

Abstract
The C-terminal domain of poly(A)-binding protein (PABC) is a peptide-binding domain found in poly(A)-binding proteins (PABPs) and a HECT (homologous to E6-AP C-terminus) family E3 ubiquitin ligase. In protein synthesis, the PABC domain of PABP functions to recruit several translation factors possessing the PABP-interacting motif 2 (PAM2) to the mRNA poly(A) tail. We have determined the solution structure of the human PABC domain in complex with two peptides from PABP-interacting protein-1 (Paip1) and Paip2. The structures show a novel mode of peptide recognition, in which the peptide binds as a pair of β-turns with extensive hydrophobic, electrostatic and aromatic stacking interactions. Mutagenesis of PABC and peptide residues was used to identify key protein–peptide interactions and quantified by isothermal calorimetry, surface plasmon resonance and GST pull-down assays. The results provide insight into the specificity of PABC in mediating PABP–protein interactions.
Keywords: EDD, eRF3, HYD, hyperplastic disks, NMR
Introduction
The poly(A)-binding protein (PABP) is an evolutionarily conserved protein, which functions as a scaffold to organize the mRNA ribonucleic acid protein (RNP) complex around the mRNA poly(A) tail (Mangus et al, 2003). Human PABP contains 636 amino acids arranged as four N-terminal RNA-recognition motifs (RRM) and a ∼70-amino-acid C-terminal domain of poly(A)-binding protein (PABC). The two parts are separated by a largely unstructured region of ∼100 amino acids. The N-terminal RRMs bind the mRNA poly(A) tail and interact with the eIF4G complex at the mRNA 5′ cap. This PABP–eIF4G interaction is important for the circularization of the mRNA in actively translating complexes (Wells et al, 1998). At the C-terminus, the PABC domain acts as a protein–protein interaction domain, recruiting various translation or mRNA processing factors to the mRNA RNP complex. Deletion of the yeast homolog gene Pab1 is lethal and its mutation leads to inhibition of translation initiation, poly(A) shortening and delay in the onset of mRNA decay (Sachs and Davis, 1989; Sachs, 1990; Amrani et al, 1997). However, these effects can be suppressed by mutations in the 60S subunit of the ribosome (Sachs and Davis, 1989; Hatfield et al, 1996; Wyers et al, 2000).
In metazoans, several binding partners of PABC have been identified. These include the PABP-interacting proteins Paip1 and Paip2, as well as eRF3/GSPT (Craig et al, 1998; Hoshino et al, 1999; Khaleghpour et al, 2001a; Cosson et al, 2002; Uchida et al, 2002). It was recently shown that a large number of novel, putative-binding partners can be identified by sequence analysis based on the presence of a consensus PABC recognition site (Kozlov et al, 2001) termed PAM2 (for PABP-interacting motif 2) (Roy et al, 2002). The C-terminus of PABP also interacts with a number of plant and yeast proteins: Pab1p-binding protein (Pbp1p), eIF4B and a viral RNA-dependent RNA polymerase, either via PABC or interactions mediated by the PABP linker region (Le et al, 1997; Mangus et al, 1998; Wang et al, 2000; Bushell et al, 2001).
In addition to PABP, PABC also occurs in a ubiquitin E3 protein ligase known as HYD (for hyperplastic discs), EDD or Rat100 (Muller et al, 1992; Mansfield et al, 1994; Huibregtse et al, 1995; Callaghan et al, 1998; Henderson et al, 2002; Honda et al, 2002; Oughtred et al, 2002). HYD is a HECT (homologous to E6-AP C-terminus) family ligase, which is involved in the targeting of ubiquitin to proteins to be modified by the addition of ubiquitin molecules to the side-chain amino groups of lysine residues. Many, but not all, such modified proteins are subsequently degraded by the proteasome (Weissman, 2001). The cellular function of HYD is under investigation; it was recently shown to bind topoisomerase IIβ-binding protein 1 (Honda et al, 2002) and the progesterone receptor (Henderson et al, 2002) in addition to possessing ubiquitin-ligase activity (Huibregtse et al, 1995; Callaghan et al, 1998). HYD has also been implicated in DNA damage response pathways (Honda et al, 2002). The proximity of the PABC and HECT domains in HYD suggests that the PABC domain may play a role in target selection by mediating protein–protein interactions.
The structures of four PABC domains from human, yeast and Trypanosoma cruzi PABP and human HYD have been determined by NMR spectroscopy and X-ray crystallography (Deo et al, 2001; Kozlov et al, 2001, 2002; Siddiqui et al, 2003). These structures are largely similar and consist of ∼70 amino acid residues arranged as a bundle of five or four α-helices. Sequence conservation is highest in helices 2, 3 and 5, which correspond to the peptide-binding site as determined by NMR chemical shift analysis (Kozlov et al, 2001).
Building on the progress in structural characterization of unliganded PABC domains, we report here the structures of the PABC domain from human PABP complexed with PAM2 peptides from Paip1 and Paip2. The peptides bind the PABC domain as a pair of β-turns with an extensive network of salt bridges, hydrophobic and aromatic stacking interactions. We use isothermal titration calorimetry (ITC), surface plasmon resonance (SPR) and GST pull-down experiments to quantify the importance of the various interactions observed by NMR and to measure the affinity of PAM2 motifs from known PABC-binding partners.
Results
The PABC-binding region extends beyond the 12-residue conserved motif
Previous NMR studies suggested that a conserved 12-residue sequence, termed the PAM2 motif, was responsible for binding to PABC domains (Kozlov et al, 2001). To determine the exact boundaries of the PABC-binding site, fragments of Paip2 (residues 106–127), Paip1 (residues 126–144) and eRF3 (residues 50–77) were cloned and expressed in media supplemented with 15NH4Cl. The 15N–1H correlation spectra of the unbound peptides showed a small dispersion of signals characteristic of unfolded peptides (Figure 1A). The addition of unlabeled PABC to the 15N-labeled Paip2 peptide caused chemical shift changes for residues involved in PABC binding (Figure 1B). This transition from unfolded to folded conformation upon binding is the signature of an induced-fit binding mechanism.
Figure 1.
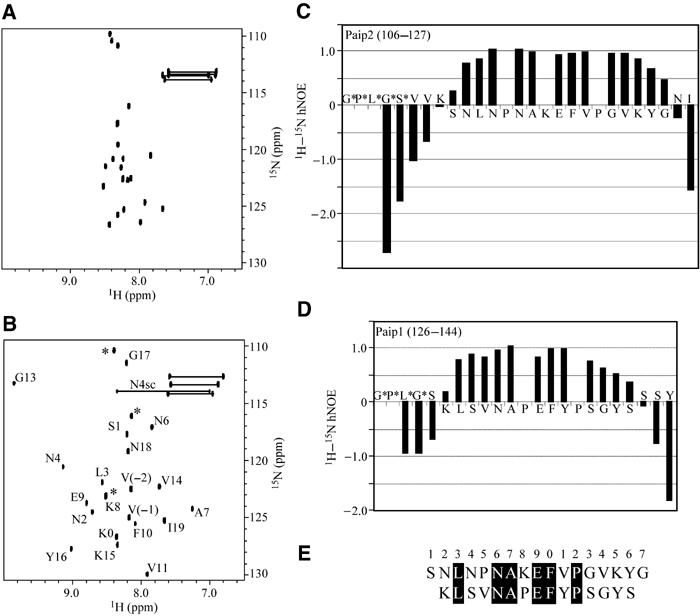
Definition of the PAM2 sites in Paip1 and Paip2 by NMR spectroscopy. Two-dimensional spectra of 15N-labeled Paip2 (106–127) in the absence (A) and presence (B) of unlabeled PABC. Residues are numbered according to the start of the PAM2 motif. N4sc refers to the side-chain resonances of Asn4. Residues originating from the expression vector are labeled with asterisks. Plot of the 1H–15N hNOEs for (C) Paip2 (106–127) and (D) Paip1 (126–144) when bound to PABC. Positive signals indicate reduced mobility reflecting binding of individual residues to the PABC domain. (E) Definition of PAM2 based on residues with positive hNOEs in panels (C) and (D). Conserved residues are blackened.
