

Abstract
The mechanisms used by checkpoints to identify DNA lesions are poorly understood and may involve the function of repair proteins. Looking for mutants specifically defective in activating the checkpoint following UV lesions, but proficient in the response to methyl methane sulfonate and double-strand breaks, we isolated cdu1-1, which is allelic to RAD14, the homolog of human XPA, involved in lesion recognition during nucleotide excision repair (NER). Rad14 was also isolated as a partner of the Ddc1 checkpoint protein in a two-hybrid screening, and physical interaction was proven by co-immunoprecipitation. We show that lesion recognition is not sufficient for checkpoint activation, but processing, carried out by repair factors, is required for recruiting checkpoint proteins to damaged DNA. Mutations affecting the core NER machinery abolish G1 and G2 checkpoint responses to UV, preventing activation of the Mec1 kinase and its binding to chromosomes. Conversely, elimination of transcription-coupled or global genome repair alone does not affect checkpoints, suggesting a possible interpretation for the heterogeneity in cancer susceptibility observed in different NER syndrome patients.
Keywords: budding yeast, cell cycle, DNA damage checkpoint, nucleotide excision repair, phosphorylation
Introduction
Checkpoints are surveillance mechanisms that monitor cell cycle progression and preserve the correct order of events (Hartwell and Weinert, 1989; Hartwell, 1992). Checkpoints are also activated in response to genomic insults, or alterations of cellular structures, and lead to temporary cell cycle arrest, slowing down of DNA replication, changes in the cellular transcriptional program and, in some instances, apoptosis (Paulovich and Hartwell, 1995; Elledge, 1996; Weinert, 1998). Alterations in these mechanisms are responsible for a number of pathologies in humans (Elledge, 1996).
In Saccharomyces cerevisiae, the DNA damage checkpoint relies on a set of proteins having structural and functional counterparts in all eukaryotic cells, indicating that checkpoint controls have been highly conserved during evolution (Foiani et al, 2000). The cellular response to DNA damage is usually described as a signal transduction cascade, where sensor proteins detect a lesion in the double helix and elicit a signal, which is relayed to several effectors through the activity of different protein kinases.
The order of function of the genes in the cascade has been mainly inferred, in checkpoint defective mutants, by monitoring the phosphorylation state of proteins acting in the pathway (Longhese et al, 1998; Lowndes and Murguia, 2000; Carr, 2002). The heterotrimeric complex, composed in yeast of Rad17, Mec3 and Ddc1, shows a structural similarity to PCNA, a clamp involved in recruiting replication proteins onto the DNA at the replication fork; from now on it will be called ‘PCNA-like complex' (Paciotti et al, 1998; Kondo et al, 1999; Thelen et al, 1999; Majka and Burgers, 2003). Yeast Rad24 has sequence similarities to replication factor C (RFC), a protein complex that loads PCNA onto DNA during replication. Rad24 interacts with the four smaller RFC subunits, forming an RFC-like complex, required to load the PCNA-like complex close to the DNA lesion (Majka and Burgers, 2003). Once the damage has been detected, Mec1 kinase is activated and triggers the signal transduction cascade.
Mec1 constitutively interacts with Ddc2/Lcd1/Pie1 and is a member of the evolutionarily conserved subfamily of phosphatidylinositol 3-kinase (Elledge, 1996; Rouse and Jackson, 2002). It is still unclear how Mec1 and its substrates are brought in close proximity onto DNA after genotoxic treatment. It has been recently shown that Mec1–Ddc2 and the PCNA-like complex can be independently recruited onto damaged DNA (Kondo et al, 2001; Melo et al, 2001; Zou et al, 2002), and it has been suggested that the PCNA-like complex might be involved in recruiting different substrates for Mec1 (Giannattasio et al, 2002; Zou et al, 2002). Critical Mec1 targets are Rad9 and Rad53; their modification allows these two proteins to interact, leading to Rad53 kinase activation. This is an essential step for the cellular response to DNA damage and can be used as a marker to monitor checkpoint activation (Sanchez et al, 1996; Sun et al, 1996; Vialard et al, 1998; Pellicioli et al, 1999).
It is still unknown how cells realize that the genome has been damaged and how all the different lesions are detected. It was proposed that the RFC-like and the PCNA-like complexes could recognize the altered DNA structure, possibly process it through some nuclease activity, and then allow Mec1 to be recruited and activated (Lydall and Weinert, 1995). On the other hand, it is hard to imagine that just one complex could recognize a plethora of potential DNA lesions in the entire genome. In this view, DNA repair machineries may specifically localize the damages and trigger the checkpoint response, by direct recruitment of checkpoint proteins or by generating intermediate DNA structures that are recognized by checkpoint factors. The Mre11/Rad50/Xrs2 complex, involved in double-strand break (DSB) processing, seems to be necessary for the activation of the DNA damage checkpoint triggered by DSBs (D'Amours and Jackson, 2001; Grenon et al, 2001; Usui et al, 2001). It has also been shown that, after a site-specific DSB, in yeast the lesion is first processed by exonuclease activities to generate long single-stranded DNA (ssDNA) regions, and this structure is likely responsible for activating the checkpoint kinase Mec1 (Lee et al, 1998; Pellicioli et al, 2001). Recently, Zou and Elledge (2003) showed that RPA-coated ssDNA seems to be critical for recruiting Ddc2 to the sites of DSBs. In a previous work, it was reported that nucleotide excision repair (NER)-defective rad14Δ cells irreversibly arrest in S phase after UV treatment in G1 (Neecke et al, 1999). While this observation could suggest a role for Rad14 in the checkpoint response, this hypothesis has recently been disputed by a paper arguing that NER is not required for checkpoint activation (Zhang et al, 2003). In this work, we have thoroughly investigated this issue, which is extremely important for understanding the crosstalk between repair mechanisms and checkpoint response.
