

Abstract
The two iron regulatory proteins IRP1 and IRP2 bind to transcripts of ferritin, transferrin receptor and other target genes to control the expression of iron metabolism proteins at the post-transcriptional level. Here we compare the effects of genetic ablation of IRP1 to IRP2 in mice. IRP1−/− mice misregulate iron metabolism only in the kidney and brown fat, two tissues in which the endogenous expression level of IRP1 greatly exceeds that of IRP2, whereas IRP2−/− mice misregulate the expression of target proteins in all tissues. Surprisingly, the RNA-binding activity of IRP1 does not increase in animals on a low-iron diet that is sufficient to activate IRP2. In animal tissues, most of the bifunctional IRP1 is in the form of cytosolic aconitase rather than an RNA-binding protein. Our findings indicate that the small RNA-binding fraction of IRP1, which is insensitive to cellular iron status, contributes to basal mammalian iron homeostasis, whereas IRP2 is sensitive to iron status and can compensate for the loss of IRP1 by increasing its binding activity. Thus, IRP2 dominates post-transcriptional regulation of iron metabolism in mammals.
Keywords: cytosolic aconitase, iron metabolism, iron regulatory protein 1 (IRP1), iron-responsive element (IRE)
Introduction
Iron regulatory protein 1 (IRP1) is a ubiquitously expressed bifunctional protein that functions either as a cytosolic aconitase or as an RNA-binding protein involved in post-transcriptional regulation of iron metabolism (Hentze and Kuhn, 1996; Rouault and Klausner, 1997; Schneider and Leibold, 2000). Similar to the related citric acid cycle enzyme (mitochondrial aconitase) and other members of the aconitase family (Gruer et al, 1997), the enzymatic form of IRP1 contains a (4Fe–4S)2+ cluster prosthetic group at its catalytic center, and interconverts citrate and isocitrate (Kennedy et al, 1992). Although IRP1 is only 22% identical to mitochondrial aconitase in peptide sequence (Rouault et al, 1991), its enzymatic specific activities with all three substrates are very similar to those of mitochondrial aconitase (Kennedy et al, 1992). However, unlike mitochondrial aconitase, the role of cytosolic aconitase in mammalian cellular metabolism remains poorly understood. IRP1 homologs are found in numerous organisms, including Caenorhabditis elegans (Gourley et al, 2003) and Drosophila melanogaster (Muckenthaler et al, 1998), but there are no IRP1 homologues in yeast.
In mammals, IRP1 is one of two iron regulatory proteins (IRP1 and IRP2) that bind to RNA stem-loop elements known as iron-responsive elements (IREs) found within transcripts that encode several iron metabolism proteins. IRP binding to IREs within 5′UTRs of transcripts such as ferritin results in translational repression, whereas binding to IREs in the 3′UTR of the transferrin receptor (TfR) results in stabilization of the transcript. Thus, IRPs coordinate the cellular response to iron depletion by decreasing iron storage and increasing iron uptake (Hentze and Kuhn, 1996; Rouault and Klausner, 1997; Schneider and Leibold, 2000). IRP1 and IRP2 share high sequence homology and exhibit very similar biochemical activities with respect to binding affinity and in vitro regulation of IRE-containing transcripts (Kim et al, 1995; Allerson et al, 1999). Overexpression of a form of IRP1 that constitutively binds IREs results in repression of ferritin synthesis and increased TfR expression (DeRusso et al, 1995; Wang and Pantopoulos, 2002), indicating that IRP1 can regulate its proposed target transcripts in cells. Furthermore, although both IRPs are ubiquitously expressed, IRP1 was the focus of much early interest because it appeared to be much more abundant than IRP2 in most cells and tissues (Rouault et al, 1990; Hirling et al, 1992; Mullner et al, 1992; Patino and Walden, 1992; Yu et al, 1992; Henderson et al, 1993). Thus, based on binding affinities, regulatory activities and abundance, IRP1 would be expected to be the predominant mammalian regulator of post-transcriptional iron metabolism.
We have previously reported that IRP2 plays an important physiological role in systemic iron homeostasis, as indicated by the fact that IRP2−/− mice develop a progressive neurodegenerative disease associated with misregulation of iron metabolism in specific areas of the brain and the small intestine (LaVaute et al, 2001). These results indicate that IRP1 alone is unable to regulate appropriately iron metabolism in mammalian tissues, and they are consistent with a report that IRP1 is not required for normal regulation of IRP targets in a lymphocyte cell line (Schalinske et al, 1997).
To evaluate the role of IRP1 in mammalian iron physiology, we have genetically ablated IRP1 in mice. IRP1−/− mice show no overt pathology and show compromise of normal iron metabolism only in tissues in which the level of IRP1 greatly exceeds that of IRP2. We demonstrate that IRP1 predominantly exists in the cytosolic aconitase form, and that iron starvation sufficient to activate IRP2 does not increase the IRE-binding activity of IRP1. Thus, although IRP1 represents a large pool of potential IRE-binding activity, IRP2 is the primary post-transcriptional regulator of mammalian iron metabolism in response to iron status.
Results
Targeted deletion of IRP1
Mice with a targeted disruption of IRP1 were generated by homologous recombination with a construct that inserted the neomycin resistance gene into exon 13. Genotyping by Southern analysis using the probe indicated in Figure 1A showed a 10.1 kb wild-type (wt) band, a 4.1 kb mutant band or both bands in heterozygous mice (Figure 1B). The absence of IRP1 can be confirmed by two functional assays. Electrophoretic separation on cellulose acetate membranes, followed by a coupled enzymatic assay for aconitase activity, revealed two distinct bands, which represent cytosolic and mitochondrial aconitase. Targeted deletion of IRP1 abolished cytosolic aconitase activity without affecting mitochondrial aconitase activity (Figure 1C). Likewise, by gel retardation assay, we distinguished IRP1 from IRP2 with a radiolabeled IRE probe and demonstrated the absence of IRP1-dependent IRE-binding activity in embryonic fibroblasts of IRP1−/− mice (Figure 1D). In addition, IRP1 was not detectable on Western blots of IRP1−/− animals (not shown).
Figure 1.