The boundaries of the regions of the Paip1 and Paip2 peptides that interact with PABC were confirmed by 15N–1H heteronuclear NOE (hNOE) experiments (Figure 1C and D). The hNOE is a measure of the reorientation rate of the amide nitrogen–hydrogen internuclear vectors and thus of the mobility of the peptide backbone. Paip2 residues Asn110 to Tyr124 showed a large positive hNOE that is indicative of the tight binding of 15 peptide residues. The Paip1 peptide gave similar results, with a slightly smaller binding site at both the N- and C-termini. Heteronuclear single quantum correlation (HSQC) spectra of eRF3-PABC were complicated due to the fact that eRF3 contains two overlapping PAM2 motifs. The eRF3-PABC NMR spectra showed intermediate exchange line broadening, suggestive of weaker binding and conformational exchange.
Relative to previously defined motifs (Khaleghpour et al, 2001a; Roy et al, 2002), the chemical shift and hNOE results demonstrate that up to five additional peptide residues are immobilized on the PABC domain. These are located at the C-terminus of the PAM2 motif and surprisingly show no conservation among known PABC ligands. Similar results were obtained with Paip2 binding to the PABC domain from yeast PABP, which suggest that the extended size of the peptide-binding sequence is a general feature of PAM2 binding (Kozlov et al, 2002).
PABC–Paip1 and PABC–Paip2 complex structures and their comparison
To address the question of how the C-termini of PAM2 peptides could bind in the absence of sequence conservation, we determined their structures in complex with PABC from PABP (Figure 2). Based on the structure of the free PABC domain and chemical shift mapping data, we previously suggested that PAM2 peptides bind to helices 2, 3 and 5 of PABC with a conserved peptide phenylalanine residue playing a critical role in binding (Kozlov et al, 2001). This is essentially correct. The peptides bind as a series of two β-turns. The N-terminus sits between helices 3 and 5 of PABC and the variable C-terminus is in contact with the N-terminus of helix 2. Hydrophobic interactions and charge complementarity give rise to the submicromolar affinity measured for PAM2 peptides.
Figure 2.
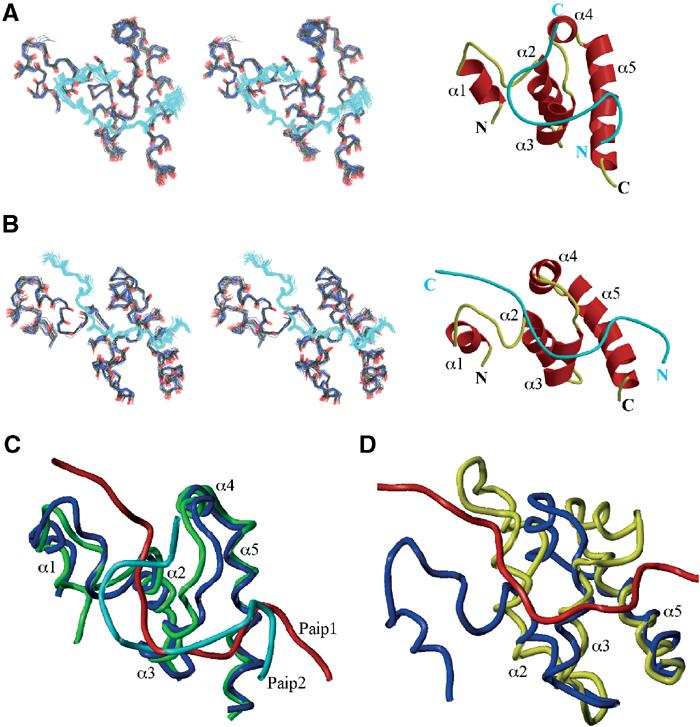
(A) Ensemble of 40 lowest-energy structures and ribbon representation of the PABC–Paip2 complex generated with MOLSCRIPT (Kraulis, 1991) and Raster3D (Merritt and Murphy, 1994). The α-helices of the PABC domain are labeled α1–α5. The peptide is shown in cyan. (B) Ensemble of 30 lowest-energy structures and ribbon representation of the PABC–Paip1 complex. (C) Superposition of the PABC (in green)–Paip2 (in cyan) structure and PABC (in blue)–Paip1 (in red) structure using Sybyl6.5 (Tripos). The superposition was carried out using secondary structure elements in the PABC domain. (D) Superposition of the unliganded PABC structure (in yellow) and structure of PABC (in blue) bound to the peptide from Paip1 (in red). The superposition was carried out using helices 2, 3 and 5 of the PABC domain.
The structure of the first β-turn is similar in the Paip1 and Paip2 complexes. In the Paip2 peptide, the turn is comprised of Asn4-Pro5-Asn6-Ala7 (numbered as in Figure 1E), which is representative of the well-known ‘Asx-Pro turn' motif (Dyson et al, 1990). A general feature of this turn is that the oxygen of the Asn4 side chain forms a hydrogen bond with the amide of Asn6, stabilizing the β-turn. In agreement, the NH2 side-chain protons of Asn4 show large chemical shift changes upon PABC binding and show positive hNOE values (Figure 1B and data not shown). The side chain of the highly conserved Ala7 at the end of the turn makes hydrophobic contact with helix 3 of PABC. Additional hydrophobic contacts are made by Leu3, which binds in the PABC hydrophobic pocket formed by Leu40, Ile43, Val62 and Ala65 (Figure 3C).
Figure 3.
