

Abstract
TRAF2 is a RING finger protein that regulates the cellular response to stress and cytokines by controlling JNK, p38 and NF-κB signaling cascades. Here, we demonstrate that TRAF2 ubiquitination is required for TNFα-induced activation of JNK but not of p38 or NF-κB. Intact RING and zinc finger domains are required for TNFα-induced TRAF2 ubiquitination, which is also dependent on Ubc13. TRAF2 ubiquitination coincides with its translocation to the insoluble cellular fraction, resulting in selective activation of JNK. Inhibition of Ubc13 expression by RNAi resulted in inhibition of TNFα-induced TRAF2 translocation and impaired activation of JNK but not of IKK or p38. TRAF2 aggregates in the cytoplasm, as seen in Hodgkin–Reed–Sternberg lymphoma cells, resulting in constitutive NF-κB activity but failure to activate JNK. These findings demonstrate that the TRAF2 RING is required for Ubc13-dependent ubiquitination, resulting in translocation of TRAF2 to an insoluble fraction and activation of JNK, but not of p38 or NF-κB. Altogether, our findings highlight a novel mechanism of TRAF2-dependent activation of diverse signaling cascades that is impaired in Hodgkin–Reed–Sternberg cells.
Keywords: JNK, NFκB, TNFα, TRAF2, ubiquitin
Introduction
Tumor necrosis factor receptor (TNF-R) adaptor factors (TRAF) play a central role in the cellular response to stress and cytokines via their regulation of stress kinases, resulting in the activation of key transcription factors, including NF-κB, c-Jun and ATF2 (Nishitoh et al, 1998; Baud et al, 1999; Bradley and Pober, 2001). With the exception of TRAF1, all family members harbor the C3HC4-type RING domain and several zinc finger motifs that are required for TRAF's ability to elicit downstream signaling events (Takeuchi et al, 1996). Both TRAF6 and TRAF2 can activate NF-κB upon stimulation by IL-1 and TNF, respectively, although they utilize different signaling components (Rothe et al, 1995; Natoli et al, 1997). Whereas deletion of TRAF2 is sufficient to abolish TNFα-induced JNK activation, deletion of both TRAF2/5 is required to abolish NF-κB activation (Nakano et al, 1999; Tada et al, 2001). Upon TNFα treatment, TRAF2 is recruited directly to TNFR2 or via TRADD to TNFR1, which results in the activation of JNK, p38 and NF-κB. However, the mechanism by which TRAF2 is capable of uncoupling these signaling pathways remains largely elusive.
Upon ligand stimulation, TNFR/TRAF2 complex is recruited to membrane rafts; yet, the regulation and the significance of such translocation remains controversial. Although TNFR1 was reported to localize exclusively in lipid rafts of unstimulated U937 or HeLa cells (Ko et al, 1999; Cottin et al, 2002), partitioning of TNFR1 in lipid rafts of fibroblasts was only observed after TNFα activation (Veldman et al, 2001; Legler et al, 2003). Of interest, ubiquitination of receptor components within the lipid rafts has often been reported; its precise significance is not well understood (Legler et al, 2003).
Using a functional in vitro screen, Deng et al (2000) identified the ubiquitin-conjugating enzymes Ubc13 and Uev1A as essential factors in TRAF6-induced MAPK/IKK activation (Deng et al, 2000). In this context, TRAF6 was shown to exhibit E3 ligase activity that results in the formation of noncanonical K-63-based polyubiquitin chains, which have been implicated in the global activation of TRAF6 downstream effectors (Wang et al, 2001). Recent studies pointed to the role of K63-based ubiquitination of TRAF2 in the activation of germinal center kinase and JNK (Shi and Kehrl, 2003), thereby supporting the role of K63-dependent ubiquitination in the regulation of TRAF2/6 activities, although the underlying mechanism for selective activation remains unknown.
We recently reported that TRAF2 is targeted for proteasome-dependent degradation by Siah2 in stress conditions that coincide with the cell's commitment to undergo apoptosis (Habelhah et al, 2002). Such degradation appeared to occur within 2–6 h after stimulation, similar to what was previously reported for TRAF6 (Takayanagi et al, 2000). Delayed degradation of TRAF2/6 suggests the possible occurrence of regulatory events that prevent faster degradation. Interestingly, ubiquitination of TRAF2 did not depend exclusively on Siah2 activity as it was also observed shortly after TNFα stimulation. Here we demonstrate that TNFα-induced, Ubc13-dependent ubiquitination of TRAF2 requires the TRAF2 RING domain and results in its translocation to the insoluble membrane/cytoskeletal fraction, and that such translocation is required for activation of JNK but not of p38 or IKK, thereby providing the first mechanistic insight into diversification of TRAF2 signaling.
Results
In vivo ubiquitination of TRAF2 requires intact RING and zinc finger domains
Earlier studies revealed that an intact RING is important for TRAF2 activities, indicating that ubiquitination may play a role in TRAF2-mediated activation of downstream signaling cascades (Hu et al, 1994; Baud et al, 1999). In contrast, the RING finger and first zinc finger domains of TRAF6 were shown to be required for activation of MAPK but not IKK (Kobayashi et al, 2001). To assess directly the role of the TRAF2 RING domain in eliciting activation of its downstream signaling, we first determined whether TRAF2 is ubiquitinated in vivo and whether such ubiquitination requires an intact RING domain. Analysis of endogenous TRAF2 revealed a time-dependent increase in its ubiquitination in response to TNFα treatment (Figure 1A). Expression of Flag-TRAF2 also caused polyubiquitination in vivo (Figure 1B). Whereas mutations of consensus C/H residues within the RING domain (TRAF2-Rm) decreased in vivo ubiquitination of TRAF2, such ubiquitination was completely abolished upon additional mutation within the fourth zinc finger domain (TRAF2-RmZm) or by deletion of 87 amino acids from its N-terminal domain (ΔN-TRAF2), which includes the RING domain (Figure 1B). These findings establish that TRAF2 is subjected to TNFα-induced polyubiquitination in vivo that is dependent on its own RING and zinc finger domains.
Figure 1.