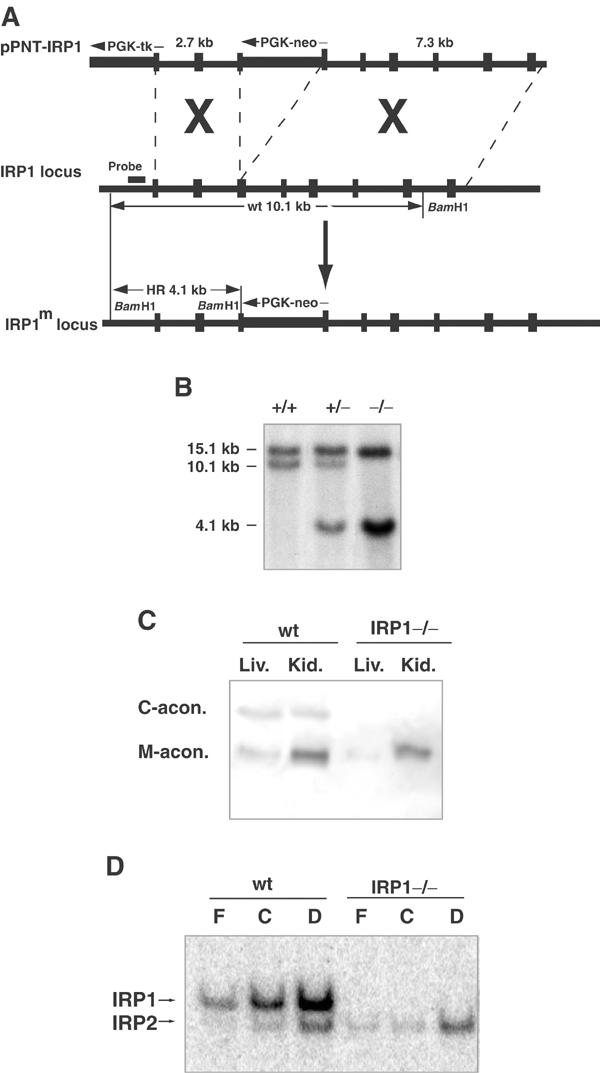
Targeted deletion of IRP1 in mouse. (A) Schematic diagram of the IRP1 deletion construct (top), IRP1 genomic clone (middle) and IRP1 deletion allele (bottom). (B) Southern analysis of BamHI digest of genomic DNA isolated from mouse tailcuts. In lane 1, wt genomic DNA shows a band of 10.1 kb. In lane 2, genomic DNA from a heterozygote mouse shows a wt band of 10.1 kb and a mutant band of 4.1 kb. In lane 3, DNA from a homozygous mutant shows only a 4.1 kb band. In all three lanes, a wt band for IRP2 is visible at 15.1 kb. (The probe used for IRP1 detection is indicated in (A).) (C) Cytosolic and mitochondrial aconitase activities in mouse liver and kidney lysates were assayed after cellulose acetate electrophoresis. Note that lysates from wt mice show both cytosolic and mitochondrial aconitase activity, while in lysates from IRP1−/− mice cytosolic aconitase activity is absent. (D) Gel retardation assays of IRPs. Lysates of wt and IRP1−/− mouse embryonic fibroblasts (15 μg protein/lane) were incubated with 32P-IRE and resolved on a 10% nondenaturing gel. Both IRPs respond to decreased iron concentration with increased IRE-binding activity. In lysates from IRP1−/− cells, IRP1 IRE-binding activity is absent (C: control; F: 100 μg/ml ferric ammonium citrate; D: 50 μM deferoxamine).
Viability and health of IRP1−/− animals
Offspring from IRP1+/− matings had a distribution of 27% wt, 27% IRP1−/− and 46% IRP1+/− (n=190), which is consistent with the genotype distribution predicted for this type of mating (25% wt, 25% IRP1−/− and 50% IRP1+/−). Litter sizes of wt matings were comparable to litter sizes of IRP1−/− matings (6.3±2.7 and 8.05±2.6, respectively). Furthermore, all major tissues and glands were histologically normal when stained with standard hematoxylin and eosin (H&E) stains or iron stains. Complete blood cell counts and serum chemistries showed no difference between wt and IRP1−/− mice. Thus, IRP1−/− mice develop and reproduce normally.
Expression patterns of IRP1 and IRP2
We have analyzed ferritin and TfR expression in primary cultures of embryonic fibroblasts of the IRP1−/− and IRP2−/− genotype, and found that each cell line is able to iron-dependently regulate ferritin and TfR expression despite the striking phenotypical difference between IRP1−/− and IRP2−/− mice (LaVaute et al, 2001; EG Meyron-Holtz, unpublished). As these results suggested that each IRP was capable of mediating iron-dependent regulation, we asked whether the difference in phenotype of the two IRP−/− mouse models was attributable to differences in tissue-specific expression levels of the two IRPs. In situ hybridization assays revealed that both IRPs were ubiquitously expressed, but the relative expression levels of the two mRNAs differed in a cell- and tissue-specific manner. In newborn pups (P0), IRP1 mRNA was highly expressed in the brown fat, liver and small intestine, whereas IRP2 mRNA expression was highest in the forebrain, cerebellum, dorsal root ganglia, thymus (insert) and retina (Figure 2A). This expression pattern was consistent with the expression pattern detected in adult tissues. IRP2 mRNA was highly expressed in many areas of the adult brain such as the substantia nigra, the granule cell layer of the cerebellum and the hippocampus (Figure 2B). In contrast, IRP1 was highly expressed in several peripheral tissues, including the liver and kidney, whereas IRP1 expression was low in the spleen and heart (Figure 2C). Notably, the expression pattern varies among cell types within specific tissues, a fact that may complicate interpretation of tissue lysate results.
Figure 2.

Expression patterns of IRP1 and IRP2 determined by in situ hybridization and Western blotting. In situ hybridization was performed using DIG-labeled cRNA probes and AP detection. Purple/brown staining develops in RNA-positive areas. (A) Fresh frozen sections of newborn pups at postpartum day 0 (P0); inset: thymus. Note that IRP1 is highly expressed in the brown fat and liver, whereas IRP2 expression is high in the brain, thymus and retina. (B) Adult brain coronal sections reveal that IRP2 is highly expressed in several brain regions. IRP1 is also expressed in these regions. (C) In adult peripheral tissues, IRP1 is highly expressed in the kidney, liver and epididymis, whereas IRP2 is most highly expressed in the testis. (D) Protein analysis by Western blotting. Lysates from each of the tissues indicated (40 μg protein/lane) were separated by SDS–PAGE, transferred to membranes, and IRP levels were compared between tissues in one gel that is representative of three separate experiments. Note that IRP1 levels are highest in the kidney, liver and brown fat, whereas IRP2 levels are highest in the cerebellum and forebrain and are hardly detectable in the brown fat.
To verify that levels of mRNA expression of IRP1 and IRP2 in different tissues correlated with levels of protein expression, we measured IRP1 and IRP2 protein levels from wt tissue lysates by Western blot. The protein levels were consistent with the results of the in situ hybridizations, showing the highest IRP1 expression predominantly in the kidney, liver and brown fat, whereas the highest IRP2 expression was in the forebrain and cerebellum (Figure 2D).
Tissue-specific misregulation of ferritin in IRP1−/− mice
To examine whether tissue-specific misregulation of ferritin in IRP−/− mice was attributable to differences in tissue-specific expression levels of the two IRPs, levels of H and L subunits of ferritin and TfR levels in various tissues of IRP1−/− or IRP2−/− mice were compared to wt mice. All tissues examined from IRP2−/− mice showed an elevation of steady-state ferritin levels compared to wt tissues, consistent with derepression of ferritin synthesis in all of these tissues (Figure 3A and B). Likewise, TfR decreased in the cerebellum and forebrain from IRP2−/− mice, consistent with lack of protection of the TfR transcript by IRP binding. These results indicate that in the absence of IRP2, IRP1 alone is unable to maintain normal TfR and ferritin levels in any of the examined tissues, and that IRP2 plays an essential role in the regulation of iron metabolism in living animals. In contrast, IRP1−/− mice showed no apparent misregulation of ferritin expression in the cerebellum, forebrain and liver. Elevated levels of ferritin were found only in the brown fat and kidney. IRP1, therefore, is needed for full regulation of iron metabolism in a few specific tissues, whereas in other tissues, IRP2 alone is sufficient for the regulation of ferritin levels (Figure 3B). In summary, we find that the high expression of IRP1 relative to IRP2 in the kidney and brown fat correlates with misregulation of ferritin in these tissues in the IRP1−/− mouse. However, loss of IRP2 disrupts iron metabolism in virtually all tissues, even including those in which its expression is very low, such as brown fat.
Figure 3.