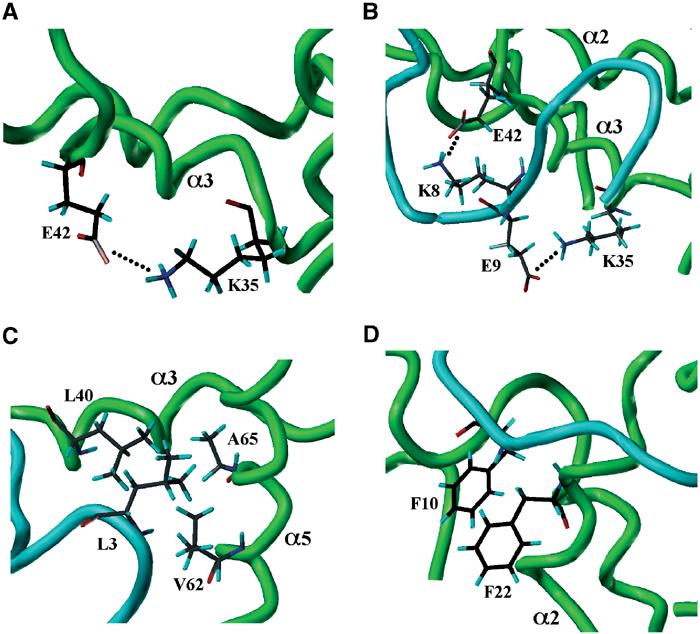
Enlarged views of PABC–peptide interactions. Helices 2, 3 and 5 of the PABC domain are labeled. (A) The intramolecular Lys35–Glu42 salt bridge in the unliganded PABC domain is replaced by (B) two intermolecular salt bridges Lys35–Glu9 and Glu42–Lys8 in the PABC–Paip2 complex. (C) Leu3 from Paip2 PAM2 is inserted into the hydrophobic pocket formed by Leu40, Val62 and Ala65 of the PABC domain. (D) In the Paip1 complex, the peptide Phe10 inserts into the hydrophobic pocket between helices 2 and 3 and stacks with Phe22 of PABC.
The second β-turn of the peptides binds between helices 2 and 3 of PABC. The invariantly conserved Phe10 of the peptides inserts into the hydrophobic pocket between these helices formed by PABC residues Leu41, Thr37 and Gly18. As predicted previously, this peptide residue stacks with Phe22 from PABC (Figure 3D). The propensity of proline residues for forming β-turns explains their conservation in the PAM2 motif.
Interestingly, the second β-turns in Paip1 and 2 are offset by one residue within the PAM2 motif. In Paip2, Val11-Pro12-Gly13-Val14 form a type II β-turn that is strikingly similar to the β-turns found in elastin. In Paip1, this turn is shifted by one residue and involves Pro12-Ser13-Gly14-Tyr15. The Paip1 β-turn is stabilized by the intramolecular stacking interactions between Tyr11 and Tyr15. As a result of the difference in the second β-turn, the C-termini of Paip1 and Paip2 exit the PABC domain in different directions (Figure 2). This explains the reduced specificity of PABC for peptide C-terminal residues. The difference in Paip1 and Paip2 binding conformations appears to originate in PAM2 residues 4, 5 and, particularly, residue 8, which is proline in Paip1. This position could determine whether a peptide binds to PABC in a Paip1- or Paip2-like mode.
In addition to the conserved turns and hydrophobic contacts, the complexes show beautiful charge complementarity, which is reflected in the sequence conservation in the PAM2 motif. In unliganded PABC, the highly conserved residues Glu42 and Lys35 of the KITGMLLE sequence in helix 3 make an intramolecular salt bridge (Figure 3A). In the complexes, this salt bridge is broken and, in the case of Paip2, it is replaced by two new intermolecular salt bridges involving Lys8 and Glu9 of PAM2 (Figure 3B). These peptide residues (especially Glu9) are highly conserved and occur individually or together in essentially all known PABC-binding peptides.
PABC undergoes a conformational change upon binding
The structures of the PABC domain in the presence and absence of the Paip1 peptide were superimposed to investigate conformational changes resulting from binding (Figure 2D). The conformational changes from Paip2 and Paip1 binding were very similar despite the different peptide backbone conformations (Figure 2C). Helices 2, 3 and 5 of PABC showed a relatively small adjustment upon the peptide binding (r.m.s.d. between free and liganded form of 1.4 Å). In contrast, helices 1 and 4 underwent a large shift to accommodate the peptide and avoid steric clashes. Overall, this leads to a significant change in the structure of the peptide-bound form (Figure 2D). These results agree well with previous studies by SPR that suggest that PABC undergoes a conformational change upon Paip1 and Paip2 binding (Khaleghpour et al, 2001a; Roy et al, 2002). In PABC domains from HYD and yeast PABP, the first helix is missing, and so we would expect that smaller helical rearrangements occur upon peptide binding to these domains.
Identification of PABC residues critical for peptide binding
The importance of PABC residues for peptide recognition was tested by site-directed mutagenesis and pull-down experiments (Figure 4). Mutation of Phe22 to alanine in PABC completely abolished the binding of His-tagged Paip1 and Paip2 (Figure 4A and B, lanes 4). The F22A PABC mutant consistently bound a small amount of eRF3 (Figure 4C, lane 4), which we suspect is due to the distributed nature of the eRF3-binding site comprised of two overlapping PAM2 motifs (see below). Binding of eRF3 was completely abolished in an F22A, L40A double mutant (data not shown). These results confirm the singular importance of the aromatic stacking interaction between the protein and peptide phenylalanine residues for PABC–peptide molecular recognition.
Figure 4.
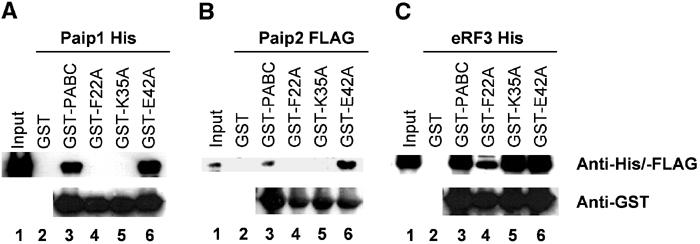
Identification of PABC residues critical for binding. Purified His- and FLAG-tagged proteins: Paip1, Paip2 and eRF3 were incubated with glutathione 4B-Sepharose immobilized GST, GST-PABC and GST-tagged F22A, K35A and E42A PABC mutants, and the resulting complexes were resolved by SDS–PAGE and immunoblotting. Comparison of experimental, anti-His/FLAG lanes 4–6 shows that Phe22 is essential for binding, Glu42 is dispensable and that loss of Lys35 differentially inhibits binding of Paip1 and Paip2. Lanes 1–3 are controls for input protein, nonspecific interactions and wild-type PABC activity. Anti-GST was used as a control for PABC loading on the glutathione beads.
Two other alanine substitutions, Lys35 and Glu42, were tested to assess the importance of the intermolecular salt bridges observed in the NMR structures. While mutation of Glu42 did not affect binding in the pull-down assay, the Lys35 mutation inhibited binding of the Paip proteins but not of eRF3 (Figure 4, lanes 5). This differential effect can be explained by the absence of the glutamate residue in the eRF3 peptide which makes the intermolecular salt bridge with Lys35 (Figure 3B).
Hydrophobic interactions are most important for PABC–peptide binding
We applied ITC to investigate the thermodynamics of PABC binding for the Paip1, Paip2 and eRF3 peptides. Measurement of the heat release upon addition of the peptides to a C-terminal fragment of PABP allowed us to determine the enthalpy, entropy and Gibbs free energy of the binding (Figure 5).
Figure 5.