All cells have developed repair mechanisms to reduce the genotoxic effects of DNA damage. In eucaryotes, NER is a major pathway devoted to the elimination of a variety of DNA lesions, including UV photoproducts (Friedberg et al, 1995). Its biological relevance is recapitulated by genetic diseases caused by a defective NER: xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD) (Hoeijmakers, 2001). NER is characterized by the recognition of the damage, likely carried out by Rad14 in budding yeast, incision of the DNA strand on both sides of the lesion, followed by the removal of the damaged oligonucleotide and refilling of the single-stranded gap. Genetic and biochemical analyses in yeast and mammalian cells indicate that NER consists of two separate pathways converging into a common core: one system, known as transcription-coupled repair (TCR), rapidly takes care of lesions affecting the transcribed strand of expressed genes; global genome repair (GGR), instead, fixes damages in nonexpressed regions and on the nontranscribed strand of expressed sequences. The different enzymatic activities involved in NER seem to exist as subcomplexes, called nucleotide excision repair factors (NEFs), consisting of several proteins that can be copurified and participate in a common function (Guzder et al, 1996). NEF1 consists of Rad14, specialized for the recognition of damaged DNA, and the Rad1–Rad10 endonuclease. NEF2, composed of Rad4 and Rad23, is involved in facilitating the identification of the lesion-containing sequence. NEF3 contains the Rad2 endonuclease and TFIIH, which includes the Rad3 helicase, while Rad7 and Rad16 are part of NEF4. In budding yeast, Rad14 and RPA, with the help of Rad4–Rad23, recognize helical distortions induced by UV lesions. The Rad3 helicase facilitates the formation of a single-stranded bubble containing the lesion, and this is the substrate for the structure-specific nucleases Rad2 and Rad1–Rad10 that incise the ssDNA on the 3′ and 5′ side of the lesion, respectively. The resulting ssDNA gap is then filled by a polymerase activity (see Prakash and Prakash, 2000).
We have found a tight functional and physical connection between NER and checkpoint proteins. With a genetic screening, aimed at isolating mutants defective in checkpoint activation specifically after UV irradiation, we identified CDU1, which is allelic to RAD14. Genetic dissection of NER revealed that recognition of the primary lesion by repair proteins and processing of the damage are prerequisites for activation of Mec1 kinase. Moreover, Rad14 was proven to interact physically with the PCNA-like complex, and its presence is required to recruit the Ddc1 and Ddc2 complexes to DNA after UV radiation, but not in response to DSBs.
Results
Isolation of a cdu1-1 mutant specifically defective in the UV-induced checkpoint response
If different types of DNA damage are detected through specific sensors, it should be possible to identify such factors by looking for mutants that lose the ability to activate the checkpoint in response to a particular lesion, while properly responding to other kinds of damage. Among mutants sensitive to UV, but resistant to the alkylating agent methyl methane sulfonate (MMS), we searched for those that displayed a specific checkpoint defect after UV irradiation. We isolated 15 independent mutants that were UV sensitive, MMS resistant and were found to have a UV-specific checkpoint defect, as monitored by looking at Rad53 phosphorylation. The mutant with the clearest phenotype was called cdu1-1 (for checkpoint deficient in UV). Figure 1A shows wild-type (wt), mec1-1 and cdu1-1 cells spotted on YEPD plates that were either mock treated, UV irradiated or contained MMS. It is clear that while mec1-1 is sensitive to both treatments, CDU1 function is specifically required for survival after UV irradiation, but not after MMS treatment. Interestingly, Figure 1B shows that cdu1-1 cells are completely defective in activating Rad53 in response to UV and 4-nitro-quinoline oxide (4NQO) treatments, both in G1- and G2-arrested cells. Conversely, Rad53 phosphorylation in exponentially growing cells treated with 0.02% MMS is unaffected, indicating that the DNA damage checkpoint does not require CDU1 to respond to MMS. We also tested the effect of the DSB-inducing agent zeocin on the activation of the G1 and G2 checkpoints in wt and cdu1-1 cells. Figure 1C shows that Rad53 phosphorylation is undistinguishable in wt and mutant strains. Our results also show that the response to zeocin is dosage dependent and seems to be stronger in G1- than in G2-arrested cells. Altogether, these data suggest that Cdu1 discriminates among different DNA alterations and is fundamental for obtaining checkpoint activation after UV-mimetic damage, but not following MMS treatment or DSB accumulation. The G1 checkpoint response in cdu1-1 was further analyzed by monitoring budding kinetics in cells UV irradiated in G1 (Figure 2A); the G2 checkpoint was studied by following nuclear division in cells treated with UV in G2 and then allowed to go through mitosis (Figure 2B). wt strains display a notable delay in emitting the bud when damaged in G1, and arrest at the metaphase–anaphase transition with undivided nuclei if the genotoxin is given in G2. A classical checkpoint mutant strain (mec3Δ), instead, buds with the same kinetics regardless of the presence of DNA lesions and does not prevent nuclear division, showing a decrease in uninucleated cells. In such an assay, cdu1-1 cells behave like a checkpoint mutant, being unable to delay bud emergence or nuclear division in response to UV treatment, indicating that CDU1 function is required for normal activation of the G1 and G2 checkpoints induced by UV lesions.