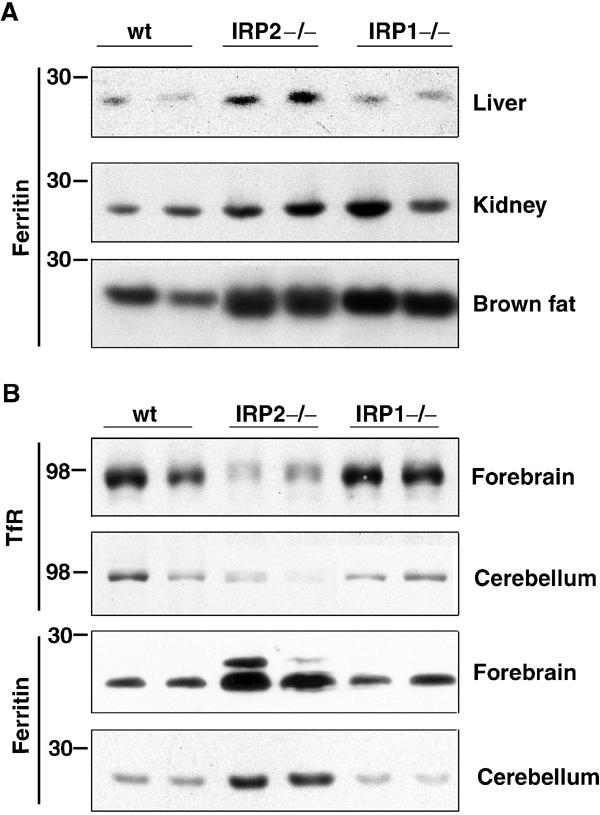
Ferritin levels are elevated in multiple tissues from IRP2−/− mice, but are elevated only in the kidney and brown fat of IRP1−/− mice. (A) Western blot analysis of ferritin in the liver, kidney and brown fat was performed on tissue lysates of wt, IRP1−/− and IRP2−/− animals using antibodies against mouse liver ferritin. (B) Western blot analysis of ferritin or TfR from the forebrain or cerebellum was performed on tissue lysates of wt, IRP1−/− and IRP2−/− animals using antibodies to mouse ferritin H-chain (forebrain), mouse liver ferritin (cerebellum) or TfR antibodies. The slower running band in forebrain ferritin is L chain that crossreacts with anti-H-chain antibody. Results are presented in duplicate and are representative of at least three experiments.
The fraction of IRP1 with cytosolic aconitase activity is much greater than the fraction with IRE-binding activity
Since Western analysis of IRP1 detects total IRP1 protein and cannot distinguish between its two functional states, we performed cytosolic aconitase and gel retardation assays to investigate the amount of IRP1 that was either a functional enzyme or an IRE-binding protein. Detection of significant amounts of cytosolic aconitase in the kidney and liver and a smaller amount in brown fat correlated well with the IRP1 expression levels determined in the Western blot (Figure 4A). In comparison, cytosolic aconitase activity was much lower in the cerebellum, forebrain, skeletal muscle and heart. Also, the gel retardation assay reflected this tissue-specific distribution. In vitro addition of 2-mercaptoethanol (2-ME) can reduce and convert IRP1 from its non-IRE-binding to its IRE-binding form, thus providing a means to measure total activatable IRP1. Comparison of IRE-binding activity of IRP1 to the total 2-ME-activatable IRE-binding activity of IRP1 indicated that between 4 and 18% of IRP1 was in an IRE-binding state in various tissues (Figure 4B).
Figure 4.
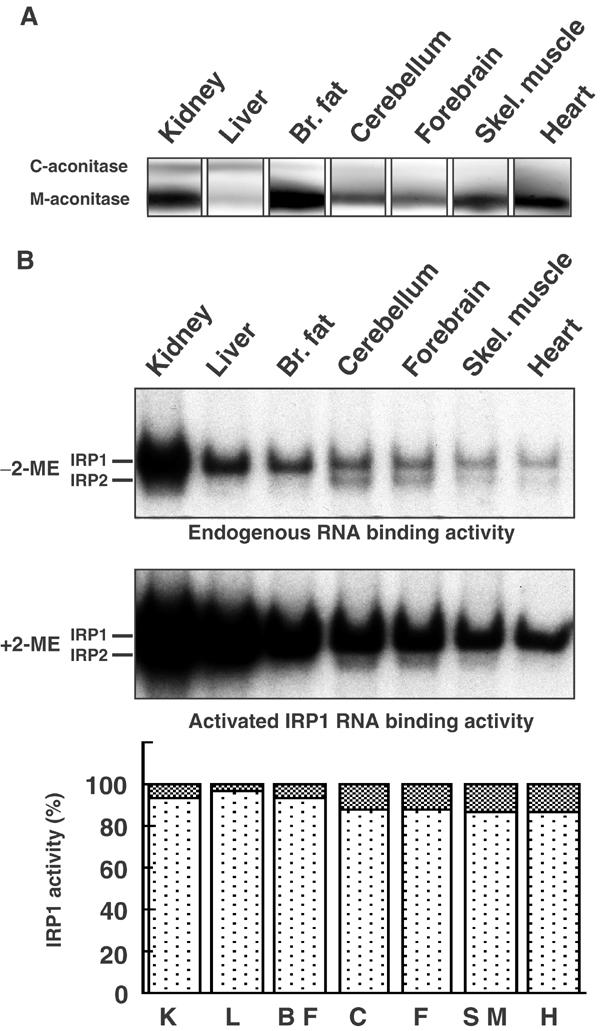
IRP1 activity levels in tissues determined by aconitase and gel retardation assays. (A) Cytosolic and mitochondrial aconitase activities in mouse tissue lysates were assayed after cellulose acetate electrophoresis. Note that relative amounts of the two aconitases vary greatly between tissues and that cytosolic aconitase is mainly detected in the kidney, liver and brown fat. (B) Gel retardation assays of IRPs. Lysates from wt tissues (9 μg protein/lane) were incubated with 32P-IRE and resolved on a 10% nondenaturing gel. Assay without 2-ME reflects IRE-binding activity in vivo (top panel). Assay with 2% 2-ME activates IRP1 that was functionally in a non-IRE-binding state to bind IREs in vitro. A representative example of at least three experiments is shown. In the bottom panel, the small percentage of IRP1 that is in the IRE-binding form is shown as cross-hatched blocks in each column. Notably, 4–18% of IRP1 is in the IRE-binding state prior to 2-ME activation in the seven tissues analyzed. Results shown are representative of three independent experiments.
Since more than 80% of endogenous IRP1 was not in the IRE-binding form, we were interested in estimating the fraction of IRP1 that functions as an active aconitase in animal tissues. To assess how much IRP1 contributes to total cellular aconitase activity, we separated mitochondrial from cytosolic aconitase on cellulose acetate membranes and calculated a mitochondrial to cytosolic aconitase ratio of 3.8 (n=3) (Figures 1C and 4A). The total aconitase activity in the kidney was measured with a coupled aconitase assay and, using the ratio of 3.8, cytosolic aconitase in the kidney lysate was calculated to be 1.1±0.3 ng/μg protein (n=5). Using IRP1 standard curves in Western blots, we calculated that kidney lysates contained 1.8±0.5 ng IRP1/μg total protein (n=12). Comparing the amount of cytosolic aconitase to the amount of total IRP1 protein detected by Western blots, we calculated that approximately 60% of all IRP1 protein in the kidney was in the cytosolic aconitase form. In the liver, a similar calculation showed that virtually all IRP1 was in the cytosolic aconitase form. These results imply that, at steady-state conditions, most IRP1 functions as cytosolic aconitase, rather than as an IRE-binding protein, or as an intermediate that does not possess either function (Brown et al, 2002).