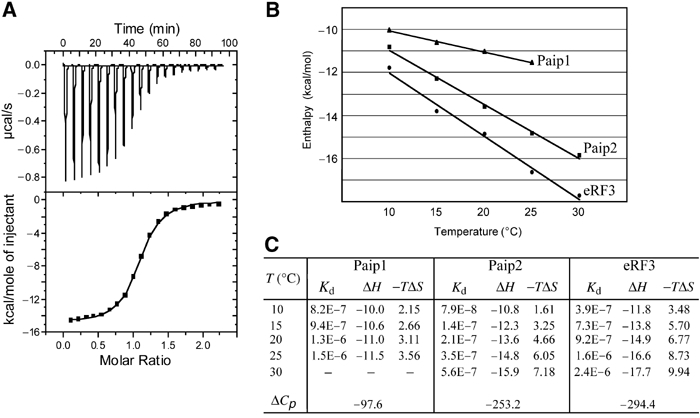
Thermodynamics of peptide binding to PABC. (A) Calorimetric titration of the C2 fragment of PABP with Paip2 (106–127). The top panel shows the heat signal for Paip2 injections into a cell containing PABC at 25°C. The bottom panel shows the integrated heat of each injection after correction for the heat of dilution of Paip2 and normalization of the amount of Paip2 injected (rectangles). The curve represents the best fit to a model involving a single set of independent sites. (B) Temperature dependence of the enthalpy change for Paip1 (triangles), Paip2 (rectangles) and eRF3 (circles) binding to PABC. (C) Thermodynamic parameters of peptides P1895 (Paip2), P1914 (Paip1) and P1913 (eRF3) as Kd (molar), ΔH (kcal/mol), TΔS (kcal/mol) and ΔCp (cal/mol/K). Peptide sequences are shown in Figure 6C.
The binding of Paip1 and Paip2 peptides to the PABC domain is dominated by favorable enthalpic effects, which give rise to the release of heat upon binding. This primarily reflects van der Waals, hydrogen bonds and electrostatic interactions. The negative entropy values for all the peptides tested indicate that the heat release is accompanied by a loss of disorder. This is due to the immobilization of the peptide backbone and a number of side chains in both peptide and protein, which dominates desolvation effects (water release upon complex formation).
The change in heat capacity upon binding (ΔCp) originates from changes in the degree of surface hydration in the free versus complexed molecules and, to a lesser extent, from changes in molecular vibrations. ΔCp has been used to estimate the amount of polar versus nonpolar surface buried upon formation of the complex. The negative ΔCp for the three complexes indicates an increase in hydrophobic interactions upon binding (Figure 5B). The more negative values for Paip2 and eRF3 binding indicate the greater importance of hydrophobic interactions for these complexes and may also reflect the larger protein/peptide contact surfaces.
Identification of peptide residues critical for PABC binding
To investigate the contribution of individual peptide residues to binding affinity, we chemically synthesized a number of peptides and tested their binding by an SPR-based sensor, the Biacore. A C-terminal fragment of PABP was covalently coupled to the biosensor surface, and the different peptide solutions were injected over it and over a control surface without PABC (Figure 6). The steady-state equilibrium values (Req) were used to determine the Kd of the interaction by Scatchard plot analysis. The Kd values, corresponding to the interactions of the peptides with PABC, are reported in Figure 6C. Very good agreement was obtained for the three Kd values measured by both ITC and SPR (compare the values for P1895, P1914 and P1913 in Figure 6C to the 25° values in Figure 5C).
Figure 6.
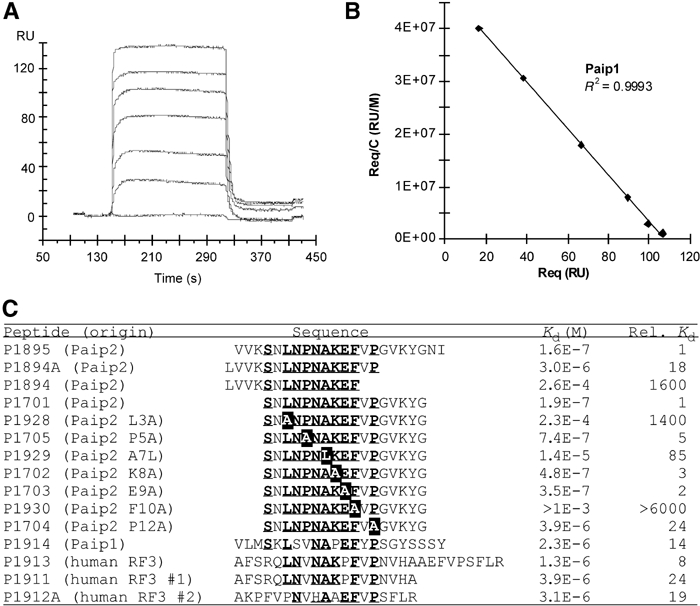
SPR studies of peptide binding to PABC. (A) Paip1 peptide injected at various concentrations (0, 0.41, 1.2, 3.7, 11.1, 33.3 and 100 μM) over a surface coated with the C2 fragment of PABP. The resonance units of each plateau (Req) were used for Scatchard analysis (B) by plotting Req/C as a function of Req. The Kd was derived as the reciprocal of the slope of the linear regression. (C) Sequence alignment and dissociation constants of peptides tested for PABC binding as determined using Biacore. Conserved PAM2 residues are underlined. Mutated Paip2 residues are shown in black.
A series of seven Paip2-derived mutants was created to dissect the relative contributions of the most conserved residues to binding affinity. Alanine substitutions at Pro5 (P1705), Lys8 (P1702) and Glu9 (P1703) had relatively small effects on binding affinity relative to the wild-type peptide (P1701). At the protein–peptide interface, introduction of the bulky amino acid, leucine, at residue 7 (P1929) decreased the affinity 85-fold. The mutant L3A (P1928) showed a significantly larger decrease of 1400-fold. This is not surprising, since Leu3 binds in the hydrophobic pocket between helices 3 and 5 and is largely responsible for the binding of the peptide N-terminus (Figure 3C). The largest effect was measured with the F10A mutant (P1930) where no binding could be detected by SPR, suggesting a Kd above 1 mM. This confirmed that the aromatic stacking interaction between Phe10 of PAM2 and Phe22 of the PABC domain is the single most important factor contributing to peptide binding.
Another Paip2-derived series of peptides probed the N- and C-terminal limits of the PAM2 motif. The full-length peptide (P1895) and N-terminal truncation (P1701) had essentially identical affinities, suggesting that the PAM2 motif starts at or after Ser1. Despite the lack of sequence conservation among Paip1, Paip2 and eRF3, removal of the seven residues following Pro12 (P1894A) decreased the binding affinity 18-fold. Deletion of two additional residues, valine and proline, lowered the affinity by another 85-fold. These results were confirmed by the P12A alanine-scanning mutant (P1704), which dropped the affinity 24-fold. The function of Pro12 is likely to direct the C-terminal PAM2 residues into the binding groove situated between helices 2 and 3 of PABC via formation of the second β-turn. The size of the PAM2 motif determined by the Biacore studies is in excellent agreement with the NMR hNOE results (Figure 1C).
Since eRF3 has two overlapping PAM2 sites, three peptides consisting of the longer eRF3 fragment (P1913) and each separate PAM2 sequence (P1911, P1912A) were synthesized and tested. The isolated PAM2 sites displayed almost identical binding affinities (3.9 and 3.1 μM), while the longer, dual sequence showed a three-fold increase in binding affinity (1.3 μM). This is in agreement with data from NMR titrations, which suggest that residues from both overlapping PAM2 sequences participate in binding to the PABC domain with a large amount of conformational exchange. The degree of cooperativity between the two sites is low since the combined affinity of the two sites is only slightly higher than the sum of the individual sites.