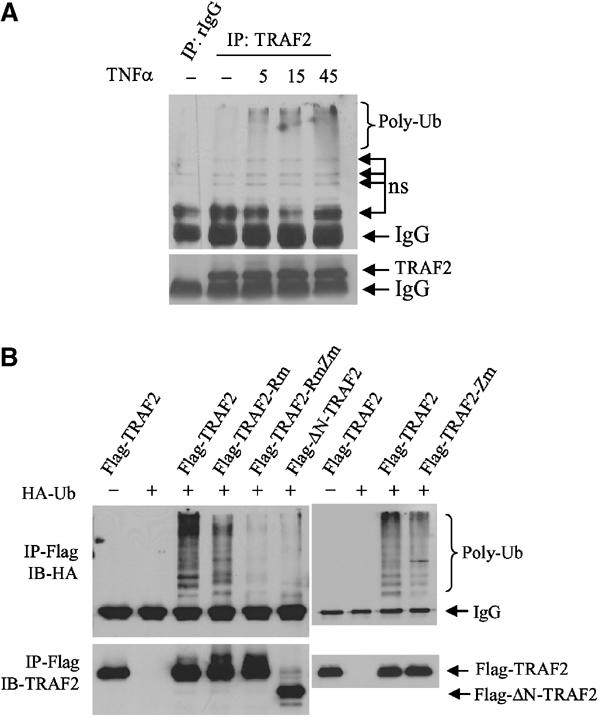
Intact RING and zinc finger domains of TRAF2 are required for its ubiquitination in vivo. (A) In vivo ubiquitination of endogenous TRAF2. HeLa cells were treated with hTNFα (40 ng/ml) at indicated time points, and proteins were extracted with RIPA buffer (supplemented with 20 mM NEM and 5 mM ubiquitin aldehyde). Protein extracts were then subjected to immunoprecipitation with anti-TRAF2 Ab or control rabbit IgG, followed by immunoblot analysis with the aid of anti-ubiquitin Ab (upper panel; ns: nonspecific bands). The same membrane was stripped and reprobed with anti-TRAF2 Ab (lower panel). (B) Intact RING and zinc finger domains of TRAF2 are required for its ubiquitination in vivo. Flag-TRAF2 (1 μg), Flag-TRAF2-Rm (1 μg, C49A, H51A, C54A and C57A), Flag-TRAF2-RmZm (1 μg, C49A, H51A, C209A and C212A) and Flag-ΔN-TRAF2 (1 μg, Δ1–87 amino acids from N-terminal) were co-transfected with HA-Ub (2 μg) to HeLa cells. At 36 h after transfection, proteins were extracted with 2% SDS/TBS, diluted with 9 vol 1% Triton X-100/TBS and subjected to immunoprecipitation (IP) using anti-Flag Ab and Western blot analysis using anti-HA Ab. The same membrane was stripped and reprobed with anti-TRAF2 polyclonal Ab (lower panel).
An intact RING domain of TRAF2 is required for TNFα-mediated activation of JNK but not of NF-κB
Given the roles of the TRAF2 RING and the fourth zinc finger domain in its ubiquitination, we determined possible roles of these domains in JNK and NF-κB activation. Forced expression of wild-type (wt) TRAF2 led to an increase in basal and inducible JNK activity. Expression of TRAF2-Rm led to a decrease in JNK activity that was also reflected in a limited degree of JNK activation following TNFα treatment (Figure 2A). Expression of TRAF2-RmZm or ΔN-TRAF2 almost completely abolished basal and inducible levels of JNK activities (Figure 2A). TRAF2 ubiquitination-dependent activation of JNK coincided with the degree of c-Jun transcriptional activities monitored by a Jun2-Luc reporter (Figure 2B). These findings provide strong evidence for the role of TRAF2 ubiquitination in eliciting signals required for activation of JNK.
Figure 2.

Intact RING and zinc finger domains of TRAF2 are required for activation of JNK but not of NF-κB. (A) TRAF2-RmZm and ΔN-TRAF2 inhibit TNFα-induced JNK activation. HA-JNK1 (0.5 μg) was co-transfected with wt or mutant forms of TRAF2 (1.0 μg) in HeLa cells. At 36 h after transfection, cells were treated with or without hTNFα (40 ng/ml) for 15 min before proteins were extracted with TNE buffer. HA-JNK1 was then immunopurified with anti-HA Ab and subjected to in vitro kinase reaction using GST-c-Jun1–87 as a substrate. Reaction mixtures were incubated at 30°C for 20 min followed by separation on SDS–PAGE, transfer onto nitrocellulose membrane and autoradiography to detect GST-c-Jun1–87 phosphorylation. The same membrane was blotted to detect the HA-JNK1 level (middle panel) using anti-HA Ab and stained with Ponceau S to monitor GST-c-Jun1–87 level (lower panel). Relative density values reflect quantification of corresponding bands by phosphorimager. (B) TRAF2-RmZm and ΔN-TRAF2 inhibit TNFα-induced c-Jun transcriptional activities. HeLa cells cultured on six-well plates were transfected with Jun2-LUC (0.2 μg), β-gal (0.1 μg), and wt or mutant Flag-TRAF2 (0.2 μg). After 36 h, cells were treated with or without hTNFα (10 ng/ml) for 6 h. Luciferase activities were then measured using a luciferase assay system (Promega) and values were normalized based on β-gal activities. The data represent triplicate experiments. (C) Whereas ΔN-TRAF2 inhibits TNFα-induced IKK activation, TRAF2-RmZm does not. HA-IKKβ (1.0 μg) was co-transfected with wt or mutant forms of TRAF2 (1.0 μg) in HeLa cells. At 36 h after transfection, cells were treated with or without hTNFα (40 ng/ml) for 5 min before proteins were extracted with TNE buffer. HA-IKKβ was then immunopurified with anti-HA Ab and subjected to in vitro kinase reaction using GST-IκB1–55 as a substrate. Reaction mixtures were incubated at 30°C for 20 min followed by separation on SDS–PAGE, transfer onto a nitrocellulose membrane and autoradiography to detect GST-IκB1–55 phosphorylation. The same membrane was blotted for HA-IKKβ level (lower panel) using anti-HA Ab and stained with Ponceau S to detect GST-IκB1–55 level (middle panel). (D) The intact TRAF2 RING domain is not required for TNFα-induced NF-κB activities. HeLa cells cultured on six-well plates were transfected with NF-κB-LUC (0.2 μg), β-gal (0.1 μg), and wt or mutant forms of TRAF2 (0.2 μg), and luciferase activities of NF-κB-Luc were analyzed as described in (B). Data represent triplicate experiments.
We next assessed whether the TRAF2 RING is required for induction of IKK/NF-κB activities after TNFα stimulation. Unlike the changes seen for JNK, TRAF2-RmZm inhibited neither IKK activation (Figure 2C) nor induction of NF-κB activities upon TNFα stimulation, monitored by an NF-κB target sequence linked to a luciferase reporter gene (Figure 2D). Along these lines, TRAF2-RmZm mutant, which was no longer ubiquitinated in vivo (Figure 1B) and failed to mediate JNK activation (Figure 2A), could further increase the degree of NF-κB activation after TNFα stimulation (Figure 2D). In contrast, ΔN-TRAF2 was no longer ubiquitinated in vivo and failed to mediate both JNK and NF-κB activation upon TNFα treatment (Figure 2C), suggesting that the actual RING structure is required for TNFα-induced activation of NF-κB. Interestingly, mutation within the fourth zinc finger alone did not affect TRAF2 activation of JNK or IKK pathways, in agreement with earlier studies (Baud et al, 1999). These findings establish that the RING domain, or its contribution to TRAF2 conformation, is required for TNFα-induced NF-κB activation, whereas the E3 ligase activity of TRAF2 is not. This highlights the difference between structural requirements for possible association of upstream kinases and the actual E3 ligase activity of the RING domain.
Ubc13 is required for TRAF2 ubiquitination and activation of JNK but not of NF-κB
The requirement for the intact RING domain of TRAF2 for JNK activation suggests that TRAF2 ubiquitination may be required for JNK but not for NF-κB activation by TNFα. Ubc13/Uev1A-mediated TRAF6 polyubiquitin chains of K63 topology have been implicated in the activation of protein kinases rather than in altered stability (Deng et al, 2000; Wang et al, 2001). Indeed, analysis of the steady-state level of TRAF2 upon exogenous expression of wt or catalytically inactive mutant forms of Ubc13 (Ubc13(C87A)) revealed that neither form affected either the steady-state level of TRAF2- or Siah2-mediated degradation of TRAF2 (Figure 3A). These observations prompted us to assess the role of Ubc13 in TRAF2 ubiquitination and concomitant activation of JNK, p38 and NF-κB. Exogenously expressed Ubc13 increased, whereas Ubc13(C87A) reduced, the level of TRAF2 ubiquitination in vivo in a dose-dependent manner (Figure 3B). This finding suggests that TRAF2 ubiquitination in vivo requires Ubc13.