Figure 1.
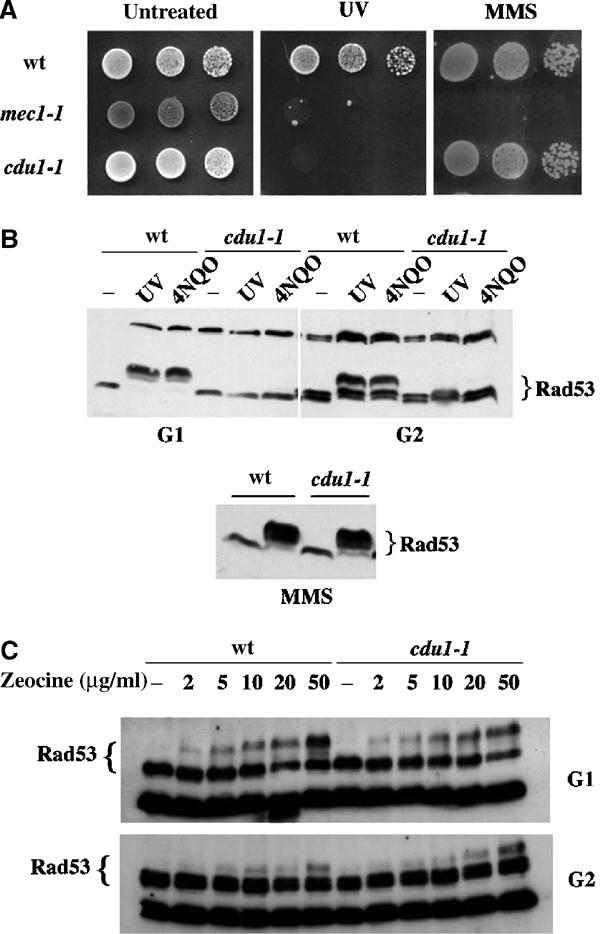
cdu1-1 is UV sensitive and shows a lesion-specific Rad53 phosphorylation defect. Strains were wt (K699), mec1-1 (DMP2697/2c) and cdu1-1 (derived from K699). (A) Serial dilutions of exponential cultures were spotted onto YEPD plates and mock treated or UV irradiated (50 J/m2); a third plate contained 0.02% MMS. The photograph was taken after 2 days of incubation. (B) Exponential cultures were treated with 0.02% MMS (bottom panel) or arrested in G1 with α-factor and in G2 with nocodazole. In panel B (top), blocked cells were either irradiated with UV (50 J/m2) or treated with 4NQO (2 μg/ml). (C) Zeocin was added at the indicated concentrations. Checkpoint activation was monitored by evaluating Rad53 phosphorylation in Western blotting.
Figure 2.
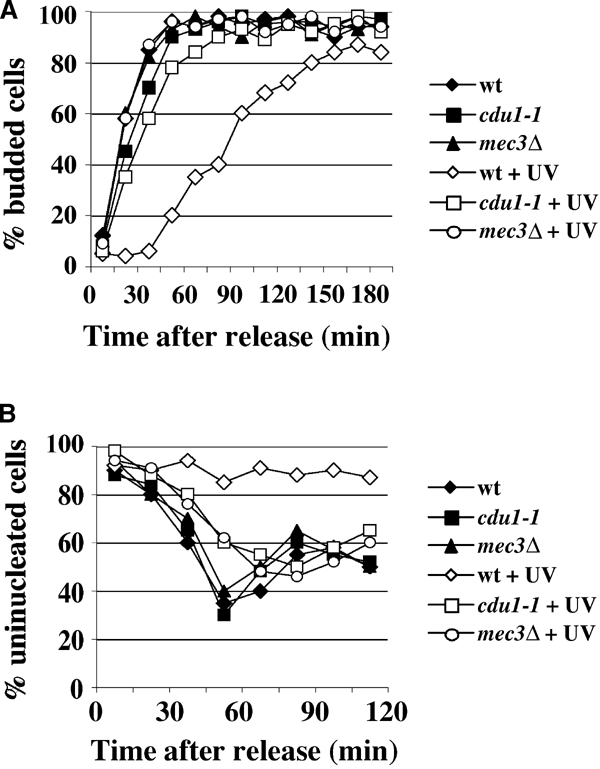
cdu1-1 exhibits defective G1/S and G2/M cell cycle arrests after UV irradiation. Strains were as in Figure 1 and mec3Δ (YMIC4E6). Exponential cultures were arrested in G1 (A) or G2 (B) and UV irradiated (40 J/m2). The cells were then allowed to progress through the cell cycle. G1/S and G2/M checkpoint arrests were monitored by evaluating, at the indicated times, the percentage of budded cells (A) or uninucleated cells (B), respectively.
