Scholar]
- Okada H, Suh WK, Jin J, Woo M, Du C, Elia A, Duncan GS, Wakeham A, Itie A, Lowe SW, Wang X, Mak TW (2002) Generation and characterization of Smac/DIABLO-deficient mice. Mol Cell Biol 22: 3509–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrovsky O, Bengal E (2003) The mitogen-activated protein Kinase cascade promotes myoblast cell survival by stabilizing the cyclin-dependent Kinase inhibitor, p21WAF1 protein. J Biol Chem 278: 21221–21231 [DOI] [PubMed] [Google Scholar]
- Phillips AC, Ernst MK, Bates S, Rice NR, Vousden KH (1999) E2F-1 potentiates cell death by blocking antiapoptotic signaling pathways. Mol Cell 4: 771–781 [DOI] [PubMed] [Google Scholar]
- Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM (2002) Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem 277: 21836–21842 [DOI] [PubMed] [Google Scholar]
- Ricci JE, Waterhouse N, Green DR (2003) Mitochondrial functions during cell death, a complex (I–V) dilemma. Cell Death Differ 10: 488–492 [DOI] [PubMed] [Google Scholar]
- Sherr CJ (2000) The Pezcoller lecture: cancer cell cycles revisited. Cancer Res 60: 3689–3695 [PubMed] [Google Scholar]
- Simpson MT, MacLaurin JG, Xu D, Ferguson KL, Vanderluit JL, Davoli MA, Roy S, Nicholson DW, Robertson GS, Park DS, Slack RS (2001) Caspase 3 deficiency rescues peripheral nervous system defect in retinoblastoma nullizygous mice. J Neurosci 21: 7089–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW (1999) Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 284: 156–159 [DOI] [PubMed] [Google Scholar]
- Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, McCombie R, Herman JG, Gerald WL, Lazebnik YA, Cordon-Cardo C, Lowe SW (2001) Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409: 207–211 [DOI] [PubMed] [Google Scholar]
- Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T (1998) Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell 2: 293–304 [DOI] [PubMed] [Google Scholar]
- Vooijs M, Berns A (1999) Developmental defects and tumor predisposition in Rb mutant mice. Oncogene 18: 5293–5303 [DOI] [PubMed] [Google Scholar]
- Wang J, Helin K, Jin P, Nadal-Ginard B (1995) Inhibition of in vitro myogenic differentiation by cellular transcription factor E2F1. Cell Growth Differ 6: 1299–1306 [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, Rosol TJ, Woollett LA, Weinstein M, Cross JC, Robinson ML, Leone G (2003) Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421: 942–947 [DOI] [PubMed] [Google Scholar]
- Yoshida H (2003) The role of apaf-1 in programmed cell death: from worm to tumor. Cell Struct Funct 28: 3–9 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739–750 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okada Y, Kinoshita N, Hara H, Sasaki M, Sawa H, Nagashima K, Mak TW, Ikeda K, Motoyama N (2002) Differential requirement for Apaf1 and Bcl-X(L) in the regulation of programmed cell death during development. Cell Death Differ 9: 1273–1276 [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E (2003) Alternative reading frame suggests an alternative model for Retinoblastoma. Cell Cycle 2: 27–30 [DOI] [PubMed] [Google Scholar]
- Zacksenhaus E, Jiang Z, Chung D, Marth J, Phillips RA, Gallie BL (1996) pRb controls cell proliferation, differentiation and death of skeletal muscle cells and other lineages during embryogenesis. Genes Dev 10: 3051–3064 [DOI] [PubMed] [Google Scholar]
- Zaidi AU, D'Sa-Eipper C, Brenner J, Kuida K, Zheng TS, Flavell RA, Rakic P, Roth KA (2001) Bcl-X(L)-caspase-9 interactions in the developing nervous system: evidence for multiple death pathways. J Neurosci 21: 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3