Figure 3.

UBC13 is required for TRAF2 ubiquitination and activation of JNK but not of NF-κB. (A) Ubc13 does not affect TRAF2 stability. HeLa cells were transfected with Flag-TRAF2 (0.5 μg), Flag-Siah2 (1.0 μg) and/or wt or mutant forms of Ubc13 (1.0 μg). At 36 h after transfection, steady-state levels of TRAF2 were analyzed with anti-Flag Ab (upper panel), and the same membrane was reprobed with anti-β-actin Ab as an internal control (lower panel). (B) Ubc13 increases TRAF2 ubiquitination in vivo. Flag-TRAF2 (1.0 μg), HA-Ub (2.0 μg), and wt-Ubc13 or mut-Ubc13 (C87A) were co-transfected into HeLa cells. At 36 h after transfection, TRAF2 ubiquitination was detected as described in Figure 1B. Ubiquitinated forms of TRAF2 were quantified by densitometry analysis and normalized to the TRAF2 level. Relative amount of TRAF2 ubiquitination is indicated. (C) Dominant-negative Ubc13 inhibits TNFα-induced JNK activation. HA-JNK1 (0.5 μg), Flag-TRAF2 (1.0 μg) and/or HA-Ubc13 or HA-Ubc13mut were co-transfected into HeLa cells. At 36 h after transfection, cells were treated with hTNFα (20 ng/ml) for 15 min, and JNK kinase activities were analyzed by the in vitro immunokinase assay as described in Figure 2A. (D) Expression of Ubc13 does not affect TNFα-induced IκB degradation or ATF2 phosphorylation. Flag-TRAF2 (1.0 μg) and HA-Ubc13 or HA-Ubc13 (C87A) mutant were co-transfected into HeLa cells. At 36 h after transfection, cells were treated with TNFα for 15 min, and degradation of endogenous IκB, phosphorylation of ATF2 and levels of Ubc13 were detected by Western blot using corresponding antibodies as indicated. (E) UBC13 has marginal effects on NF-κB activities. HeLa cells cultured on six-well plates were transfected with NF-κB-Luc (0.2 μg), β-gal (0.2 g), Flag-TRAF2 (0.2 μg) and/or HA-Ubc13 or HA-Ubc13 (C87A). After 24 h, cells were treated with TNFα (10 ng/ml) for 6 h, and luciferase activities were measured as described in Figure 2B.
Expression of TRAF2 was sufficient to increase JNK activity, monitored by immunokinase reactions carried out in vitro using immunopurified HA-JNK and bacterially expressed GST-Jun as a substrate, and such activity was further increased upon TNFα stimulation (Figure 3C). Both basal and inducible levels of JNK activity were inhibited upon expression of the mutant form of Ubc13 (Figure 3C; compare lanes 4 and 10 and lanes 5 and 11). These data suggest an efficient, albeit incomplete, effect of mutant Ubc13 on JNK activation. That expression of wt Ubc13 failed to increase JNK activity may be attributed to the already high levels of endogenous Ubc13, or to the possible existence of a feedback loop mechanism whereby excess TRAF2 ubiquitination may serve to limit further activation of JNK upon TNFα stimulation. Consistent with these observations is the finding that endogenous c-Jun phosphorylation can be induced by wt but not by the mutant form of Ubc13 (data not shown). These results establish that Ubc13-dependent TRAF2 ubiquitination is required for TNFα-induced, TRAF2-mediated activation of JNK signaling.
In contrast to the effects on JNK signaling, neither wt nor mutant forms of Ubc13 affected the degree of IκB phosphorylation (not shown) or degradation following TNFα stimulation (Figure 3D). Similarly, neither form of Ubc13 affected the degree of ATF2 phosphorylation (Figure 3D). These data suggest that neither p38 nor IKK is affected by the extent of TRAF2 ubiquitination or Ubc13 expression.
To further assess possible effects of Ubc13 expression on NF-κB, we monitored NF-κB-Luc activity. Ectopic expression of Ubc13, which increases TRAF2 ubiquitination, caused a small decrease, whereas expression of the mutant form of Ubc13 led to an insignificant increase in NF-κB transcriptional activities (Figure 3E), which is in contrast to changes seen in JNK activity (Figure 3C). Given the mild changes in NF-κB activity, we conclude that neither Ubc13 nor TRAF2 ubiquitination is required for TNFα/TRAF2-dependent activation of NF-κB.
An intact RING domain and Ubc13 are required for translocation of TRAF2 to the insoluble membrane/cytoskeletal fraction
RING-dependent localization of TRAF2 in lipid rafts was shown to be required for TRAF2-mediated signaling (Hostager et al, 2000). To identify possible mechanisms underlying the ability of Ubc13-dependent TRAF2 ubiquitination to induce activation of JNK but not of p38 or IKK signaling pathways, we monitored changes in TRAF2 localization within nonionic detergent soluble and insoluble fractions that represent cytoplasmic and membrane/cytoskeletal (including lipid rafts) complexes, respectively. Whereas endogenous TRAF2 was present in the soluble fraction, exogenously expressed wt-TRAF2 localized in both soluble and insoluble fractions (Figure 4A). Only trace amounts of TRAF2-Rm, and no TRAF2-RmZm or ΔN-TRAF2, were found in the insoluble fraction (Figure 4A). The degree of TRAF2 translocation to the insoluble fraction correlates with the level of TRAF2 ubiquitination (Figure 1B), suggesting that the intact RING is required for TRAF2 ubiquitination and for its translocation to the insoluble membrane/cytoskeletal fraction (Figure 4A).
Figure 4.

Ubc13 and an intact RING domain are required for TRAF2 translocation to the insoluble fraction. (A) TRAF2 translocation to the insoluble fraction requires its intact RING domain. Wt or mutant forms of TRAF2 were expressed in HeLa cells, and 36 h later nonionic detergent soluble and insoluble fractions were prepared as described in Materials and methods. Both transfected and endogenous TRAF2 levels were analyzed by Western blot with anti-TRAF2 Ab. The same membranes were stripped and reprobed for anti-RIP1 and anti-Src antibodies for control of fractionation. (B) Translocation of endogenous TRAF2 to the insoluble fraction is induced by Ubc13. HeLa cells were transfected with Ubc13 (2.0 μg) or Ubc13mut. After 36 h, cells were treated with or without hTNFα (40 ng/ml) for 15 min, and nonionic detergent soluble and insoluble fractions were prepared as described in Materials and methods. The distribution of endogenous TRAF2, RIP1 and Src proteins was analyzed by immunoblotting using the corresponding antibodies. (C) Polyubiquitinated TRAF2 is located within the insoluble fraction. HeLa cells were co-transfected with Flag-TRAF2 (1.0 μg) and HA-Ub (2.0 μg). After 36 h, soluble and insoluble fractions were prepared as described in Materials and methods, and protein samples were subjected to immunoprecipitation with anti-Flag Ab followed by immunoblot analysis with anti-HA Ab to detect ubiquitinated TRAF2. The same membrane was reprobed for TRAF2 level with anti-TRAF2 Ab. The same protein samples were separated on another SDS–PAGE, and distributions of caveolin-1, RIP1 and Src were analyzed with corresponding antibodies. (D) Polyubiquitinated TRAF2 is localized both in membrane rafts and in the insoluble pellet. HeLa cells (3 × 100 mm plates) were co-transfected with Flag-TRAF2 (1.0 μg) and HA-Ub (2.0 μg). After 36 h, cells were lysed in TNPN buffer (containing 20 mM NEM) on ice for 20 min, and membrane rafts and the insoluble pellet were prepared as described in Materials and methods. Protein samples were then subjected to immunoprecipitation with anti-Flag Ab followed by immunoblot analysis with anti-HA Ab to detect the distribution of ubiquitinated TRAF2. The same membrane was reprobed to determine TRAF2 levels with anti-TRAF2 Ab. (E) Inhibition of Ubc13 expression inhibits TNFα-induced JNK activation. HeLa cells were infected with 5 ml of high-titer retroviral supernatants of pRS (control) or pRS-Ubc13 in the presence of 4 μg/ml polybrene overnight followed by further incubation in fresh medium for 72 h before exposure to hTNFα (40 ng/ml) for 10 min. Protein samples were extracted in TNE buffer (containing phosphatase inhibitors) and subjected to immunoblot analysis with anti-Ubc13, anti-phospho-JNK, anti-phospho-p38, anti-IκB and control antibodies as indicated. (F) Inhibition of Ubc13 expression inhibits TNFα-induced TRAF2 translocation to insoluble fraction. HeLa cells were infected with retroviral supernatants as in (E). Cells were treated with or without hTNFα (40 ng/ml) for 10 min, and soluble and insoluble fractions were extracted. Protein samples were then subjected to immunoblot analysis using anti-TRAF2, anti-RIP1 and anti-Src antibodies.