Discussion
Compared to well-known peptide-binding domains such as PDZ, SH3 or PTB domains, PABC is much more specific. This is a reflection of both the longer recognition sequence and PABC's occurrence in just two proteins: PABP and HYD. Here, we define the intermolecular interactions responsible for the high specificity of PABC domains. We show that the domain from PABP binds a conserved motif of 12–15 amino acids, termed PAM2, with submicromolar affinity.
Structurally, we show that the peptide-binding interactions are highly conserved among PABC domains including PABC from HYD (Deo et al, 2001; Kozlov et al, 2001, 2002; Siddiqui et al, 2003). This suggests that crosstalk exists between HYD and PABP. Indeed, in vitro studies have shown that functional overlap exists both in GST pull-down assays and peptide-binding assays (Deo et al, 2001; NS Lim, unpublished results).
The role of PABC in PABP is to recruit proteins to the mRNP complex (Craig et al, 1998; Khaleghpour et al, 2001b; Uchida et al, 2002). Experimentally, one reported effect of PABC deletion is nuclear localization of PABP (Afonina et al, 1998). Intriguingly, in yeast, the HECT-family ubiquitin ligase Tom1p was shown to be required for efficient export of the nuclear PABP Nab2p (Duncan et al, 2000), suggesting a parallel link between nuclear export and ubiquitination. In HYD, PABC seems likely to function similarly to recruit proteins containing PAM2 motifs.
Outside of its PABC domain, PABP is involved in several critical protein–protein interactions. The N-terminal RRM domains bind eIF4G and induce circularization of the mRNA (Wells et al, 1998). The RRM domains also bind Paip1 and Paip2 via a conserved PAM1 motif composed of acidic and hydrophobic amino acids (Khaleghpour et al, 2001b; Figure 7). The physical basis of the RRM–PAM1 interaction is not known and appears to be different for Paip1 and Paip2. Paip1 binding to PABP stimulates mRNA translation and can occur in the presence of poly(A) RNA (Craig et al, 1998). Although containing two binding sites (PAM1 and PAM2), Paip1 binds with an obligate 1:1 stoichiometry to PABP (Roy et al, 2002). In contrast, Paip2 binding inhibits translation, is competitive with poly(A) binding, and can form 2:1 complexes with PABP (Khaleghpour et al, 2001a, 2001b).
Figure 7.
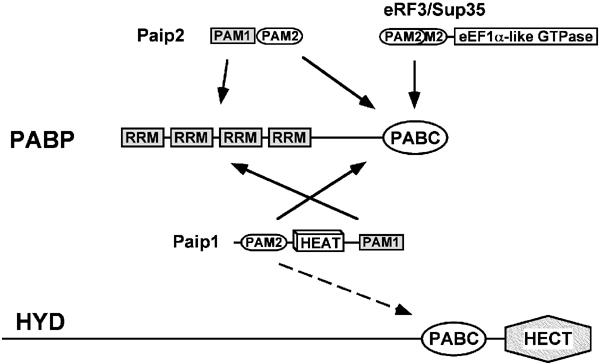
Interactions between PABP or HYD and proteins containing PAM2 motifs. The C-terminal PABC domain of PABP binds the PAM2 motifs of Paip1, Paip2, RF3 and a number of partners (PAM2 proteins) identified by peptide-binding studies. PABP also binds Paip1 and Paip2 through interactions of their PAM1 motifs with the RRMs of PABP. The ubiquitin E3 ligase HYD contains a PABC domain proximal to its catalytic HECT domain. HYD PABC was shown to interact in vitro with GST-Paip1 (Deo et al, 2001) and may play a role in target selection by HYD. Among PABC ligands, the middle segment of Paip1 is notable in that it contains an eIF4G-like HEAT domain that binds eIF4A. eRF3/Sup35 has two overlapped PAM2 motifs and an eEF1A-like domain responsible for its translation termination activity.
Paip1 and Paip2 also differ in their interaction with PABC. The affinity of Paip2 for PABC is unchanged by deletion mutagenesis so that the affinity of intact Paip2 and its isolated PAM2 motif are essentially the same (Khaleghpour et al, 2001a; Figure 5C and 6C). This is consistent with structural studies that found Paip2 unfolded (Kozlov et al, 2001). On the other hand, deletions of Paip1 outside of its PAM2 motif strongly decrease its affinity for PABC, and the isolated PAM2 motif binds several hundred fold less well than intact Paip1 (Roy et al, 2002; Figure 5C and 6C). Either intact Paip1 preorganizes the PAM2 motif for higher affinity binding (decreasing the entropic penalty identified by ITC experiments) or Paip1 makes additional, as yet unidentified, contacts with PABP. In addition to PAM1 and PAM2 motifs, Paip1 contains an eIF4G-like HEAT domain which binds the RNA helicase eIF4A (Craig et al, 1998; Marcotrigiano et al, 2001). Following the RRMs, PABP contains a poorly conserved, unfolded linker sequence of ∼100 amino acids. This linker appears to mediate some protein–protein associations, most notably PABP binding to Pbp1p in yeast (Mangus et al, 1998), and is the site of PABP methylation (Lee and Bedford, 2002). The C-terminal portion of PABP has been implicated in a number of other processes. It is cleaved by viral proteases (Kerekatte et al, 1999; Kuyumcu-Martinez et al, 2002) and has been implicated in PABP dimerization, mRNA stability, structuring of PABP on the mRNA and enhanced translation efficiency (Kuhn and Pieler, 1996; Mangus et al, 1998; Wang and Kiledjian, 2000).
The studies reported here expose the structural and molecular details of the PABC–PAM2 interaction. They further our understanding of the interactions of PABP with known binding partners, Paip1, Paip2 and eRF3, and suggest new perspectives in the study of their divergent functions in translation.
Materials and methods
PABC expression and purification
The PABC domain of human PABP (residues 544–636) was cloned into BamHI and EcoRI restriction sites of the pGEX-6P-1 vector (Amersham Pharmacia Biotech, Piscataway, NJ), and the plasmid was transformed into the Escherichia coli expression host BL21 Gold (DE3) (Stratagene). The protein was expressed and purified by affinity chromatography to yield the 93 C-terminal residues of PABP plus a five-residue (Gly-Pro-Leu-Gly-Ser) N-terminal extension. The amino acid composition was confirmed by mass spectrometry. For NMR analysis, the protein was exchanged into NMR buffer (50 mM KHPO4, 100 mM NaCl, 1 mM NaN3, pH 6.3). The final yield of purified protein was 6 mg/l of M9 culture media.
For binding studies, the C2 terminal domain of PABP (residues 498–636) was used (Khaleghpour et al, 2001a). This corresponds to the fragment used in previous structural studies (Kozlov et al, 2001). PABC mutants were created by overlap extension polymerase chain reaction using the Expand High Fidelity PCR System (Roche Diagnostics, Indianapolis, IN). The reverse primer oligonucleotide sequences used to generate PABC mutants were TGA ATA AGA GGA GCC AGC CGT TCA CC for the F22A mutant, ATG CCA GTG ATT GCA CCA GCA AGA GTA GGG for K35A and GAA TTA TCA ATC GCC AAC AAC ATG CC for E42A. The authenticity of the substitutions and the absence of undesired mutations were confirmed by DNA sequence analysis.