The cdu phenotype is caused by nucleotide excision repair defects
We rescued the UV-sensitive phenotype with a yeast genomic library (Jansen et al, 1993) and cloned the CDU1 gene, which, by classical genetic analysis, turned out to be identical to RAD14. Expression of RAD14 recovered both the UV sensitivity and the Rad53 phosphorylation defect of the cdu1-1 mutant, and sequence analysis revealed that the cdu1-1 mutation causes truncation of the RAD14 open reading frame (ORF) after the third codon (not shown). Rad14 is a factor required for the initial steps of NER, being involved in the recognition of the primary lesion. This finding establishes a tight link between DNA damage checkpoint and the repair machinery, and is in agreement with an observation that suggested a possible involvement of Rad14 in the checkpoint response (Neecke et al, 1999). On the other hand, it was recently argued that NER functions are not required for UV-induced checkpoint activation in G1 and G2 (Zhang et al, 2003 and see Discussion).
We decided to identify the step of the DNA damage response cascade affected by a RAD14 deficiency, and to test whether this effect could be extended to other NER functions, besides RAD14.
Rad53 activation requires prior hyperphosphorylation of Rad9. This event is Mec1 dependent and can be observed in response to any kind of damage, both in G1 and G2, where Rad9 is already modified at a basal level (Emili, 1998; Sun et al, 1998; Vialard et al, 1998; Giannattasio et al, 2002). We compared the status of Rad9 after 4NQO treatment in wt and in cells lacking RAD14 or RAD2, a gene coding for a factor of the structure-specific endonuclease. Figure 3A shows that damage-induced hyperphosphorylation of a myc-tagged Rad9 protein is abolished in G1- and G2-arrested cells, if RAD14 or RAD2 is deleted. This result suggests that the checkpoint response to UV-mimetic lesions requires both Rad14 and Rad2 and that, in NER mutant backgrounds, the signal transduction cascade is interrupted upstream of Rad9.
Figure 3.
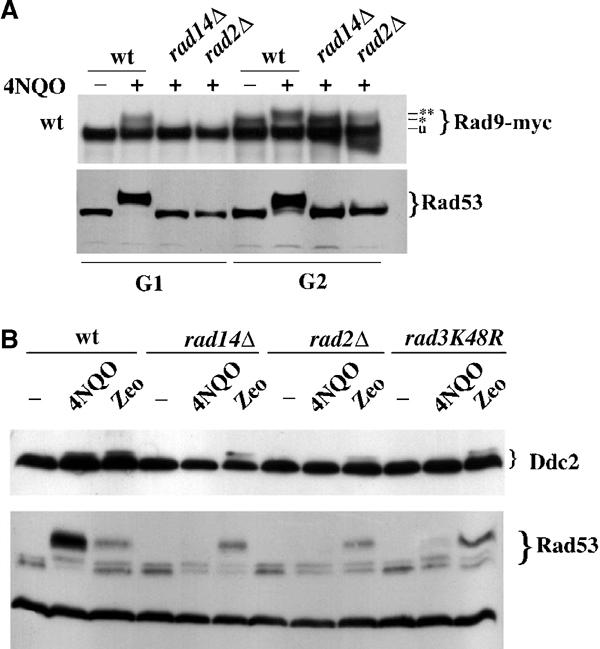
Inactivation of genes involved in the early steps of NER prevents Mec1-dependent phosphorylation of Rad9 and Mec1 kinase activation, which requires processing of the primary lesion. (A) Strains were wt (YMIC5B5), rad14Δ (YMIC8B3) and rad2Δ (YMIC8C2). Exponential cells were arrested in G1 and G2 and treated with 4NQO. TCA extracts were analyzed by Western blotting with antibodies against the MYC tag or Rad53, in order to evaluate Rad9 and Rad53 phosphorylation, respectively. The * and ** symbols indicate the cell cycle-dependent phosphorylation of Rad9, normally observed in G2, and the damage-dependent hyperphosphorylation, respectively. (B) Strains were wt (YLL683.8/3b), rad14Δ (YMIC8B9), rad2Δ (YMIC8B7) and rad3K48R (YMIC12D3). Cells were blocked in G2 with nocodazole and treated with 4NQO (2 μg/ml) or zeocin (200 μg/ml). Ddc2 and Rad53 phosphorylation was assayed by Western blotting with anti-HA tag or anti-Rad53 antibodies, after TCA protein extraction. Similar results were obtained with UV irradiation (not shown).
It is worth mentioning that the experiments shown in Figure 3 were performed on cells that were kept arrested in G1 or G2 after genotoxin treatments. In a previous work, rad14Δ cells were shown to phosphorylate Rad53 after UV treatment in G1 and release into the cell cycle. This event required firing of replication origins and it was due to activation of the intra-S checkpoint (Neecke et al, 1999).