Similar changes in the localization of endogenous TRAF2 provided important confirmation of the observations made with the exogenously expressed forms. Endogenous TRAF2 could be found in the insoluble fraction following TNFα stimulation or following expression of exogenous Ubc13, and even more so using their combination (Figure 4B). The mutant form of Ubc13 reduced the translocation of endogenous TRAF2 upon TNFα stimulation (based on normalization to Src levels, TNFα caused a three-fold increase in TRAF2 localized to the insoluble fraction, compared with 1.8-folds in the presence of TNFα and mutant Ubc13; Figure 4B). In line with these findings, the polyubiquitinated form of TRAF2 was found primarily in the insoluble fraction (Figure 4C). The translocation of TRAF2 to the insoluble fraction also coincided with its ability to induce JNK activities (Figure 3C) and directly correlated with the degree of TRAF2 ubiquitination (Figure 1B). Together, these data provide strong evidence in support of the role of TRAF2 RING-dependent ubiquitination in translocation to the insoluble portion of the cell membrane and confirmation of Ubc13's role in this process.
Ubiquitinated TRAF2 is primarily found in the insoluble membrane/cytoskeletal fraction
The experiments performed so far monitored the distribution of TRAF2 based on its solubility in nonionic detergent, thereby distinguishing between the cytoplasmic (soluble) and membrane/cytoskeletal (insoluble) fractions, which also consist of lipid rafts. To distinguish between lipid rafts and the membranal as well as cytoskeletal fractions, we followed the distribution of ubiquitinated TRAF2 in sucrose gradient fractionation. Although a portion of ubiquitinated TRAF2 was found within the light buoyant lipid rafts, most (80%) ubiquitinated TRAF2 was found within the insoluble pellet, which consists of membrane/cytoskeletal proteins (Figure 4D). These findings indicate that lipid rafts may not be the ultimate destination of ubiquitinated TRAF2.
Inhibition of Ubc13 expression by siRNA inhibits TNFα-induced TRAF2 translocation to the insoluble fraction and attenuates JNK but not IKK or p38 activation
To further confirm the role of Ubc13 in TNFα-induced TRAF2 translocation and JNK activation, we generated a Ubc13 RNAi construct (pRS-Ubc13) that could efficiently decrease the expression of endogenous Ubc13 (Figure 4E). Inhibition of Ubc13 expression resulted in selective inhibition of TNFα-induced JNK activation but did not affect IκB degradation or p38 phosphorylation. These findings provide important direct support for the conclusion that Ubc13 is required for activation of JNK but not of IKK or p38 in response to TNFα stimulation. Further, we monitored changes in the translocation of endogenous TRAF2 after TNFα treatment in cells that express pRS-Ubc13. Indeed, TRAF2 localization within the insoluble cellular fraction was no longer seen in cells that express pRS-Ubc13 (Figure 4F). These data strongly support the role of Ubc13 in TNFα-induced, TRAF2 translocation-dependent activation of JNK and further indicate that IKK and p38 activation is subject to another regulatory pathway.
Disruption of lipid rafts does not impair TNFα-induced activation of JNK or IκB degradation
To further assess the relationship between ubiquitinated TRAF2 and lipid rafts, we determined changes of TRAF2 signaling to the JNK and IKK pathways in cells subjected to treatments known to disrupt lipid rafts. To this end, we used methyl-β-cyclodextrin (MCD), which efficiently depletes cholesterol, a major component of membrane rafts, and which has often been used in the analysis of signal transduction pathways. Using either low (5–10 mM) or high (20–30 mM) MCD, we monitored changes in TNFα-dependent activation of JNK and IKK. Surprisingly, disruption of lipid rafts by low concentrations of MCD alone led to the induction of JNK and IκB phosphorylation in HeLa cells (Figure 5A, compare lane 1 with lanes 4–6). Nevertheless, upon TNFα treatment, phosphorylation of JNK and IκB increased further (Figure 5A, compare lanes 2, 3 and 7, 8). Pretreatment with a higher MCD dose (20 mM) caused greater induction of JNK phosphorylation and IκB degradation, both in NIH 3T3 (Figure 5B) and in HeLa (Supplementary Figure S3) cells, which no longer responded to TNFα treatment, probably because of their full activation prior to cytokine stimulation. Since cholesterol-enriched membrane rafts are found in cytoplasmic and Golgi/ER membranes, its depletion by MCD is expected to disrupt the integrity and trafficking of membrane vehicles between the Golgi apparatus and cytoplasmic membrane, in addition to disrupting membrane rafts.
Figure 5.
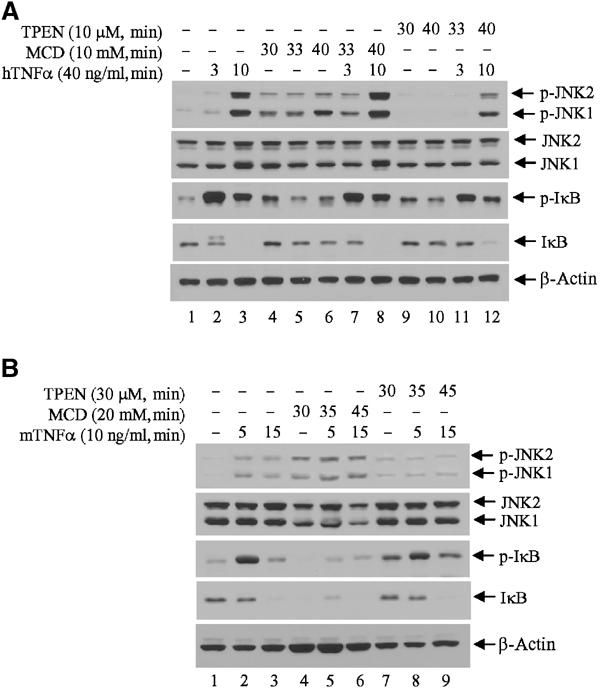
Effect of lipid raft disruption and zinc depletion on TNFα-induced JNK and IκB phosphorylation. (A) Zinc depletion inhibits TNFα-induced JNK activation but not IκB phosphorylation and degradation. HeLa cells were cultured in 1% FBS/DMEM for 1 h and further incubated in the presence of MCD or TPEN for 30 min before exposure to TNFα treatment in the same medium. Protein samples were extracted in TNE buffer (containing phosphatase inhibitor) and subjected to immunoblot analysis with anti-phospho-JNK, anti-phospho-IκB and control antibodies. (B) Rapid disruption of membrane rafts activates JNK and IKK signaling pathways. NIH 3T3 cells were cultured in 1% FBS/DMEM for 1 h and further incubated in the presence of MCD or TPEN for 30 min before exposure to TNFα treatment in the same medium. Protein samples were extracted in TNE buffer (containing phosphatase inhibitor) and subjected to immunoblot analysis with anti-phospho-JNK, anti-phospho-IκB and control antibodies as indicated.