IRE-binding activity of IRP1 is not activated by dietary-induced iron deficiency in animals
The substantial fraction of IRP1 that is in the cytosolic aconitase form represents an enormous store of potential regulatory power. To test if this pool of latent IRE-binding activity can be activated by iron starvation, mice were weaned to low- or high-iron diets, and IRP activity and ferritin levels were assessed after 4 months. Surprisingly, the IRE-binding activity of IRP1 was not activated in any tissue examined from mice maintained on a low-iron diet in comparison to the IRE-binding activity in tissues of mice fed a high-iron diet (Figure 5, top and center panels). In contrast, IRP2 in these mice was activated in a diet-dependent manner as expected (Figure 5, top and bottom panels). The diet-induced activation of IRP2 was especially evident in the gel shift assays of IRP1−/− tissues.
Figure 5.

IRE-binding activity of IRP1 is not recruited by a low-iron diet. Mice were maintained on a low (no iron added)- or high (0.3 g ferric ammonium citrate/kg chow)-iron diet after weaning for a period of 4 months prior to killing. Tissues were prepared, and gel retardation assays and Western blot analysis for ferritin were carried out as described previously. Duplicates are shown of lysates from age-, sex- and diet-matched mice. Note that although IRP1 IRE-binding activity is not recruited by the low-iron diet, IRP2 binding activity increases significantly under the same conditions. Results are presented in duplicate and are representative of at least three experiments.
To assess whether the ability to regulate IRP targets was intact, we assessed whether iron-deficient IRP1−/− and IRP2−/− mice were able to repress ferritin synthesis. In peripheral tissues of wt mice, ferritin levels were significantly decreased in animals on the low-iron diet (Figure 5). In agreement with the results in Figure 3, IRP2−/− animals on a low-iron diet did not appropriately repress ferritin synthesis in the liver, kidney and brown fat. Ferritin levels were markedly elevated in the cerebellum of IRP2−/− mice compared to wt on both the low- and high-iron diets. These results imply that IRP1 in IRP2−/− animals is not only unable to maintain the steady-state level of ferritin found in wt animals (Figure 3), but it is also unable to register and respond to iron deficiency by inhibiting ferritin synthesis. In contrast to IRP2−/− animals, elevated ferritin levels in IRP1−/− mice were found only in the kidney and brown fat of animals that were on the low-iron diet. In the liver of IRP1−/− mice, ferritin regulation was comparable to wt regulation, and in the cerebellum, ferritin levels were similar to those of wt. Thus, in tissues such as brain and liver, lack of IRP1 does not result in misregulation of ferritin synthesis, whereas in brown fat and kidney, lack of IRP1 compromises but does not completely abolish appropriate ferritin repression under iron starvation conditions. It is interesting to note that the impact of the low-iron diet on ferritin in the cerebellum was minimal, perhaps because the blood–brain barrier protects the brain from extreme iron conditions.
IRP2 levels increase in the cerebellum and spleen of IRP1−/− animals, suggesting that IRP2 compensates for loss of IRP1
On multiple occasions, levels of IRP2 detected in gel retardation assays appeared to be elevated in tissues of IRP1−/− animals relative to wt animals. To better assess whether IRP2 levels increase in cells that lack IRP1, we quantitated total IRP2 levels by Western blot in tissues of wt, IRP1−/− and IRP2−/− animals. We consistently observed an approximately two-fold increase in IRP2 levels in the cerebellum and spleen of IRP1−/− animals (Figure 6A). Results of four separate experiments for the cerebellum and three separate experiments for the spleen were quantitated, and a statistically significant increase (P=0.0001 and 0.035) of IRP2 in IRP1−/− cerebellum and spleen, respectively, is shown in Figure 6B. As IRP2 constitutes over one-third of the total IRE-binding activity in the cerebellum (Figure 4B, lane 4), the measured increase of IRP2 in the cerebellum of IRP1−/− mice substantially compensates for the loss of IRE-binding activity normally provided by IRP1 in the cerebellum.
Figure 6.
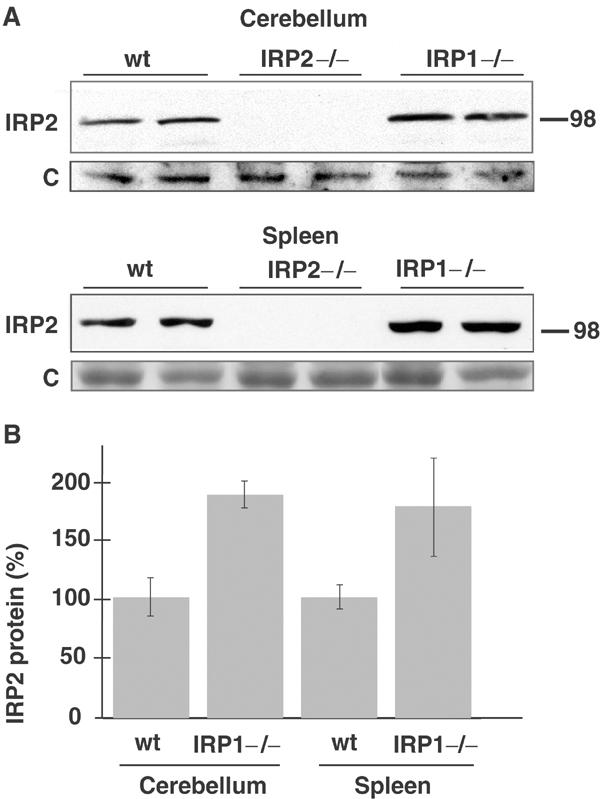
Steady-state levels of IRP2 increase in the cerebellum and spleen of IRP1−/− mice. (A) Western blots of equal amounts of cerebellar or spleen lysate from two wt (lanes 1,2), IRP2−/− (lanes 3,4) and IRP1−/− (lanes 5,6) mice show a two-fold increase in IRP1−/− animals compared to control. The blot shows duplicates that are representative of four separate experiments from the cerebellum and three separate experiments from the spleen, and results are represented in a bar graph in (B) after quantitation and statistical comparison (P=0.0001 for cerebellum and P=0.035 for spleen).
Discussion
Analyses of IRP1−/− and IRP2−/− mice allow us to make a distinction between the major in vivo functions of each IRP. Unlike the IRP2−/− mice, the IRP1−/− mice do not develop neurodegeneration or profound aberrations of iron homeostasis. Our results indicate that IRP1 in animal tissues is mostly in the form of a cytosolic aconitase that is not recruited to regulate iron metabolism in iron-deficient animals. The viability and health of IRP1−/− animals indicate that IRP1 is not critical for either of its activities under normal physiological conditions. It is likely that the viability of the IRP1−/− animals can be attributed to redundancy for each of the two functions of IRP1. In the absence of cytosolic aconitase, mitochondrial aconitase can interconvert citrate and isocitrate, and precursors and products of this reaction can cross the mitochondrial membrane to the cytosol if they are needed (Chen et al, 1998). Similarly, our results reveal that IRP2 appears to be able to regulate fully post-transcriptional iron metabolism in most tissues.