Peptide preparation and purification
Unlabeled peptides were synthesized by Fmoc solid-phase peptide synthesis and purified by reverse phase chromatography on a C18 column (Vydac, Hesperia, CA). The composition and purity of the peptides were verified by ion-spray quadrupole mass spectroscopy.
The C-terminal fragment of human Paip2 (residues 106–127) was cloned into BamHI and EcoRI restriction sites of the vector pGEX-6P-1. 15N-labeled peptide was expressed in BL21 Gold (DE3), grown in M9 media containing 15NH4Cl, and the fusion protein was purified as described previously for PABC to yield a 27-residue peptide consisting of the 22 C-terminal residues of human Paip2 plus a five-residue (Gly-Pro-Leu-Gly-Ser) N-terminal extension. The peptide was desalted using C18 reverse phase chromatography and lyophilized. Residues 126–144 of human Paip1 were cloned, expressed and purified as described for the Paip2 peptide. The final product consisted of 23 residues including a four-residue (Gly-Pro-Leu-Gly) N-terminal extension. The composition and purity of the peptides were verified by ion-spray quadrupole mass spectroscopy.
The human eRF3 (50–77) construct was generated by annealing the oligonucleotide sequences 5′-GAT CCC AAC AAT CCA TGT TCA TAG GAC TAG GAT TAG AAC AAT TAT CAC AGT TGA TAA ACA TTA ATT TAT TAA GTA GTG CTT CAA CTA AA-3′ and 5′-TCG AGT TAT TTA GTT GAA GCA CTA CTT AAT AAA TTA ATG TTT ATC AAC TGT GAT AAT TGT TCT AAT CCT AGT CCT ATG AAC ATG GAT TGT TG-3′. The annealed sequence was then cloned into BamHI and XhoI sites of pGEX-6P-1. The 15N-labeled eRF3 peptide was expressed in BL21 Gold grown in M9 media containing 15NH4Cl. The fusion protein was purified as described previously for PABC, yielding a 33-residue peptide consisting of 28 residues from eRF3 and a five-residue (Gly-Pro-Leu-Gly-Ser) N-terminal extension.
Peptide titrations were carried out by adding either unlabeled peptides into labeled PABC or unlabeled PABC into labeled peptides. Titrations were monitored by 15N–1H HSQC spectra of the labeled species at concentrations of 0.5–1.0 mM.
NMR spectroscopy
NMR resonance assignments of the PABC–peptide complexes were determined using standard triple-resonance techniques on a 13C, 15N-labeled sample (Bax and Grzesiek, 1993) with a Bruker DRX500 NMR spectrometer. All NMR experiments were recorded at 303 K. Hα resonance assignments and 3JHNHα coupling constants were obtained from an HNHA experiment (Kuboniwa et al, 1994) for both the PABC domain and peptides. 15N–1H dipolar couplings were measured with an IPAP-HSQC experiment on an isotropic sample (without phage) and on a sample containing 18 mg/ml Pf1 phage (Hansen et al, 1998; Ottiger et al, 1998). Other backbone and side-chain signal assignments were obtained from three-dimensional (3D) heteronuclear NOESY and TOCSY experiments at 500 and 800 MHz and a homonuclear 2D NOESY experiment at 500 MHz. NOESY constraints for the structure determination were obtained from 15N-edited NOESY and 13C-edited NOESY 3D experiments and 2D homonuclear NOESY at 100 ms mixing times. For the Paip2–PABC complex, NMR samples contained 15N-labeled (15N-NOESY) and 13C,15N-labeled (13C-NOESY) PABC domain and unlabeled Paip2 peptide or 15N-labeled and 13C,15N-labeled Paip2 peptide and unlabeled PABC domain. The Paip1–PABC samples for 15N-NOESY contained 15N-labeled protein and peptide. The 13C-NOESY spectra were recorded on a Varian Inova 800 MHz spectrometer at the Canadian National High Field NMR Centre (NANUC). The 15N–1H hNOEs were measured at 500 MHz on a 15N-labeled Paip1 (126–144) and Paip2 (106–127) complexed with unlabeled PABC (Peng and Wagner, 1994). NMR spectra were processed with GIFA (Malliavin et al, 1996) and XWINNMR software version 2.5 (Bruker Biospin) and analyzed with XEASY (Bartels et al, 1995).
Structure calculation
For the structure determination, NOE distance constraints were derived from homonuclear and 15N- and 13C-edited NOESY spectra of the PABC–peptide complexes acquired at 500 and 800 MHz. NOE constraints for the unliganded PABC domain and manually assigned intermolecular protein–peptide NOEs were used to generate starting structures for ARIA (Nilges et al, 1997), which were subsequently used for automated NOE assignments. The structure was refined using standard protocols in CNS v. 1.1 (Brünger et al, 1998). The final calculations included 15N–1H dipolar couplings. PROCHECK-NMR was used to verify the protein stereochemical geometry (Laskowski et al, 1996). The structural statistics for Paip1–PABC and Paip2–PABC complexes are shown in Table I. The coordinates have been deposited in the RCSB under PDB accession codes 1JGN (Paip2–PABC) and 1JH4 (Paip1–PABC) and the NMR assignments under BMRB accession numbers 5084 (Paip1–PABC) and 5085 (Paip2–PABC).
GST pull-downs and Western blotting
Purified GST-C2 fragments of PABP (2 μg) were incubated with a 50% slurry of glutathione 4B-Sepharose (10 μl; Amersham Pharmacia Biotech) for 15 min at 4°C with 200 μl of pull-down buffer (PDB; 20 mM HEPES, pH 7.5, 100 mM KCl, 1 mM dithiothreitol, 0.5 mM EDTA, 10% glycerol and 0.5% NP-40). His-tagged or FLAG-tagged recombinant proteins (5 μg) were then added in 100 μl of PDB and the mixture was incubated for 2 h at 4°C. The resin was washed three times with 300 μl of PDB. Proteins were eluted with 30 μl of 1 × Laemmli sample buffer, boiled for 5 min, resolved by SDS–PAGE and transferred to a nitrocellulose membrane (0.45 μm pores; Schleicher & Schuell).
For Western blot analysis, nitrocellulose membranes were incubated for 1 h at RT with either anti-His (1:1000) antibody diluted in phosphate-buffered saline-Tween (PBST) supplemented with 3% BSA or anti-FLAG (1:2000) antibody diluted in PBST supplemented with 5% skim milk. Membranes were washed in PBST and incubated with a peroxidase-coupled secondary antibody (1:5000) in either PBST with 3% BSA or 5% skimmedmilk. Membranes were washed again in PBST and the signals were detected using an ECL kit (Amersham Pharmacia Biotech) and exposure to X-ray film (DuPont). The antibodies were stripped off the membranes by incubating them in 0.2 M glycine, pH 2.8, 0.5 M NaCl for 5 min at RT and then neutralized for 5 min with 1 M Tris–HCl, pH 8.5. The membranes were then incubated with rabbit polyclonal anti-GST antibody (1:1000), washed in PBST and incubated with a peroxidase-coupled secondary antibody (1:5000). The signals were detected as described above.