Inactivation of RING E3 ligase activity impairs TNFα-mediated activation of JNK but not IκB degradation
To further assess the observation that the intact TRAF2 RING is required for activation of JNK but not of IKK or p38, we assessed changes in TNFα-induced signaling under conditions that interfere with RING activity as an E3 ligase. Since zinc is essential for E3 ligase activity of all RING finger proteins (Joazeiro and Weissman, 2000), we depleted zinc from HeLa cells using N,N,N-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN; 10 μM) prior to TNFα treatment. Depletion of zinc significantly reduced TNFα-induced JNK phosphorylation but did not affect IκB phosphorylation or its degradation (Figure 5A). Rapid depletion of zinc with high doses of TPEN (30 μM) prior to TNFα treatment sufficed to increase basal JNK phosphorylation but abolished further induction of JNK activity by TNFα (Figure 5B). These treatments, however, failed to interfere with TNFα-induced IκB phosphorylation and degradation. These results strongly support the conclusion that TRAF2's own E3 ligase activity is required for JNK's, but not IKK's, activation.
Hodgkin–Reed–Sternberg cells exhibit TRAF2 cytoplasmic aggregates that coincide with constitutive NF-κB but impaired JNK activities
Our findings suggest that TRAF2 localization is a major determinant in its ability to activate diverse signaling cascades. Accordingly, improper localization of TRAF2 is expected to alter basal or inducible activity of TRAF2 and its downstream effectors. Hodgkin–Reed–Sternberg (HRS) cells were reported to express constitutively activated NF-κB (Staudt, 2000), which coincided with colocalization of TRAF2 and TRAF5 with IKK and NIK in cytoplasmic aggregates (Horie et al, 2002). Therefore, we analyzed TRAF2 localization, JNK phosphorylation and IκB degradation in HRS cells after TNFα treatment. Indeed, in the HRS cells tested (L-428), TRAF2 failed to translocate to the insoluble fraction following TNFα treatment, whereas such translocation was seen in cells obtained from an anaplastic large cell lymphoma (Figure 6A). Basal levels of IκB were almost undetectable in L-428 cells (Figure 6B), corresponding to the observation that NF-κB is constitutively active in HRS cells. Conversely, JNK activity was barely detected and not induced following TNFα treatment in HRS cells (Figure 6B). These findings exemplify changes in TRAF2-elicited signaling that are due to its altered subcellular localization, further supporting the role of TRAF2 subcellular localization in its ability to diversify activities of downstream effectors.
Figure 6.

Impaired translocation of TRAF2 into the insoluble fraction in HRS lymphoma cells coincides with failure to activate JNK. (A) Impaired translocation of TRAF2 into insoluble membrane rafts in HRS cells. HRS lymphoma (L-428) and anaplastic large cell lymphoma (SU-DHL-1) cells were treated with or without TNFα (20 ng/ml) for the indicated time points, and soluble and insoluble fractions were extracted. Protein samples were then subjected to immunoblot analysis using anti-TRAF2, anti-RIP1 and anti-Src antibodies. (B) Constitutive IκB degradation and impaired JNK activation in response to TNFα treatment in HRS cells. L-428 and SU-DHL-1 cells were treated with or without TNFα (20 ng/ml) for the indicated time points, and proteins were extracted with TNE buffer. Protein samples were then subjected to immunoblot analysis with anti-phospho-JNK and anti-IκB antibodies. The same membranes were stripped and blotted with anti-α-tubulin and anti-JNK antibodies as a loading control and for the detection of the level of JNK expression.
Discussion
Our results highlight distinct requirements for activation of JNK, p38 and NF-κB by TNFα–TRAF2 signaling. The requirement of the TRAF2's own RING domain for its in vivo ubiquitination and the link between such ubiquitination and the activation of JNK, but not of p38 or NF-κB, offers a novel mechanistic insight into the regulation of TRAF2 activities, reflected in its activation of distinct downstream pathways (Figure 7). Consistent with our findings, the RING finger and first zinc finger domains of TRAF6 were shown to be required for activation of MAPK but not IKK (Kobayashi et al, 2001). By demonstrating that both Ubc13 and an intact RING are required for translocation of TRAF2 to the insoluble membrane/cytoskeletal fraction, our findings also identify the mechanism underlying ubiquitination-dependent activation of JNK. The ubiquitinated TRAF2 found in the insoluble pellet is likely to be associated with the membrane–cytoskeletal complex, since only a small fraction of it was found in lipid rafts under the same fractionation protocol. Since the presence of TRAF2 within lipid rafts is well documented (Arch et al, 2000; Arron et al, 2002; Legler et al, 2003), our findings point to the dynamic translocation of TRAF2 from membrane rafts to the submembrane/cytoskeletal protein complex. Importantly, we demonstrate that such translocation is required for activation of JNK, but not of p38 or NF-κB, thereby pointing to different mechanisms used by TRAF2 for activation of distinct downstream signaling cascades. Consistent with our finding are the observations that ASK1 and RIP, the upstream kinases for p38 and IKK, respectively, are primarily found within the soluble fractions of the cell, whereas MEKK1 is found within the insoluble fraction following ligand stimulation (Baud et al, 1999; Legler et al, 2003).
Figure 7.

Proposed model. Based on our findings, the following model is proposed for TNFα-induced TRAF2-mediated activation of diverse signaling pathways: first, upon TNFα treatment, TRAF2 is recruited to TNFR through TRADD where TRAF2, in turn, recruits upstream kinase(s) and activates the IKK signaling pathway; independently, TRAF2 undergoes Ubc13-dependent ubiquitination, which induces its translocation to the insoluble membrane rafts, where it is likely to recruit upstream kinases (e.g. MEKK1 and GCK). Although the complex assembled within the lipid rafts may suffice for the activation of JNK per se, further translocation of TRAF2 to insoluble cytoskeletal (or other cytosolic positioned) fraction occurs to bring JNK to necessary substrates (i.e. paxillin). It is also possible that localization within the insoluble portion serves to limit the duration of JNK activity. Neither ubiquitination, nor TRAF2 RING activity or translocation is required for the activation of p38 or IKK, indicating that the latter are subjected to alternate regulatory quos.
The importance of the cytoskeletal domain in TRAF2's ability to activate JNK is further supported by the finding that TNFα fails to activate JNK in human melanoma cell lines deficient in filamin, an actin-binding protein (Leonardi et al, 2000). Interestingly, most TRAF2-interacting proteins bind to the C-terminus of TRAF2, whereas filamin binds to the TRAF2 RING and zinc finger domains (Leonardi et al, 2000), which were shown in the present study to be important for JNK activation.