A surprising conclusion of our studies is that IRP1 is not recruited to the IRE-binding form in iron-deficient animals. Although the low-iron diet led to markedly increased IRP2 activity in the wt and IRP1−/− mice, IRP1 was not activated by iron starvation, even in mice that expressed no IRP2. Moreover, expression of both subunits of ferritin was misregulated in IRP2−/− animals in all tissues examined. These results strongly imply that IRP2 is poised to sense iron status in animal tissues, whereas IRP1 is not. Interestingly, most IRP1 remains in the non-IRE-binding form, even when iron starvation is sufficient to stabilize and thereby activate IRP2. The inability of IRP1 to convert to the IRE-binding form in iron-deficient animal tissues differs from results that have been repeatedly observed in tissue culture, where IRP1 is usually the major IRE-binding protein activated by treatment of cells with the iron chelator desferal (Pantopoulos and Hentze, 1995, and Figure 1D of this paper). One major difference between animal tissues and tissue culture may be the degree to which the iron–sulfur cluster of IRP1 is destabilized by exposure to oxygen (Rouault and Klausner, 1996; Beinert et al, 1997). Once formed, iron–sulfur clusters may be more stable in cells exposed to low tissue oxygen concentrations than in cell cultures, which are exposed to atmospheric oxygen concentrations (Beckman and Ames, 1998). In addition, the iron–sulfur cluster assembly machinery may continue to function even when cellular iron deficiency is sufficient to activate IRP2. The iron concentration threshold necessary to trigger changes in regulation of proteins involved with iron uptake and sequestration through IRP2 activation may be set differently than the threshold for certain vital functions such as iron–sulfur cluster assembly. A hierarchy in which regulatory responses to iron deficiency precede functional compromise could be achieved through calibration of the affinities of various iron proteins and enzymes for iron. In contrast to the IRP1 IRE-binding activity, IRP2 is relatively stabilized under the low-oxygen conditions that are found in animal tissues (Bourdon et al, 2003; Hanson et al, 2003), and this large pool of IRP2 is apparently responsible for registering iron deficiency and regulating iron homeostasis.
A major difference between IRP1 and IRP2 can be observed in the cerebellum. Although both IRP1 and IRP2 contribute substantially to total IRE-binding activity in wt animals, abnormalities of post-transcriptional gene regulation are found only in the IRP2−/− brain. It is likely that normal post-transcriptional regulation depends on optimizing the ratio between numbers of IRE-binding proteins and IRE-containing transcripts, and that significant decreases in the number of IRE-binding proteins can result in loss of regulatory range. With this concept in mind, it is interesting to note that we consistently see two-fold elevations of IRP2 binding activity in cerebellar lysates of IRP1−/− mice, whereas we do not see comparable activation of IRP1 in IRP2−/− mice. The mechanism that allows this compensatory increase in IRP2 is unknown, but it could arise from decreased iron-dependent degradation of IRP2 (Guo et al, 1995; Iwai et al, 1998; Bourdon et al, 2003) caused by increased sequestration of iron by excess ferritin (Cozzi et al, 2000). Similarly, IRP2 levels increased significantly in spleen lysates of IRP1−/− animals. To characterize fully the mechanism for compensatory increases of IRP2 in IRP1−/− mice, we need to evaluate homogeneous populations of cells from each genotype. However, at this point, the compensatory increase of IRP2 in response to the loss of IRP1 potentially explains why normal regulation of iron metabolism is maintained in IRP1−/− mice, whereas iron metabolism in IRP2−/− animals is abnormal in every tissue we have examined.
Even though IRP1 is not responsive to iron deficiency, it may still play a role in iron metabolism by contributing to the basal IRE-binding capacity of tissues. We found that in two peripheral tissues that highly express IRP1, kidney and brown fat, IRP1−/− animals cannot repress ferritin synthesis fully. These results imply that IRP1 is essential for the maintenance of normal baseline levels of ferritin in these tissues. It is possible that in a tissue such as brown fat, which expresses very little IRP2, the loss of IRP1 is proportionally too great to be fully compensated by an increase of IRP2 binding activity.
The role of IRP1 in iron homeostasis is further emphasized by a pronounced worsening of neurodegenerative disease in IRP1+/− IRP2−/− mice, which we observe both clinically and pathologically, compared to IRP2−/− mice (Smith et al, Ann NY Acad Sci, in press). In multiple tissues of IRP1+/− IRP2−/− mice, ferritin is further increased and TfR is further decreased compared to IRP2−/− animals. Moreover, IRP1−/− IRP2−/− animals are not viable past the blastocyst stage (Smith et al, in preparation), and we suspect that complete failure to repress ferritin synthesis and a severe deficiency of TfR in the developing embryo may be the cause of this early lethality.
Although our data indicate that IRP1 is not regulated by dietary iron levels, it is possible that other factors can modulate IRP1 activity in vivo. Many studies have demonstrated a destabilization of the iron–sulfur cluster of IRP1 by oxidative stress and by exposure to nitric oxide (Pantopoulos and Hentze, 1995; Recalcati et al, 1998; Hanson and Leibold, 1999; Caltagirone et al, 2001; Mueller et al, 2001; Kim and Ponka, 2002; Bouton and Drapier, 2003). Although our experiments do not address whether activation of IRP1 takes place under pathologic conditions, we find that there is a vast reservoir of potential IRE-binding activity in tissue. Activation of IRP1 by infection or other stresses could profoundly affect tissue iron homeostasis. Thus, the IRP1−/− and IRP2−/− mouse models may allow us to better characterize individual IRP responses to stress and inflammation.
In situ hybridizations indicate high IRP1 RNA levels in brown fat, kidney, and in the epithelium of the epididymis. These tissues do not have major roles in systemic iron metabolism, but in each case we can postulate a metabolic role for cytosolic aconitase activity, which can catalyze the interconversion of citrate and isocitrate in either direction. In the case of embryonic brown fat, which is mainly involved in body temperature regulation, citrate may be synthesized in the cytosol from glutamate (Belfiore and Iannello, 1995) and consumed in the citrate lyase reaction, which yields the products acetyl CoA, a precursor in fatty acid synthesis, and oxaloacetate (Swierczynski et al, 2000). However, we do not find a significant difference in body weight or appearance of the brown fat in IRP1−/− animals. As adult mice in temperature-controlled animal facilities lose brown fat activity, we tested the ability of newborn IRP1−/− animals to maintain body temperature when exposed to cold temperatures, but found no difference between IRP1−/− and wt animals (data not shown). In the kidney, serum citrate is freely filtered at the glomerulus, but the proximal tubule reabsorbs most of the citrate (Hamm, 1990), and cytosolic aconitase may be high in these cells to reduce the levels of cytosolic citrate, which may otherwise inhibit glycolysis and may cause iron toxicity similar to that reported for citrate–iron complexes in yeast (Chen et al, 2002). Nevertheless, there is no evidence for compromised renal function or abnormal pathology in IRP1−/− mice as evaluated by blood chemistry and tissue pathology. Glutamate is secreted by epididymal cells (Hinton, 1990) and is believed to serve as an important nitrogen source for developing spermatozoa. In the epididymis, cytosolic aconitase could facilitate synthesis of glutamate in a cytosolic metabolic pathway that includes the conversion of citrate to isocitrate (cytosolic aconitase) to alpha ketoglutarate (via cytosolic isocitrate dehydrogenase) (Nekrutenko et al, 1998) to glutamate (via cytosolic alanine aminotransferase) (ALT, EC 2.6.1.2) (Belfiore and Iannello, 1995). But we find that IRP1−/− males are fertile, and litter sizes of IRP1−/− mating pairs are not smaller than wt litter sizes. Thus, we do not find obvious defects in IRP1−/− mice, which suggests that mitochondrial aconitase can substitute for the lack of IRP1 enzymatic activity under basal metabolic conditions. It is possible that phenotypic abnormalities caused by the lack of cytosolic aconitase activity could be elicited in IRP1−/− mice in specific stress situations.