Nucleotide excision repair is required to activate Mec1 kinase
Mec1-dependent phosphorylation of Rad9 is mediated by the PCNA-like complex (Vialard et al, 1998; Giannattasio et al, 2002). The deficiency reported in Figure 3A could be due to a problem affecting recruitment of such complex to the lesion or to a defective activation of Mec1. Ddc2 phosphorylation, which requires only active Mec1 and no other known checkpoint function (Paciotti et al, 2000; Rouse and Jackson, 2000; Wakayama et al, 2001), was used to investigate the status of Mec1 kinase in rad14Δ and rad2Δ cells treated with 4NQO or zeocin. In the absence of NER functions, there is no detectable activation of Mec1 kinase, leading to Ddc2 modification (Figure 3B). This finding strongly supports the notion that NER is required for the initial steps of checkpoint activation.
The mutations analyzed so far cause the complete loss of NER proteins, possibly affecting the structure of the complexes involved in lesion recognition. Moreover, under these circumstances, we cannot distinguish between a requirement for damage recognition and a requirement for damage processing. We thus repeated some of the experiments using rad3-K48R, a point mutant defective in the helicase activity necessary to actually repair the UV damage (Sung et al, 1988). An in vitro system assembled with a Rad3K48R mutant protein was reported to display just a background (approximately 3%) of incision activity of damaged DNA, while there was no measurable excision of the lesion (Sung et al, 1996). We compared Mec1 activation in rad3K48R, rad14Δ and rad2Δ cells. Figure 3B shows that the rad3K48R mutation greatly reduces Rad53 phosphorylation and Ddc2 modification in response to 4NQO treatment, much like the deletion of RAD14 or RAD2. On the other hand, none of these NER functions is required for Mec1 activation if cells are damaged with a DSB-inducing agent. These results argue that the lesion recognition function of NER is not sufficient to achieve checkpoint activation, and that processing of the primary lesion is likely required.
It has been suggested that all yeast NER proteins exist together in a complex, forming a repairosome (Svejstrup et al, 1995), but the existence of tightly associated subcomplexes, called NEFs, that can be purified intact, has also been demonstrated (Guzder et al, 1996). We then tested more NER-defective strains in order to evaluate the effect of deficiencies in the various NEFs on checkpoint activation. Cells deleted in RAD4 (NEF2) or RAD7 (NEF4) were arrested in G1 and G2, treated with UV and activation of the checkpoint was monitored by evaluating phosphorylation of Rad53, via Western blotting and by measuring its kinase activity using an in situ kinase assay. Figure 4A confirms that a rad3K48R mutant has a greatly reduced checkpoint function. These data also show that loss of NEF2 (rad4Δ) causes a complete deficiency in G1 and G2 checkpoint activity, while elimination of NEF4 (rad7Δ) does not affect the checkpoint response. NER can fix lesions on both transcribed and nontranscribed regions of the genome using two different, but overlapping, sets of proteins. Rad7 is solely required for GGR; in fact, rad7 mutants can repair lesions affecting transcribed sequences (Bang et al, 1992). On the contrary, RAD26 is specifically involved in TCR, and rad26Δ cells are able to fix damages affecting transcriptionally silent DNA (van Gool et al, 1994). As shown in Figure 4B, Rad53 phosphorylation and kinase activity in rad26Δ cells is undistinguishable from that observed in wt and rad7Δ strains, while rad26Δrad7Δ double-mutant cells are completely deficient in the G1 and G2 checkpoint responses following UV treatment (Figure 4C). Altogether, these results indicate that residual NER activity, due to TCR in rad7Δ or to GGR in rad26Δ, is sufficient to allow Mec1 activation. Moreover, they suggest that recognition and processing of just some lesion may be enough to trigger the signal transduction cascade. Likewise, the 3% residual incision activity described in rad3K48R may be responsible for the low level of Rad53 kinase activity detected in such a mutant.
Figure 4.
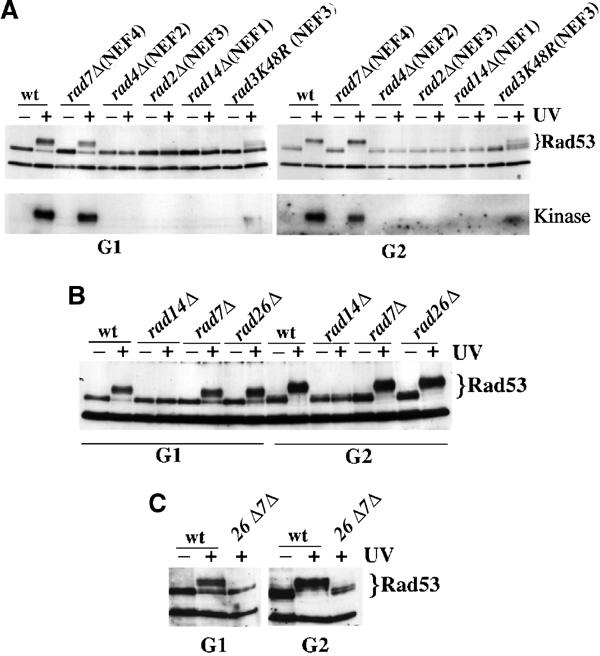
Either GGR or TCR alone is sufficient to trigger properly the checkpoint response following UV radiation. Strains were wt (K699), rad7Δ (YMIC12I1), rad4Δ (YMIC12H6), rad2Δ (YMIC8B7), rad14Δ (YMIC12H6), rad3K48R (YMIC10H2), rad26Δ (YMG30) and rad7Δrad26Δ (YMG48/9d). Exponential cultures were arrested in G1 and G2 and mock or UV (100 J/m2) treated. Rad53 phosphorylation was analyzed by Western blotting. Rad53 kinase activity was monitored by in situ kinase assay, as previously described (Pellicioli et al, 1999).