Isothermal titration calorimetry measurements
Experiments were carried out on a MicroCal VP-ITC titration calorimeter (MicroCal Inc., Northampton, MA) using the VPViewer software for instrument control and data acquisition. The buffer used for ITC experiments contained 50 mM phosphate buffer, 100 mM NaCl, pH 6.5. During a titration experiment, samples of the C2 fragment of PABP were thermostated at the desired temperature in a stirred (300 rpm) reaction cell of 1.45 ml. Nineteen injections, each of 15 μl volume and 30 s duration with a 5-min interval between injections, were carried out using a 250 μl syringe filled with the peptide solution. Titration experiments were performed with 16–18 μM PABC solution in the cell and 147–160 μM peptide solution in the syringe to ensure a final peptide:PABC molar ratio of 2:1 in the reaction cell. The calorimetric data were processed using the software package ORIGIN (version 5.0) provided by the manufacturer. The binding isotherm was fitted by an iterative nonlinear least-squares algorithm (Marquadt method) to a binding model employing a single set of independent sites. The Gibbs free energy of binding, molar binding stoichiometry (N), molar binding entropy (So) and molar binding enthalpy (Ho) were determined directly from the fitted curve. The dissociation constant (Kd) was calculated as Go=RT ln Kd, where R=1.99 kcal/mol/K is the gas constant. From calorimetric titrations at different temperatures, the heat capacity change (ΔCp) was determined as the slope of ΔH versus temperature.
SPR-based biosensor analysis
The interaction of PABC with the different synthetic peptides was analyzed by SPR using both BIACORE 1000 and BIACORE 3000 optical biosensors (Biacore AB, Uppsala, Sweden). The C2 fragment of PABP was immobilized onto a CM5 biosensor chip using amine coupling chemistry as described previously (Johnsson et al, 1991). For each peptide interaction, the biosensor experiment was repeated three times over different PABC surfaces (between 2000 and 4000 RU coupled) using the two biosensors. A separate control flow cell was activated and blocked to correct for refractive index changes. The experiments were performed at 25°C using a flow rate of 5 μl/min. For each peptide, solutions at different concentrations (a minimum of five concentrations) were injected over the PABC and control flow cells. All peptide dilutions were performed in running buffer, which contained 10 mM HEPES at pH 7.4, 150 mM NaCl, 3.4 mM EDTA and 0.005% Tween 20. After each injection, regeneration was performed using two pulses of HCl (120 mM, 15 μl), followed by an EXTRACLEAN and a RINSE procedure.
Thermodynamic dissociation constants (Kd) were determined by Scatchard plot analysis, Req/C=f(Req), using the control corrected plateau values (Req) corresponding to the injection of the peptide at concentration C. The Kd was derived as the reciprocal of the slope of the linear regression of the plot.
Acknowledgments
We thank the Canadian National High Field NMR Centre (NANUC) for assistance and use of their facilities. Operation of NANUC is funded by the Canadian Institutes of Health Research (CIHR), the NSERC and the University of Alberta. GDC was supported by PENCE. NS is a CIHR Distinguished Scientist and an HHMI International Scholar. KG is a Senior Boursier of the FRSQ. This study was supported by NIH grant 1R01 GM66157-01 to NS and CIHR grant MA14219 to KG NRC publication no. 46169.
References
- Afonina E, Stauber R, Pavlakis GN (1998) The human poly(A)-binding protein 1 shuttles between the nucleus and the cytoplasm. J Biol Chem 273: 13015–13021 [DOI] [PubMed] [Google Scholar]
- Amrani N, Minet M, Le Gouar M, Lacroute F, Wyers F (1997) Yeast Pab1 interacts with Rna15 and participates in the control of the poly(A) tail length in vitro. Mol Cell Biol 17: 3694–3701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels C, Xia T-H, Billeter M, Guntert P, Wüthrich K (1995) The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J Biomol NMR 5: 1–10 [DOI] [PubMed] [Google Scholar]
- Bax A, Grzesiek S (1993) Methodological advances in protein NMR. Acc Chem Res 26: 131–138 [Google Scholar]
- Brünger AT, Adams PD, Clore GM, Gros P, Grosse-Kuntsleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ (1998) Crystallography and NMR system: a new software system for macromolecular structure determination. Acta Crystallogr D54: 905–921 [DOI] [PubMed] [Google Scholar]
- Bushell M, Wood W, Carpenter G, Pain VM, Morley SJ, Clemens MJ (2001) Disruption of the interaction of mammalian protein synthesis eukaryotic initiation factor 4B with the poly(A)-binding protein by caspase- and viral protease-mediated cleavages. J Biol Chem 276: 23922–23928 [DOI] [PubMed] [Google Scholar]
- Callaghan MJ, Russell AJ, Woollatt E, Sutherland GR, Sutherland RL, Watts CK (1998) Identification of a human HECT family protein with homology to the Drosophila tumor suppressor gene hyperplastic discs. Oncogene 17: 3479–3491 [DOI] [PubMed] [Google Scholar]
- Cosson B, Couturier A, Chabelskaya S, Kiktev D, Inge-Vechtomov S, Philippe M, Zhouravleva G (2002) Poly(A)-binding protein acts in translation termination via eukaryotic release factor 3 interaction and does not influence [PSI(+)] propagation. Mol Cell Biol 22: 3301–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AW, Haghighat A, Yu AT, Sonenberg N (1998) Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature 392: 520–523 [DOI] [PubMed] [Google Scholar]
- Deo RC, Sonenberg N, Burley SK (2001) X-ray structure of the human hyperplastic discs protein: an ortholog of the C-terminal domain of poly(A)-binding protein. Proc Natl Acad Sci USA 98: 4414–4419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan K, Umen JG, Guthrie C (2000) A putative ubiquitin ligase required for efficient mRNA export differentially affects hnRNP transport. Curr Biol 10: 687–696 [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Satterthwait AC, Lerner RA, Wright PE (1990) Conformational preferences of synthetic peptides derived from the immunodominant site of the circumsporozoite protein of Plasmodium falciparum by 1H NMR. Biochemistry 29: 7828–7837 [DOI] [PubMed] [Google Scholar]
- Hansen MR, Mueller L, Pardi A (1998) Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat Struct Biol 5: 1065–1074 [DOI] [PubMed] [Google Scholar]
- Hatfield L, Beelman CA, Stevens A, Parker R (1996) Mutations in trans-acting factors affecting mRNA decapping in Saccharomyces cerevisiae. Mol Cell Biol 16: 5830–5838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson MJ, Russell AJ, Hird S, Munoz M, Clancy JL, Lehrbach GM, Calanni ST, Jans DA, Sutherland RL, Watts CK (2002) EDD, the human hyperplastic discs protein, has a role in progesterone receptor coactivation and potential involvement in DNA damage response. J Biol Chem 277: 26468–26478 [DOI] [PubMed] [Google Scholar]
- Honda Y, Tojo M, Matsuzaki K, Anan T, Matsumoto M, Ando M, Saya H, Nakao M (2002) Cooperation of HECT-domain ubiquitin ligase hHYD and DNA topoisomerase II-binding protein for DNA damage response. J Biol Chem 277: 3599–3605 [DOI] [PubMed] [Google Scholar]