Our results indicate that IRP1 in animal tissues functions mostly as cytosolic aconitase. It is interesting to note that the major IRP1 homologs identified in bacteria (Prodromou et al, 1992), plants (Peyret et al, 1995) and nonvertebrate animals (Gourley et al, 2003) also appear to function mainly as cytosolic aconitases. The major and unexpected finding of this study is that IRP1 does not appear to be recruited to regulate iron metabolism in iron-deficient animals under conditions that are sufficient to activate IRP2. We suggest that the small fraction of IRP1 that is in the IRE-binding form contributes to basal regulation of iron metabolism, as indicated by findings in the IRP1+/− IRP2−/− mouse (Smith et al, Ann NY Acad Sci, in press), but that it has a minimal role in direct sensing of cellular iron status. IRP2 appears to be the protein responsible for the sensing and regulation of acute fluctuations in cellular iron concentration. Therefore, the targeted deletion of IRP2 leads to severe misregulation of the target proteins of IRPs, while the targeted deletion of IRP1 adversely affects iron metabolism in only a few tissues. Thus, the targeted deletions of IRPs in mice permit us to distinguish the specific role of each member of this duplicated gene pair in vivo.
Materials and methods
Targeted deletion of IRP1
Embryonic stem cells (ES) were transfected with an IRP1 mutant construct in the targeting vector pPNT (Love et al, 1993). We generated the 5′ arm of the construct by subcloning a 2.7 kb genomic fragment from the AvaII site in exon 11 to the BstEII site in exon 13 from a genomic IRP1 clone derived from a 129/Sv library. The 3′ arm was created by subcloning a 6.6 kb fragment of the IRP1 clone containing parts of exon 13 and exons 14–18. The targeting construct was linearized with NotI before electroporation and insertion into ES cells. Chimeric mice were bred, backcrossed to C57Bl/6 mice and genotyped by Southern blot analysis (LaVaute et al, 2001). Embryonic fibroblasts of 16-day embryos were isolated as described (Ishino et al, 2000).
Tissue and lysate preparation
Animals were killed and tissues were quick frozen in liquid N2 and stored at −80°C under argon. Immediately before experiments, tissues were ground in liquid-N2-cooled mortars in an anaerobic chamber and lysed in degassed lysis buffer for 3 min. Nuclei and debris were removed by centrifugation. Both activity assays for IRP1, namely aconitase assays and the gel retardation assay, were performed anaerobically. Also for protein analysis by Western blotting, lysate preparation was carried out anaerobically.
Aconitase assay
Total aconitase activity was detected by the coupled assay, monitoring reduction of NADP at 340 nm (Rose and O'Connell, 1967). Tissue lysis buffer contained 25 mM Tris–HCl (pH 7.5), 40 mM KCl, 2% Triton X-100, 0.6 mM MnCl2, 2 mM citric acid, 5 mM DTT, 1 mM AEBSF, 10 μg/ml Leupeptin and Complete™ EDTA free protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Protein concentrations were determined using the Bradford assay (BIO-RAD, Hercules, California). Samples were diluted to 10–50 ng protein/ml into a reaction mix containing 33 mM Tris–HCl (pH 7.4), 1.3 mM MnSO4, 0.2 mM citric acid, 0.5 mM NADP and 10 U/ml isocitrate dehydrogenase. Production of NADPH was monitored at 340 nm every second for 180 s, and aconitase activity was calculated from the linear slope using an extinction coefficient of 6220/M/cm. The amount of aconitase was then calculated taking into account that the specific activity for both aconitases is about 21.4 μmol/min/mg (Kennedy et al, 1992). Mitochondrial and cytosolic aconitase activities were assayed in parallel in tissue lysates after electrophoretic separation. Lysates were electrophoresed on Sepraphore III membranes (Pall) with an electrophoresis buffer containing 20 mM KH2PO4/K2HPO4 (pH 6.5) and 3.6 mM citric acid. Aconitase activity was detected chromogenically by incubating the membrane in 100 mM KH2PO4/K2HPO4 (pH 6.5), 1 mM NADP, 2 mM cis-aconitic acid, 1.2 mM 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, 0.3 mM phenazine methosulfate, 25mM MgCl2 and 5 U/ml isocitrate dehydrogenase. All chemicals were purchased from Sigma (Missouri). Quantitation of mitochondrial and cytosolic aconitase on cellulose acetate membranes was performed using ScionImage software.
RNA mobility shift assays
Gel retardation assays were performed as described (Allerson et al, 1999). Tissue lysates were prepared as described above in lysis buffer consisting of 10 mM HEPES (pH 7.2), 3 mM MgCl2, 40 mM KCl, 5% glycerol, 0.2% Nonidet P-40, 5 mM DTT, 1 mM AEBSF, 10 μg/ml Leupeptin and Complete™ EDTA free protease inhibitor cocktail (Roche Applied Science, Indiana). Equal amounts of about 10 μg of total protein were added to a final volume of 12.5 μl buffer containing 25 mM Tris–HCl (pH 7.5) and 40 mM KCl with or without 2% 2-ME, which activates IRP1 in vitro. The samples were incubated for 5 min at room temperature (RT) with 12.5 μl of a reaction cocktail containing 20% glycerol, 0.2 U/μl Super RNAsine (Ambion, Texas), 0.6 μg/μl yeast tRNA, 5 mM DTT and 20 nM 32P-labelled IRE from the human ferritin H-chain gene in 25 mM Tris–HCl (pH 7.5) and 40 mM KCl. A measure of 20 μl of this reaction mixture was loaded onto a 10% acrylamide/TBE gel, which was run at 200 V for 2.25 h, and then the gel was fixed, dried and exposed for autoradiography.
Western blotting and antibodies
Protein analysis was carried out as previously described (LaVaute et al, 2001). Equal amounts of protein (40 μg/lane) were separated on 13% SDS–PAGE and transferred to nitrocellulose membranes. The membrane was blocked with 5% non-fat milk, 0.1% Triton X-100 in PBS and probed at RT in the same blocking buffer. IRP1 antibody was prepared against purified hIRP1 and used at 1:5000 dilution. IRP2 antibodies were prepared against aa 137–209 of hIRP2 and used at 1:2000 dilution. For IRP1 quantitation, a standard curve of purified IRP1 ranging from 2 to 64 ng was generated on the same membranes as the samples, and quantitations were performed on a Typhoon 9200 Imager from Molecular Dynamics using Image Quant software. Ferritin antibodies raised against mouse liver ferritin were a kind gift from Prof. AM Konijn (Hebrew University, Jerusalem) and we raised antibodies to the mouse ferritin H chain using an overexpression construct generously provided by Dr Paolo Santambrogio (Santambrogio et al, 2000). Both were used at 1:5000 dilutions. A mouse monoclonal TfR antibody from Zymed was used at 1:2000 dilution, and rabbit anti-mouse antibody from Jackson Lab was used as secondary antibody at 1:1000 dilution. Secondary/tertiary antibody used at 1:200 dilution was biotinylated anti-rabbit antibody (Amersham Biosciences, New Jersey), and finally the blots were incubated with 125I-streptavidin at 1:500 dilution (Amersham). Some Western blots were performed with a secondary/tertiary horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Amersham) (1:5000 dilution) and developed using enhanced chemiluminescence (ECL kit, Pierce, Illinois).