Rad14 physically interacts with the Rad17–Mec3–Ddc1 complex
All our findings could be explained if NER factors had a role in recruiting checkpoint proteins to UV-induced DNA lesions. According to this hypothesis, a physical interaction between NER and checkpoint factors could be predicted. Indeed, Rad14 was isolated as a strong interactor in a two-hybrid screening using Ddc1 as bait.
We isolated several RAD14-containing plasmids and restricted the interaction domain to a region between amino acids 103 and 318 of the Rad14 protein. Figure 5A shows a panel of yeast strains expressing the Rad14 prey and harboring several baits, representing different checkpoint proteins. Drops of the relevant cultures were spotted on plates containing either glucose (prey not expressed) or galactose (prey expressed). On XGAL medium, there is a clear indication of a strong interaction between Rad14 and Ddc1 and of a milder connection of Rad14 with Mec3. These data are confirmed by plating on selective medium (−LEU), which allows growth only if bait and prey interact. The −LEU plating also shows a mildly positive result with Rad14 and Rad24, which is more evident with longer incubations and has been reconfirmed several times (not shown). In order to obtain biochemical evidence for these interactions, we performed co-immunoprecipitation experiments using strains expressing myc-tagged Rad14. Figure 5B shows that, by using anti-myc antibodies, Ddc1 co-immunoprecipitates with Rad14 only in extracts obtained from myc-tagged Rad14 cells, but not from the untagged control. In the same experiment, Mec3 was also found to co-immunoprecipitate with Rad14 (not shown), suggesting that Rad14 physically interacts with the whole PCNA-like complex. These interactions are not affected by 4NQO treatment and were further confirmed by in vitro GST pull-down experiments (Figure 5C). The RAD14 coding sequence was amplified by PCR, and this fragment was in vitro transcribed and translated in the presence of 35S-methionine. Labeled Rad14 was incubated with the different GST fusions indicated in the figure, and the bound peptides were analyzed by autoradiography after SDS–PAGE. The data in Figure 5C suggest that Rad14 interacts directly with Ddc1 and particularly with its N-terminal half. The modest interaction detected with the C-terminal half of Ddc1 may suggest that Rad14 could contact the central region of Ddc1. Altogether, these findings demonstrate a requirement for NER factors in checkpoint activation. This new function may involve a role in the recruitment of checkpoint proteins to the site of lesions following UV treatment.
Figure 5.
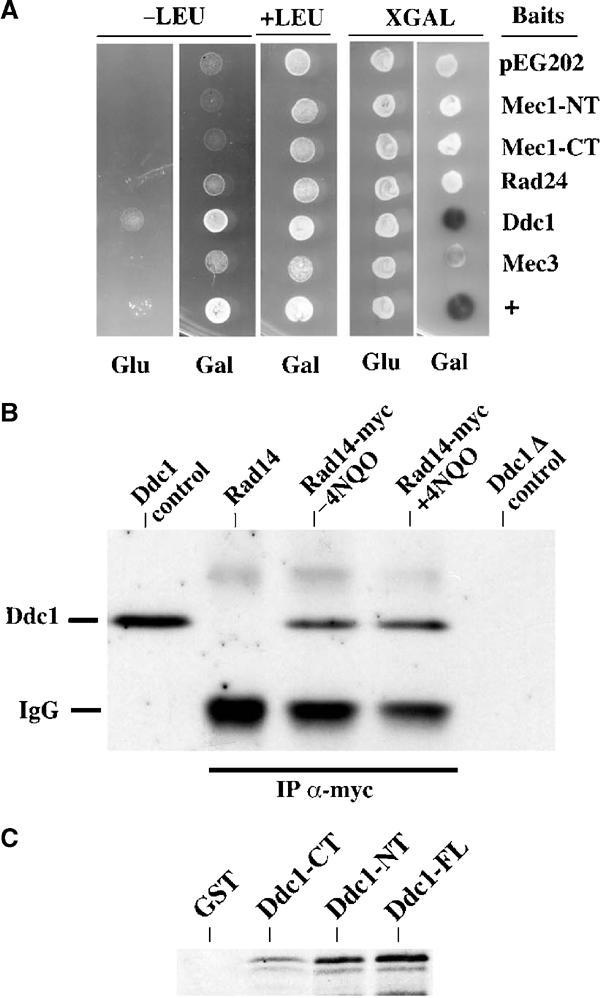
Rad14 physically interacts with the PCNA-like complex. (A) EGY48 cells containing the Rad14 prey plasmid (pJG4-5 Rad14TH1) were transformed with the plasmids expressing the indicated baits. Cells were spotted on plates (SC-HIS, TRP, URA) containing (+LEU) or lacking (−LEU) leucine to select for interactors. The same cells were also spotted on SC-HIS, TRP, URA plates, containing XGAL to monitor lacZ reporter expression. The sugar source was as indicated (Glu: 2% glucose, to shut off prey expression; Gal: 2% raffinose, 2% galactose, to turn on prey expression). + indicates the positive control (p53 bait, SV40 Tag prey). (B) myc-tagged Rad14 was immunopurified, using anti-myc antibodies crosslinked to protein G–sepharose, from Rad14-Myc or Rad14 cells treated or mock treated with 4NQO. The recovered samples were separated by SDS–PAGE and analyzed by Western blotting with Ddc1-specific antibodies. Control lanes (Ddc1 control and Ddc1Δ control) contain TCA extracts from wt and ddc1Δ cells, respectively. Strains were RAD14 (K699), RAD14-myc (YMIC7E5) and ddc1Δ (YLL244). (C) The GST fusion proteins were purified from E. coli cells and incubated with 35S-labeled in vitro-translated Rad14. Proteins bound to the fusions were recovered, separated by SDS–PAGE and analyzed by autoradiography.