Disruption of membrane rafts by MCD is sufficient for activation of JNK/IKK signaling, possibly as a result of upstream kinases otherwise concentrated within these domains. Altered distribution of these kinases may suffice to induce their activity, which would be otherwise controlled within the lipid structures. It is possible that rerouting or regrouping of select kinases may be achieved by TNFα-induced ubiquitination of TRAF2. Accordingly, ubiquitinated TRAF2 would recruit GCK/MEKK1 to its partition (which could also be mediated by lipid raft, see below) in the insoluble cytoskeletal fraction, where proper conformation or additional recruitment of downstream kinases would enable JNK activation (Figure 7). Consistent with this model is the finding that fusion of the N-terminal myristoylation domain from src tyrosine kinases (which translocate to membrane rafts) with TRAF3 or TRAF2 whose RING domains have been deleted enabled their translocation to membrane rafts, resulting in concomitant activation of JNK, but not IKK signaling (Dadgostar and Cheng, 2000; Arron et al, 2002). We cannot exclude the possibility that routing of ubiquitinated TRAF2 to the insoluble cytoskeletal domains also serves to limit TRAF2 signaling, somewhat similar to ubiquitination of active transcription factors that limits the duration of transcriptional output (Fuchs and Ronai, 1999). Alternatively, since rafts have also been implicated in endoplasmic transport routes, as in transport between endosomes and the Golgi apparatus (Ikonen, 2001), they may possibly have a role in sorting and recycling of regulatory signaling components, thereby altering their activity.
Consistent with this is the finding that cells derived from HRS patients exhibit cytoplasmic aggregation of TRAF2 along with constitutive NF-κB activation (Horie et al, 2002) but impaired activation of JNK. This example highlights a novel correlation between impaired TRAF2 translocation which results in poor JNK activation and a pathological situation. The nature of altered TRAF2 distribution in HRS patients is yet to be determined.
Our studies also distinguish between the need for RING structure versus RING activity. Whereas the RING structure is required for activation of NF-κB signaling, RING activity that mediates TRAF2 ubiquitination-dependent translocation is required for JNK activation. These results are in line with the finding regarding TRAF2A, a naturally occurring splice variant of TRAF2 including a 7-amino-acid insert within the RING that no longer mediates activation of NF-κB (Brink and Lodish, 1998).
The role of Ubc13 in TRAF6-dependent signaling was previously demonstrated (Deng et al, 2000; Wang et al, 2001). Our finding supports this model, in part, as we demonstrate the role of Ubc13 in TRAF2 activation of JNK. Unlike global TRAF6 signaling requiring Ubc13, however, Ubc13-dependent ubiquitination of TRAF2 serves as the mechanism for activation of JNK, but not of p38 or IKK/NF-κB, as clearly demonstrated by our results. In particular, our findings highlight that Ubc13-dependent ubiquitination enables TRAF2 activation of certain downstream targets. Accordingly, one would expect that TRAF2-dependent activation of p38 and NFκB would be subjected to additional layers of regulation, independent of those required for JNK signaling. Although TRAF2/6 ubiquitination was reported to be mediated by a K63-based polyubiquitin chain (Deng et al, 2000), our results, while supporting the role of Ubc13 in TRAF2 ubiquitination in vivo, also suggest that TRAF2 ubiquitination requires both K48 and K63 residues of ubiquitin (Supplementary Figure S1), and that such a combination is also necessary for TNFα-mediated activation of JNK (Supplementary Figure S2), implying a topology different from those of homogeneous K63 or K48 polychains.
The TRAF2 ubiquitination studied here is different from Siah2 targeting of TRAF2 ubiquitination-dependent degradation (Habelhah et al, 2002). Interestingly, Siah2-mediated degradation of TRAF2 takes place within 2–6 h after exposure to stress stimuli, similar to what was observed for TRAF6 (Takayanagi et al, 2000). This probably reflects localization of TRAF2 within the membrane or the need for removal of K63-based ubiquitin polychains by a deubiquitinating enzyme before K48 polychains can be assembled. Along this line, the deubiquitinating enzyme for p53 (Li et al, 2002) was originally identified as a TRAF-associated protein (Zapata et al, 2001) and may be required for deubiquitination of TRAF2 before it can be targeted for ubiquitination-mediated degradation by Siah2.
Materials and methods
Cell lines, plasmids, regent and transfection
HeLa and NIH 3T3 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (10%) and antibiotics. Cells were transfected with LipofectAMINE PLUS Reagent (Invitrogene/Lifetech) according to the manufacturer's protocol for 12 h followed by medium replacement for an additional 24 h. Antibodies and reagents were purchased as follows: anti-TRAF2, anti-JNK, anti-ATF2 and anti-caveolin-1 antibody (Ab) (Santa Cruz), anti-phospho-JNK Ab (Promega), anti-phospho-IκB, IκB and p38 Ab (Cell Signaling), anti-phospho ATF2 Ab (CALBIOCHEM), anti-RIP1 Ab (BD Biosciences), anti-Src Ab (Upstate), phospho-p38 Ab, MCD and TPEN (Sigma), mTNF (Roche) and hTNF (R&D), HA Ab and Ubc13 Ab (Zymed Lab) and Ub aldehyde (Boston Biochem). Mutations on Flag-TRAF2 and HA-Ub expression vector were introduced using the Quick Change Site-Directed Mutagenesis Kit (Stratagene) and were confirmed by DNA sequencing. Flag-TRAF2-Rm was mutated on the RING domain at C49A, H51A, C54A and C57A; Flag-TRAF2-RmZm was mutated on the RING (C49A, H51A) and fourth zinc finger domains (C209A and C212A), and Flag-ΔN-TRAF2 was deleted 87 amino acids from the N-terminal including the RING domain. HA-IKKβ plasmid was kindly provided by Michael Karin (UCSD), and HA-Ubc13 plasmid by Zhijian J Chen (UT Southwestern Medical Center).
In vivo ubiquitination
wt or mutant forms of Flag-TRAF2 (1.0 μg) and/or HA-tagged ubiquitin (2.0 μg) were transfected into HeLa cells. After 36 h, cells were harvested and cell pellets lysed as previously described (Habelhah et al, 2002). Beads-bound proteins were then eluted in SDS sample buffer and subjected to immunoblot analysis with anti-HA Ab. The same membrane was stripped and reprobed with anti-TRAF2 polyclonal Ab.
JNK and IKK immunokinase assays
HA-JNK1 or HA-IKKβ was co-transfected with wt or mutant forms of Flag-TRAF2 in HeLa cells. After 36 h, cells were treated with or without hTNFα (40 ng/ml), and HA-JNK1 or HA-IKKβ was extracted, immunopurified with anti-HA Ab and subjected to in vitro kinase assays using fusion proteins GST-Jun1–87 (for JNK) and GST-IκB1–55 (for IKK) as substrates. Kinase reactions were carried out in 1 × kinase buffer (Habelhah et al, 2002) in the presence of 2 μCi [γ-32P]ATP and 25 μM cold ATP for 30 min at 30°C. Reaction mixtures were then separated on SDS–PAGE, transferred onto a nitrocellulose membrane, and the phosphorylation states of substrates were detected by autoradiography and quantified via a phosphorimager. The same membranes were then used for immunoblotting of HA-JNK1 or HA-IKKβ, and for Ponceau S staining of substrates.