In situ analysis
Nonisotopic in situ hybridization was performed using digoxigenin (DIG)-labeled cRNA probes and alkaline phosphatase (AP) detection as described (Berger and Hediger, 1998). Cryostat sections of fresh frozen tissues or newborn pups were cut at 10 μm thickness, fixed in 4% paraformaldehyde and acetylated. Hybridization was performed in slide mailers by total immersion in hybridization buffer that contained 50% formamide, 5 × SSC, 2% blocking reagent (Roche Applied Science, Indiana), 0.02% SDS, 0.1% sarcosine and approximately 100 ng/ml of cRNA probe. Sections were hybridized at 68°C over 72 h with the full-length mouse IRP1 or IRP2 probe that had been alkali-hydrolyzed to around 500 bases in length. Washing steps included incubations in 2 × SSC and 0.2 × SSC at 68°C. Sections were incubated at RT in 1% blocking reagent in maleic acid buffer, then in AP-conjugated anti-DIG Fab fragments (1:5000 dilution, Roche), and developed overnight with BCIP/NBT substrate (Kierkegard and Perry Laboratories, Gaithersburg, MD). Sections were rinsed several times in 100 mM Tris, 150 mM NaCl, 20 mM EDTA (pH 9.5), and coverslipped with glycerol gelatin (Sigma, Missouri). Control sections were incubated in an identical concentration of the sense probe transcript.
Diet description
Newly weaned mice were maintained on a diet containing either 2–10 mg iron/kg chow (low-iron diet) or 70 mg iron/kg chow (high-iron diet) from Harlan Teklad for a period of 4 months prior to killing. Weights of mice on the low-iron diet were slightly lower but not significantly different from mice on the high-iron diets (28.5±6.9 and 31.5±7.2 g, respectively, P=0.38).
Pathology
The Veterinary Resources Program of the National Institutes of Health performed histology and blood work, including chemistries and complete blood counts. Animals were autopsied and tissues were blocked in paraffin and analyzed by H&E and Prussian blue stains.
Acknowledgments
We thank Wing-Hang Tong, Fanis Missirlis and other members in our lab and Caroline Philpott for helpful discussions and a critical reading of the manuscript. We thank the Veterinary Resources Program of NIH for measuring complete blood counts and serum chemistries and for histological evaluations. This work was supported by the Intramural Program of the National Institute of Child Health and Human Development and in part by the Lookout Foundation.
References
- Allerson CR, Cazzola M, Rouault TA (1999) Clinical severity, thermodynamic effects of iron-responsive element mutations in hereditary hyperferritinemia–cataract syndrome. J Biol Chem 274: 26439–26447 [DOI] [PubMed] [Google Scholar]
- Beckman KB, Ames BN (1998) The free radical theory of aging matures. Physiol Rev 78: 547–581 [DOI] [PubMed] [Google Scholar]
- Beinert H, Holm RH, Munck E (1997) Iron–sulfur clusters: nature's modular, multipurpose structures. Science 277: 653–659 [DOI] [PubMed] [Google Scholar]
- Belfiore F, Iannello S (1995) Fatty acid synthesis from glutamate in the adipose tissue of normal subjects, obese patients: an enzyme study. Biochem Mol Med 54: 19–25 [DOI] [PubMed] [Google Scholar]
- Berger UV, Hediger MA (1998) Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol (Berl) 198: 13–30 [DOI] [PubMed] [Google Scholar]
- Bourdon E, Kang DK, Ghosh MC, Drake SK, Wey J, Levine RL, Rouault TA (2003) The role of endogenous heme synthesis and degradation domain cysteines in cellular iron-dependent degradation of IRP2. Blood Cells Mol Dis 31: 247–255 [DOI] [PubMed] [Google Scholar]
- Bouton C, Drapier JC (2003) Iron regulatory proteins as NO signal transducers. Sci STKE, (2003) pe17 [DOI] [PubMed] [Google Scholar]
- Brown NM, Kennedy MC, Antholine WE, Eisenstein RS, Walden WE (2002) Detection of a [3Fe-4S] cluster intermediate of cytosolic aconitase in yeast expressing iron regulatory protein 1. Insights into the mechanism of Fe-S cluster cycling. J Biol Chem 277: 7246–7254 [DOI] [PubMed] [Google Scholar]
- Caltagirone A, Weiss G, Pantopoulos K (2001) Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J Biol Chem 276: 19738–19745 [DOI] [PubMed] [Google Scholar]
- Chen OS, Hemenway S, Kaplan J (2002) Genetic analysis of iron citrate toxicity in yeast: implications for mammalian iron homeostasis. Proc Natl Acad Sci USA 99: 16922–16927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XZ, Shayakul C, Berger UV, Tian W, Hediger MA (1998) Characterization of a rat Na+-dicarboxylate cotransporter. J Biol Chem 273: 20972–20981 [DOI] [PubMed] [Google Scholar]
- Cozzi A, Corsi B, Levi S, Santambrogio P, Albertini A, Arosio P (2000) Overexpression of wild type, mutated human ferritin H-chain in HeLa cells: in vivo role of ferritin ferroxidase activity. J Biol Chem 275: 25122–25129 [DOI] [PubMed] [Google Scholar]
- DeRusso PA, Philpott CC, Iwai K, Mostowski HS, Klausner RD, Rouault TA (1995) Expression of a constitutive mutant of iron regulatory protein 1 abolishes iron homeostasis in mammalian cells. J Biol Chem 270: 15451–15454 [DOI] [PubMed] [Google Scholar]
- Gourley BL, Parker SB, Jones BJ, Zumbrennen KB, Leibold EA (2003) Cytosolic aconitase, ferritin are regulated by iron in Caenorhabditis elegans. J Biol Chem 278: 3227–3234 [DOI] [PubMed] [Google Scholar]
- Gruer MJ, Artymiuk PJ, Guest JR (1997) The aconitase family: three structural variations on a common theme. Trends Biochem Sci 22: 3–6 [DOI] [PubMed] [Google Scholar]
- Guo B, Phillips JD, Yu Y, Leibold EA (1995) Iron regulates the intracellular degradation of iron regulatory protein 2 by the proteasome. J Biol Chem 270: 21645–21651 [DOI] [PubMed] [Google Scholar]
- Hamm LL (1990) Renal handling of citrate. Kidney Int 38: 728–735 [DOI] [PubMed] [Google Scholar]
- Hanson ES, Leibold EA (1999) Regulation of the iron regulatory proteins by reactive nitrogen, oxygen species. Gene Expression 7: 367–376 [PMC free article] [PubMed] [Google Scholar]
- Henderson BR, Seiser C, Kuhn LC (1993) Characterization of a second RNA-binding protein in rodents with specificity for iron-responsive elements. J Biol Chem 268: 27327–27334 [PubMed] [Google Scholar]
- Hanson ES, Rawlins ML, Leibold EA (2003) Oxygen and iron regulation of iron regulatory protein 2. J Biol Chem 278: 40337–40342 [DOI] [PubMed] [Google Scholar]
- Hentze MW, Kuhn LC (1996) Molecular control of vertebrate iron metabolism: mRNA-based regulatory circuits operated by iron, nitric oxide, and oxidative stress. Proc Natl Acad Sci USA 93: 8175–8182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton BT (1990) The testicular, epididymal luminal amino acid microenvironment in the rat. J Androl 11: 498–505 [PubMed] [Google Scholar]