RAD14 function is needed for Ddc1 and Ddc2 loading onto chromosomes
It has been shown that the Mec1–Ddc2 kinase complex is localized at specific foci in response to DSBs (Kondo et al, 2001; Melo et al, 2001). This localization does not require any other checkpoint functions, but is dependent on RPA-coated ssDNA (Zou and Elledge, 2003). It is possible that NER is required to recognize UV-induced DNA lesions and activates the checkpoint kinase by increasing the local concentration of checkpoint factors, through direct recruitment, and by generating an ssDNA intermediate, which allows Mec1 kinase binding to DNA. Thus, NER-deficient cells may be defective in loading the Ddc2 and Ddc1 complexes onto damaged DNA. It has been reported that in human cells, ATR cosediments with chromatin (Zou et al, 2002), so we looked at association of Mec1–Ddc2 with bulk chromatin. Figure 6A shows that, after 4NQO treatment, the amount of Ddc2 that enters the chromatin fraction increases only when RAD14 function is present. On the other hand, after zeocin treatment, Ddc2 is DNA bound irrespectively of the presence of RAD14. DNA binding is confirmed by the loss of Ddc2 signal upon DNase treatment (not shown). These results suggest that NER is required to recruit Ddc2 to DNA when cells are damaged with 4NQO, but not when the primary lesions are DSBs.
Figure 6.
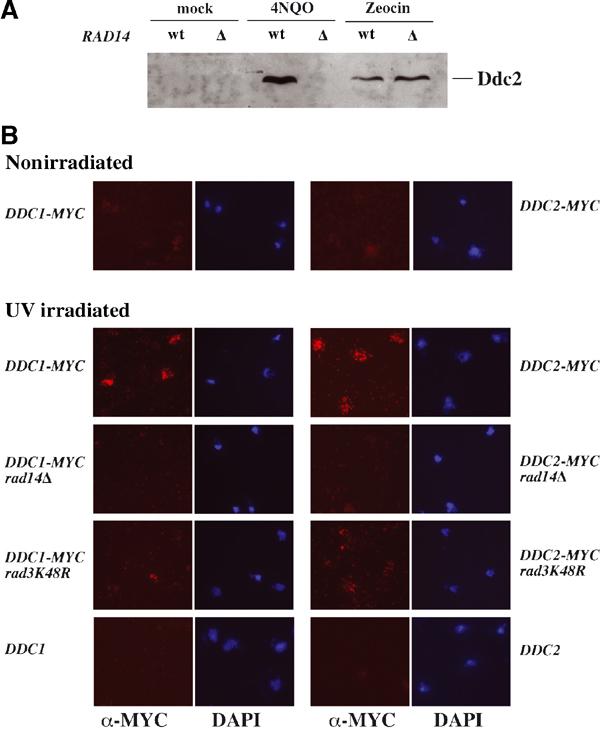
NER is required for loading Mec1–Ddc2 kinase and Ddc1 complexes onto damaged chromosomes. (A) Strains were wt (YLL683.8/3b) and rad14Δ (YMIC8B9). Cultures were arrested in G1 and treated with 4NQO or zeocine. Cells were lysed and processed as described in Materials and methods, and proteins associated with bulk chromatin were analyzed by SDS–PAGE and Western blotting with anti-HA tag antibodies. (B) All strains, except for untagged controls, contained either DDC1-27MYC or DDC2-18MYC, as indicated. Chromosome spreads were prepared from G1 cells that had been mock treated or UV treated (100 J/m2), as described in Materials and methods. Samples were examined by indirect immunofluorescence and by DAPI staining. Similar experiments performed with G2 cells gave similar results. Strains were DDC1-MYC (YLL444.9), DDC2-MYC (YLL733.1), DDC1-MYC rad14Δ (YMG44), DDC2-MYC rad14Δ (YMG39), DDC1-MYC rad3K48R (YMG36), DDC2-MYC rad3K48R (YMG38), DDC1 (K699) and DDC2 (K699).
Since chromatin binding experiments with Ddc1 were unsuccessful, we analyzed the role of Rad14 in loading Ddc1 by chromosome spreading (Michaelis et al, 1997). Figure 6B shows that both Ddc1 and Ddc2 bind to chromosomes upon UV irradiation, while only a low basal signal is barely detectable in untreated samples (Figures 6B and 1). This association is dependent on RAD14 and RAD3 functions, further supporting the notion that binding of checkpoint proteins to UV-damaged DNA requires NER activity.