Immunoblotting
HeLa or NIH 3T3 cells were treated with or without hTNFα (20–40ng/ml) or mTNFα (10 ng/ml) at the indicated time points, harvested and lysed in TNE buffer (20 mM Tris–HCl, pH 7.5, 350 mM NaCl, 1.0% NP-40, 2 mM EDTA, 2 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 50 mM NaF, 1.0 mM sodium vanadate, 10 mM β-glycerolphosphate and 1 × cocktail protease inhibitors) for 30 min on ice followed by centrifugation at 12 500 g for 20 min at 4°C. Protein samples were separated on SDS–PAGE and transferred onto nitrocellulose membranes. For analysis of JNK, ATF2 and IκB phosphorylation, blots were blocked with TBS containing 3% BSA and 10% goat serum for 4 h before immunoblot analysis using anti-phospho Ab. The same membranes were then stripped and reprobed with corresponding anti-JNK, ATF2 or IκB Ab.
Luciferase assays
Cells cultured in six-well plates were transiently transfected with NF-κB or AP1 target sequence linked-luciferase reporter plasmid (NF-kB-Luc or Jun2-Luc; 0.2 μg) together with different TRAF2 or Ubc13 expression vectors (0.2–0.6 μg) and pCMV-β-gal (0.1 μg). At 24 h after transfection, cells were treated with or without TNFα (10 ng/ml), and protein samples were prepared 6 h after treatment. Luciferase activity was measured using the luciferase assay system (Promega) in a luminometer and normalized to the β-galactosidase activity in the same sample.
Cell fractionation
HeLa cells (5 × 106) were washed and harvested with ice-cold PBS, and cell pellets were lysed with 200 l of TNPN buffer (20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.5% NP-40, 10% glycerol, 0.5 mM PMSF, 20 mM N-ethyl-maleimide (NEM) and 1 × cocktail protease inhibitor) on ice for 20 min followed by spindown at 10 000 g for 15 min. The supernatant was taken as soluble protein and the pellets were washed once with 300 μl of the TNPN buffer, and the remaining pellets were subsequently solubilized in 100 μl of RIPA buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM DTT, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 0.5 mM PMSF, 1 × cocktail protease inhibitor, 20 mM NEM) on ice for 30 min. Extracts were spun down at 12 000 g for 15 min and the supernatant served as the insoluble fraction.
Sucrose gradient isolation of lipid rafts
HeLa cells (2 × 100 mm plates) were transfected with Flag-TRAF2 (1.0 μg) and HA-Ub (2.0 μg). After 36 h, cells were washed and harvested with ice-cold PBS. The cell pellet was then lysed in 0.5 ml of TNPN buffer on ice for 20 min and mixed well by vortexing. The same amount of 80% sucrose in TNPN buffer (NP-40 free) was added, mixed and transferred to a centrifuge tube. The sample was then overlaid with 2 ml of 30% sucrose, 1 ml of 5% sucrose in TNPN buffer (NP-40 free) and centrifuged at 200 000 g in a Beckman (SW60 Ti) centrifuge for 16 h. The opaque band migrating at 5–15% sucrose was harvested, diluted with detergent-free TE buffer and pelleted in the microcentrifuge. The pellet from the opaque band (light buoyant lipid rafts) and the pellet from the sucrose gradient tube (insoluble pellet) were then solublized in RIPA (containing 20 mM NEM) buffer by sonication, and Flag-TRAF2 was immunopurified with anti-Flag Ab and subjected to immunoblot analysis of ubiquitination stats of TRAF2 with anti-HA Ab.
Generation of pRETRO-SUPER-Ubc13 (pRS-Ubc13)
A pair of oligonucleotides (nt) containing 19 bp of human Ubc13 from nt 191–210 (TGGCAGCCCCTAAAGTACGT) were generated as follows: 5′gatccccTGGCAGCCCCTAAAGTACGttcaagagaCGTAC TTTAGGGGCTGCCAttttggaaa3′ and 5′agcttttccaaaaGGCAGCCCCTAAAGTACGtctcttga aCGTACTTTAGGGGCTGCCggg3′. The oligos were then annealed and ligated into BglII and HindIII sites of pRS vector. Construct integrity was confirmed by direct sequencing of the plasmid.
Preparation of amphotropic retroviral supernatant
In all, 60–70% confluent 293T cells were co-transfected with 6 μg of pMD.OGP, 5 μg of pMD.G and 8 μg of pBabe-puro-EGFP, pRS or pRS-Ubc13 by calcium phosphate precipitation overnight. The viral supernatant was collected after 48 h and filtered (0.45 μm) followed by immediate use for infection (in the presence of 4 g/ml polybrene). pBabe-puro-EGFP was used for monitoring the efficiency of transfection to 293T and infection of HeLa cells.
Supplementary Material
Supplementary Figure S1 TRAF2 ubiquitination is mediated by both K48 and K63 lysine residue of ubiquitin. HeLa cells were transfected with Flag-TRAF2 (1 μg), HA-Ub (2 μg), HA-Ub-K48R (2 μg), HA-Ub-K63R (2 μg) or HA-Ub-K48/63R (2 μg). 36 h after transfection proteins were extracted and subjected to immunoprecipitation using anti-Flag Ab followed by and western blot analysis using anti-HA Ab. The same membrane was stripped and re-probed with anti-TRAF2 polyclonal Ab (lower panel).
Supplementary Figure S2 Double mutant HA-Ub-K48/63R inhibits TNFα-induced JNK activation. HeLa cells were co-transfected with HA-JNK1 (0.5 μg), Flag-TRAF2 (1.0 μg) and HA-Ub (2 μg), HA-Ub-K48R (2 μg), HA-Ub-K63R (2 μg) or HA-Ub-K48/63R (2 μg). 36 h after transfection cells were treated with or without hTNF-α (40 ng/ml) for 15 min before proteins were extracted with TNE buffer supplemented with phosphatase inhibitor. HA-JNK1 was then immunopurified with anti-HA Ab and subjected to in vitro kinase reaction using GST-c-Jun1–87 as a substrate. The same membrane was blotted for HA-JNK1 level (lower panel) using anti-HA Ab and stained with Ponceau S for GST-c-Jun1–87 level (middle panel).
Supplementary Figure S3 High dose of MCD treatment activates JNK and IKK signaling pathway. HeLa cells were cultured in 1%FBS/DMEM for 1 h and further incubated in the presence of MCD (20 mM) or TPEN (30 μM) for 30 min before exposed to TNF-α treatment in the same medium. Protein samples were extracted in TNE buffer (containing phosphatase inhibitor) and subjected to immunoblot analysis with anti-phospho-JNK, anti-phospho-IκB and control antibodies as indicated.
Acknowledgments
We thank Michael Karin for the TRAF2, JNK and NF-κB constructs, Cecile Pickart and James Chen for Ubc13 constructs, Reuven Agami for pRS plasmid, and Adrian Ting for pMD.G and pMD.OGP plasmids. We also thank Zhen Q Pan and members of the Ronai Lab for discussions, and Ravi Iyengar and Serge Fuchs for valuable comments. Support by NCI grant CA78419 (to ZR) is gratefully acknowledged.