- Hirling H, Emery-Goodman A, Thompson N, Neupert B, Seiser C, Kuhn LC (1992) Expression of active iron regulatory factor from a full-length human cDNA by in vitro transcription/translation. Nucleic Acids Res 20: 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino K, Kaneyama J, Shibanuma M, Nose K (2000) Specific decrease in the level of Hic-5, a focal adhesion protein, during immortalization of mouse embryonic fibroblasts, and its association with focal adhesion kinase. J Cell Biochem 76: 411–419 [DOI] [PubMed] [Google Scholar]
- Iwai K, Drake SK, Wehr NB, Weissman AM, LaVaute T, Minato N, Klausner RD, Levine RL, Rouault TA (1998) Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: implications for degradation of oxidized proteins. Proc Natl Acad Sci USA 95: 4924–4928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MC, Mende-Mueller L, Blondin GA, Beinert H (1992) Purification, characterization of cytosolic aconitase from beef liver, its relationship to the iron-responsive element binding protein (IRE-BP). Proc Natl Acad Sci USA 89: 11730–11734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Klausner RD, Rouault TA (1995) Translational repressor activity is equivalent and is quantitatively predicted by in vitro RNA binding for two iron-responsive element binding proteins, IRP1 and IRP2. J Biol Chem 270: 4983–4986 [DOI] [PubMed] [Google Scholar]
- Kim S, Ponka P (2002) Nitrogen monoxide-mediated control of ferritin synthesis: implications for macrophage iron homeostasis. Proc Natl Acad Sci USA 99: 12214–12219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, Switzer R III, Grinberg A, Love P, Tresser N, Rouault TA (2001) Targeted deletion of iron regulatory protein 2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet 27: 209–214 [DOI] [PubMed] [Google Scholar]
- Love PE, Shores EW, Johnson MD, Tremblay ML, Lee EJ, Grinberg A, Huang SP, Singer A, Westphal H (1993) T cell development in mice that lack the zeta chain of the T cell antigen receptor complex. Science 261: 918–921 [DOI] [PubMed] [Google Scholar]
- Muckenthaler M, Gunkel N, Frishman D, Cyrklaff A, Tomancak P, Hentze MW (1998) Iron-regulatory protein-1 (IRP-1) is highly conserved in two invertebrate species—characterization of IRP-1 homologues in Drosophila melanogaster and Caenorhabditis elegans. Eur J Biochem 254: 230–237 [DOI] [PubMed] [Google Scholar]
- Mueller S, Pantopoulos K, Hubner CA, Stremmel W, Hentze MW (2001) IRP1 activation by extracellular oxidative stress in the perfused rat liver. J Biol Chem 276: 23192–23196 [DOI] [PubMed] [Google Scholar]
- Mullner EW, Rothenberger S, Muller AM, Kuhn LC (1992) In vivo and in vitro modulation of the mRNA-binding activity of iron-regulatory factor. Tissue distribution and effects of cell proliferation, iron levels and redox state. Eur J Biochem 208: 597–605 [DOI] [PubMed] [Google Scholar]
- Nekrutenko A, Hillis DM, Patton JC, Bradley RD, Baker RJ (1998) Cytosolic isocitrate dehydrogenase in humans, mice, and voles and phylogenetic analysis of the enzyme family. Mol Biol Evol 15: 1674–1684 [DOI] [PubMed] [Google Scholar]
- Pantopoulos K, Hentze MW (1995) Rapid responses to oxidative stress mediated by iron regulatory protein. EMBO J 14: 2917–2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino MM, Walden WE (1992) Cloning of a functional cDNA for the rabbit ferritin mRNA repressor protein: demonstration of a tissue specific pattern of expression. J Biol Chem 267: 19011–19016 [PubMed] [Google Scholar]
- Peyret P, Perez P, Alric M (1995) Structure, genomic organization, and expression of the Arabidopsis thaliana aconitase gene. Plant aconitase show significant homology with mammalian iron-responsive element-binding protein. J Biol Chem 270: 8131–8137 [DOI] [PubMed] [Google Scholar]
- Prodromou C, Artymiuk PJ, Guest JR (1992) The aconitase of E. Coli: nucleotide sequence of the aconitase gene and amino acid sequence similarity with mitochondrial aconitases, the iron-responsive-element-binding protein and isopropylmala. Eur J Biochem 204: 599–609 [DOI] [PubMed] [Google Scholar]
- Recalcati S, Taramelli D, Conte D, Cairo G (1998) Nitric oxide-mediated induction of ferritin synthesis in J774 macrophages by inflammatory cytokines: role of selective iron regulatory protein-2 downregulation. Blood 91: 1059–1066 [PubMed] [Google Scholar]
- Rose IA, O'Connell EL (1967) Mechanism of aconitase action. I. The hydrogen transfer reaction. J Biol Chem 242: 1870–1879 [PubMed] [Google Scholar]
- Rouault TA, Klausner RD (1996) Iron–sulfur clusters as biosensors of oxidants and iron. Trends Biochem Sci 21: 174–177 [PubMed] [Google Scholar]
- Rouault T, Klausner R (1997) Regulation of iron metabolism in eukaryotes. Curr Top Cell Regul 35: 1–19 [DOI] [PubMed] [Google Scholar]
- Rouault TA, Stout CD, Kaptain S, Harford JB, Klausner RD (1991) Structural relationship between an iron-regulated RNA-binding protein (IRE-BP) and aconitase: functional implications. Cell 64: 881–883 [DOI] [PubMed] [Google Scholar]
- Rouault TA, Tang CK, Kaptain S, Burgess WH, Haile DJ, Samaniego F, McBride OW, Harford JB, Klausner RD (1990) Cloning of the cDNA encoding an RNA regulatory protein—the human iron-responsive element-binding protein. Proc Natl Acad Sci USA 87: 7958–7962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santambrogio P, Cozzi A, Levi S, Rovida E, Magni F, Albertini A, Arosio P (2000) Functional and immunological analysis of recombinant mouse H- and L-ferritins from Escherichia coli. Protein Expr Purif 19: 212–218 [DOI] [PubMed] [Google Scholar]
- Schalinske KL, Blemings KP, Steffen DW, Chen OS, Eisenstein RS (1997) Iron regulatory protein 1 is not required for the modulation of ferritin and transferrin receptor expression by iron in a murine pro-B lymphocyte cell line. Proc Natl Acad Sci USA 94: 10681–10686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider BD, Leibold EA (2000) Regulation of mammalian iron homeostasis. Curr Opin Clin Nutr Metab Care 3: 267–273 [DOI] [PubMed] [Google Scholar]
- Smith SR, Cooperman S, LaVaute T, Tresser N, Ghosh M, Meyron-Holtz EG, Land W, Ollivierre H, Jortner B, Switzer R III, Messing A, Rouault TA (in press) Severity of neurodegeneration correlates with compromise of post-transcriptional iron metabolism regulation in genetically engineered mouse models. Ann NY Acad Sci [DOI] [PubMed] [Google Scholar]
- Swierczynski J, Goyke E, Wach L, Pankiewicz A, Kochan Z, Adamonis W, Sledzinski Z, Aleksandrowicz Z (2000) Comparative study of the lipogenic potential of human and rat adipose tissue. Metabolism 49: 594–599 [DOI] [PubMed] [Google Scholar]
- Wang J, Pantopoulos K (2002) Conditional derepression of ferritin synthesis in cells expressing a constitutive IRP1 mutant. Mol Cell Biol 22: 4638–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Radisky E, Leibold EA (1992) The iron-responsive element binding protein: purification, cloning and regulation in rat liver. J Biol Chem 267: 19005–19010 [PubMed] [Google Scholar]