- Hoshino S, Imai M, Kobayashi T, Uchida N, Katada T (1999) The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3′-poly(A) tail of mRNA. Direct association of erf3/GSPT with polyadenylate-binding protein. J Biol Chem 274: 16677–16680 [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S, Howley PM (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc Natl Acad Sci USA 92: 2563–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson B, Lofas S, Lindquist G (1991) Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal Biochem 198: 268–277 [DOI] [PubMed] [Google Scholar]
- Kerekatte V, Keiper BD, Badorff C, Cai A, Knowlton KU, Rhoads RE (1999) Cleavage of poly(A)-binding protein by coxsackievirus 2A protease in vitro and in vivo: another mechanism for host protein synthesis shutoff? J Virol 73: 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaleghpour K, Kahvejian A, De Crescenzo G, Roy G, Svitkin YV, Imataka H, O'Connor-McCourt M, Sonenberg N (2001a) Dual interactions of the translational repressor Paip2 with poly(A) binding protein. Mol Cell Biol 21: 5200–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaleghpour K, Svitkin YV, Craig AW, DeMaria CT, Deo RC, Burley SK, Sonenberg N (2001b) Translational repression by a novel partner of human poly(A) binding protein, Paip2. Mol Cell 7: 205–216 [DOI] [PubMed] [Google Scholar]
- Kozlov G, Siddiqui N, Coillet-Matillon S, Trempe JF, Ekiel I, Sprules T, Gehring K (2002) Solution structure of the orphan PABC domain from Saccharomyces cerevisiae poly(A)-binding protein. J Biol Chem 277: 22822–22828 [DOI] [PubMed] [Google Scholar]
- Kozlov G, Trempe JF, Khaleghpour K, Kahvejian A, Ekiel I, Gehring K (2001) Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc Natl Acad Sci USA 98: 4409–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 24: 946–950 [Google Scholar]
- Kuboniwa H, Grzesiek S, Delaglio F, Bax A (1994) Measurement of HN-H alpha J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J Biomol NMR 4: 871–878 [DOI] [PubMed] [Google Scholar]
- Kuhn U, Pieler T (1996) Xenopus poly(A) binding protein: functional domains in RNA binding and protein–protein interaction. J Mol Biol 256: 20–30 [DOI] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Joachims M, Lloyd RE (2002) Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J Virol 76: 2062–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486 [DOI] [PubMed] [Google Scholar]
- Le H, Tanguay RL, Balasta ML, Wei CC, Browning KS, Metz AM, Goss DJ, Gallie DR (1997) Translation initiation factors eIF-iso4G and eIF-4B interact with the poly(A)-binding protein and increase its RNA binding activity. J Biol Chem 272: 16247–16255 [DOI] [PubMed] [Google Scholar]
- Lee J, Bedford MT (2002) PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep 3: 268–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malliavin T, Pons J-L, Delsuc M-A (1996) Gifa V4: a complete package for NMR data-set processing. J Biomol NMR 8: 445–452 [DOI] [PubMed] [Google Scholar]
- Mangus DA, Amrani N, Jacobson A (1998) Pbp1p, a factor interacting with Saccharomyces cerevisiae poly(A)-binding protein, regulates polyadenylation. Mol Cell Biol 18: 7383–7396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangus DA, Evans MC, Jacobson A (2003) Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol 4: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield E, Hersperger E, Biggs J, Shearn A (1994) Genetic and molecular analysis of hyperplastic discs, a gene whose product is required for regulation of cell proliferation in Drosophila melanogaster imaginal discs and germ cells. Dev Biol 165: 507–526 [DOI] [PubMed] [Google Scholar]
- Marcotrigiano J, Lomakin IB, Sonenberg N, Pestova TV, Hellen CU, Burley SK (2001) A conserved HEAT domain within eIF4G directs assembly of the translation initiation machinery. Mol Cell 7: 193–203 [DOI] [PubMed] [Google Scholar]
- Merritt EA, Murphy MEP (1994) Raster3D version 2.0: a program for photorealistic molecular graphics. Acta Crystallogr D50: 869–873 [DOI] [PubMed] [Google Scholar]
- Muller D, Rehbein M, Baumeister H, Richter D (1992) Molecular characterization of a novel rat protein structurally related to poly(A) binding proteins and the 70K protein of the U1 small nuclear ribonucleoprotein particle (snRNP). Nucleic Acids Res 20: 1471–1475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilges M, Macias MJ, O'Donoghue SI, Oschkinat H (1997) Automated NOESY interpretation with ambiguous distance restraints: the refined NMR solution structure of the pleckstrin homology domain from beta-spectrin. J Mol Biol 269: 408–422 [DOI] [PubMed] [Google Scholar]
- Ottiger M, Delaglio F, Bax A (1998) Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson 131: 373–378 [DOI] [PubMed] [Google Scholar]
- Oughtred R, Bedard N, Adegoke OA, Morales CR, Trasler J, Rajapurohitam V, Wing SS (2002) Characterization of rat100, a 300-kilodalton ubiquitin-protein ligase induced in germ cells of the rat testis and similar to the Drosophila hyperplastic discs gene. Endocrinology 143: 3740–3747 [DOI] [PubMed] [Google Scholar]
- Peng JW, Wagner G (1994) Investigation of protein motions via relaxation measurements. Methods Enzymol 239: 563–596 [DOI] [PubMed] [Google Scholar]
- Roy G, De Crescenzo G, Khaleghpour K, Kahvejian A, O'Connor-McCourt M, Sonenberg N (2002) Paip1 interacts with poly(A) binding protein through two independent binding motifs. Mol Cell Biol 22: 3769–3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs A (1990) The role of poly(A) in the translation and stability of mRNA. Curr Opin Cell Biol 2: 1092–1098 [DOI] [PubMed] [Google Scholar]
- Sachs AB, Davis RW (1989) The poly(A) binding protein is required for poly(A) shortening and 60S ribosomal subunit-dependent translation initiation. Cell 58: 857–867 [DOI] [PubMed] [Google Scholar]
- Siddiqui N, Kozlov G, D'Orso I, Trempe J-F, Gehring K (2003) Solution structure of the C-terminal domain from poly(A)-binding protein in Trypanosoma cruzi: a vegetal PABC domain. Protein Sci 12: 1925–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Hoshino S, Imataka H, Sonenberg N, Katada T (2002) A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in cap/poly(A)-dependent translation. J Biol Chem 277: 50286–50292 [DOI] [PubMed] [Google Scholar]
- Wang X, Ullah Z, Grumet R (2000) Interaction between zucchini yellow mosaic potyvirus RNA-dependent RNA polymerase and host poly-(A) binding protein. Virology 275: 433–443 [DOI] [PubMed] [Google Scholar]
- Wang Z, Kiledjian M (2000) The poly(A)-binding protein and an mRNA stability protein jointly regulate an endoribonuclease activity. Mol Cell Biol 20: 6334–6341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman AM (2001) Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2: 169–178 [DOI] [PubMed] [Google Scholar]
- Wells SE, Hillner PE, Vale RD, Sachs AB (1998) Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2: 135–140 [DOI] [PubMed] [Google Scholar]
- Wyers F, Minet M, Dufour ME, Vo LT, Lacroute F (2000) Deletion of the PAT1 gene affects translation initiation and suppresses a PAB1 gene deletion in yeast. Mol Cell Biol 20: 3538–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]