References
- Arch RH, Gedrich RW, Thompson CB (2000) Translocation of TRAF proteins regulates apoptotic threshold of cells. Biochem Biophys Res Commun 272: 936–945 [DOI] [PubMed] [Google Scholar]
- Arron JR, Pewzner-Jung Y, Walsh MC, Kobayashi T, Choi Y (2002) Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J Exp Med 196: 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M (1999) Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev 13: 1297–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JR, Pober JS (2001) Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 20: 6482–6491 [DOI] [PubMed] [Google Scholar]
- Brink R, Lodish HF (1998) Tumor necrosis factor receptor (TNFR)-associated factor 2A (TRAF2A), a TRAF2 splice variant with an extended RING finger domain that inhibits TNFR2-mediated NF-kappaB activation. J Biol Chem 273: 4129–4134 [DOI] [PubMed] [Google Scholar]
- Cottin V, Doan JE, Riches DW (2002) Restricted localization of the TNF receptor CD12a to lipid rafts: a novel role for the death domain. J Immunol 168: 4095–4102 [DOI] [PubMed] [Google Scholar]
- Dadgostar H, Cheng GH (2000) Membrane localization of TRAF3 enables JNK activation. J Biol Chem 275: 2539–2544 [DOI] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ (2000) Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103: 351–361 [DOI] [PubMed] [Google Scholar]
- Fuchs SY, Ronai Z (1999) Ubiquitination and degradation of ATF2 are dimerization dependent. Mol Cell Biol 19: 3289–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habelhah H, Frew IJ, Laine A, Janes PW, Relaix F, Sasson D, Bowtell DD, Ronai Z (2002) Stress-induced decrease in TRAF2 stability is mediated by Siah2. EMBO J 21: 5756–5765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie R, Watanabe T, Ito K, Morisita Y, Watanabe M, Ishida T, Higashihara M, Kadin M, Watanabe T (2002) Cytoplasmic aggregation of TRAF2 and TRAF5 protein in the Hodgkin–Reed–Sternberg cells. Am J Pathol 160: 1647–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostager BS, Catlett IM, Bishop GA (2000) Recruitment of CD40 and tumor necrosis factor receptor-associated factors 2 and 3 to membrane microdomains during CD40 signaling. J Biol Chem 275: 15392–15398 [DOI] [PubMed] [Google Scholar]
- Hu HM, O'Rourke K, Boguski MS, Dixit VM (1994) A novel RING finger protein interacts with the cytoplasmic domain of CD40. J Biol Chem 269: 30069–30072 [PubMed] [Google Scholar]
- Ikonen E (2001) Role of lipid rafts in membrane transport. Curr Opion Cell Biol 13: 470–477 [DOI] [PubMed] [Google Scholar]
- Joazeiro CA, Weissman AM (2000) RING finger proteins: mediators of ubiquitin ligase activity. Cell 102: 49–52 [DOI] [PubMed] [Google Scholar]
- Ko YG, Lee JS, Kang YS, Ahn JH, Seo JS (1999) TNFα-mediated apoptosis is initiated in caveolae-like domains. J Immunol 162: 7217–7223 [PubMed] [Google Scholar]
- Kobayashi N, Kadono Y, Naito A, Matsumoto K, Yamamoto T, Tanaka S, Inoue J (2001) Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J 20 (6): 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi A, Ellinger-Ziegelbauer H, Franzoso G, Brown K, Siebenlist U (2000) Physical and functional interaction of filamin (actin-binding protein-280) and tumor necrosis factor receptor-associated factor 2. J Biol Chem 275: 271–278 [DOI] [PubMed] [Google Scholar]
- Legler DF, Micheau O, Doucey MA, Tschopp J, Bron C (2003) Recruitment of TNF receptor 1 to lipid rafts is essential for TNFa-mediated NF-kB activation. Immunity 18: 655–664 [DOI] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416: 648–653 [DOI] [PubMed] [Google Scholar]
- Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M, Munechika E, Sakai T, Shirasawa T, Akiba H, Kobata T, Santee SM, Ware CF, Rennert PD, Taniguchi M, Yagita H, Okumura K (1999) Targeted disruption of Traf5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc Natl Acad Sci USA 96: 9803–9808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C, Levrero M (1997) Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science 275: 200–203 [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Saito M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H (1998) ASK1 is essential for JNK.SAPK activation by TRAF2. Mol Cell 2: 389–395 [DOI] [PubMed] [Google Scholar]
- Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV (1995) The TNFR2–TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 83: 1243–1252 [DOI] [PubMed] [Google Scholar]
- Shi CS, Kehrl JH (2003) Tumor necrosis factor (TNF)-induced germinal center kinase related (GCKR) and stress-activated protein kinase (SAPK) activation depends upon the E2/E3 complex Ubc13–Uev1A/TNF receptor-associated factor 2 (TRAF2). J Biol Chem 278: 15429–15434 [DOI] [PubMed] [Google Scholar]
- Staudt LM (2000) The molecular and cellular origins of Hodgkin's disease. J Exp Med 191: 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S, Hashimoto H, Mak TW, Yagita H, Okumura K, Yeh WC, Nakao H (2001) Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-κB activation and protection from cell death. J Biol Chem 276: 36530–36534 [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, Nakamura K, Taniguchi T (2000) T-cell-mediated regulation of osteoclastogenesis by signaling cross-talk between RANKL and IFN-γ. Nature 408: 600–605 [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Rothe M, Goeddel DV (1996) Anatomy of TRAF2. Distinct domains for nuclear factor-kappaB activation and association with tumor necrosis factor signaling proteins. J Biol Chem 271: 19935–19942 [DOI] [PubMed] [Google Scholar]
- Veldman RJ, Maestre N, Aduib OM, Medin JA, Salvayre T, Levade T (2001) A neutral sphingomyelinase resides in sphingolipid-enriched microdomains and is inhibited by the caveolin-scaffolding domain: potential implications in tumour necrosis factor signaling. Biochem J 355: 859–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J-I, Chen ZJ (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412: 346–351 [DOI] [PubMed] [Google Scholar]
- Zapata JM, Pawlowski K, Haas E, Ware CF, Godzik A, Reed JC (2001) A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J Biol Chem 276: 24242–24252 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 TRAF2 ubiquitination is mediated by both K48 and K63 lysine residue of ubiquitin. HeLa cells were transfected with Flag-TRAF2 (1 μg), HA-Ub (2 μg), HA-Ub-K48R (2 μg), HA-Ub-K63R (2 μg) or HA-Ub-K48/63R (2 μg). 36 h after transfection proteins were extracted and subjected to immunoprecipitation using anti-Flag Ab followed by and western blot analysis using anti-HA Ab. The same membrane was stripped and re-probed with anti-TRAF2 polyclonal Ab (lower panel).
Supplementary Figure S2 Double mutant HA-Ub-K48/63R inhibits TNFα-induced JNK activation. HeLa cells were co-transfected with HA-JNK1 (0.5 μg), Flag-TRAF2 (1.0 μg) and HA-Ub (2 μg), HA-Ub-K48R (2 μg), HA-Ub-K63R (2 μg) or HA-Ub-K48/63R (2 μg). 36 h after transfection cells were treated with or without hTNF-α (40 ng/ml) for 15 min before proteins were extracted with TNE buffer supplemented with phosphatase inhibitor. HA-JNK1 was then immunopurified with anti-HA Ab and subjected to in vitro kinase reaction using GST-c-Jun1–87 as a substrate. The same membrane was blotted for HA-JNK1 level (lower panel) using anti-HA Ab and stained with Ponceau S for GST-c-Jun1–87 level (middle panel).
Supplementary Figure S3 High dose of MCD treatment activates JNK and IKK signaling pathway. HeLa cells were cultured in 1%FBS/DMEM for 1 h and further incubated in the presence of MCD (20 mM) or TPEN (30 μM) for 30 min before exposed to TNF-α treatment in the same medium. Protein samples were extracted in TNE buffer (containing phosphatase inhibitor) and subjected to immunoblot analysis with anti-phospho-JNK, anti-phospho-IκB and control antibodies as indicated.