Discussion
DNA damage checkpoints ensure a proper cellular response to genotoxic insults. Malfunctioning of such systems is linked to premature aging, uncontrolled cellular proliferation, genome instability and, ultimately, tumor development (Hartwell, 1992). Failure to detect the primary lesion results in defective activation of the checkpoint response and in failure to arrest cell cycle progression and DNA replication. Moreover, if the primary damage is not detected by the checkpoint, it may be channeled into other cellular processes (e.g. aberrant recombination) that can cause DNA instability. The biochemical mechanisms involved in the sensing step of the checkpoint response are still poorly understood. How do cells realize that their DNA has been damaged, how are the lesions found within the genome and how is the first kinase in the cascade, Mec1, activated? A number of studies in S. cerevisiae allowed the positioning of three protein complexes near damaged DNA. A modified RFC complex, where Rad24 substitutes Rfc1, is needed to load the PCNA-like complex in proximity of the lesion (Lowndes and Murguia, 2000; Rouse and Jackson, 2002; Majka and Burgers, 2003). Moreover, a Mec1–Ddc2 complex is independently recruited on damaged DNA, and this seems to allow Mec1 to phosphorylate several targets (Kondo et al, 2001; Melo and Toczyski, 2002). It was proposed that the PCNA-like complex might possess a nuclease activity that by processing the primary DNA lesion and generating ssDNA would lead to Mec1 recruitment and activation (Lydall and Weinert, 1995). However, we have purified the yeast PCNA-like complex and have not detected any nuclease activity (unpublished). It has been recently shown that DNA binding of the Ddc2–Mec1 complex in response to DSBs requires RPA-coated ssDNA (Zou and Elledge, 2003); however, the RFC- and PCNA-like complexes must play additional roles (such as recruitment of Mec1 substrates or reorganization of protein–DNA interactions at the site of DNA damage) since localization of Ddc2–Mec1 alone at DNA lesions is not sufficient to activate fully the checkpoint response. Moreover, it is unlikely that checkpoint proteins are capable of recognizing directly all sorts of DNA lesions, which, despite being of an ample structural variety, trigger the same checkpoint response. An attractive possibility relies on DNA repair proteins attacking the lesions and providing common substrates to checkpoint factors. This hypothesis gets support from different works (Neecke et al, 1999; D'Amours and Jackson, 2001; Grenon et al, 2001; Usui et al, 2001), but it has recently been disputed (Zhang et al, 2003). We reasoned that if cells developed specialized checkpoint factors devoted to the recognition of specific lesions, it should be possible to isolate mutations impairing checkpoint activation following specific genotoxic treatments, while leaving the cellular response to other kinds of damages unaffected. We performed a genetic screen looking for mutants that were hypersensitive to UV radiation, while being normally resistant to treatment with the alkylating agent MMS. Among these mutants, we were interested in those that could not activate the checkpoint kinase Rad53 following UV treatment, but were still proficient in the response to MMS. The cdu1-1 mutant is sensitive to UV, resistant to MMS treatment, and it is completely defective in phosphorylating Rad53 and arresting cell cycle progression, in G1 and G2, specifically in response to UV or UV-mimetic agents. CDU1 function is thus essential for activation of the G1 and G2 checkpoints following treatment with UV, and this requirement is lesion specific.
Cloning the corresponding ORF revealed that CDU1 is allelic to RAD14, the yeast homolog of mammalian XPA. Rad14 is a NER factor involved in the recognition of UV-induced lesions (Prakash and Prakash, 2000). Previous work had shown that loss of RAD14 caused UV-irradiated cells to arrest irreversibly at the beginning of S phase (Neecke et al, 1999), and we found that exponentially growing rad14Δ cells exhibit a certain level of Rad53 phosphorylation, after UV irradiation (not shown). This is likely due to the contribution of cells that are in S phase, when replication forks, encountering DNA lesions, activate the intra-S checkpoint.
To shed some light on the crosstalk between NER and checkpoint mechanisms, we analyzed at what step of the signal transduction cascade NER intervenes and we investigated what NER function is required for checkpoint activation. Since phosphorylation of Rad53 following DNA damage is a late event in the pathway and requires previous modification of Rad9, we tested the NER dependency of Rad9 phosphorylation. We show that a rad14 mutant fails to hyperphosphorylate Rad9 following 4NQO treatment, suggesting that the G1 and G2 checkpoint signal transduction cascade is blocked upstream of RAD9. Loss of Rad2, a nuclease required for excision of the lesion, also causes a complete loss of phosphorylated Rad9, suggesting that processing of the damage is likely required for checkpoint activity. Then we looked at Ddc2 phosphorylation, which does not need any checkpoint function other than Mec1, and is the first biochemical indication of Mec1 activity (Paciotti et al, 2000; Rouse and Jackson, 2000; Wakayama et al, 2001). Loss of RAD14 or RAD2 leads to a complete disappearance of phosphorylated Ddc2 after UV or 4NQO treatment, but not after DSB induction. This finding indicates that NER functions are required, in response to specific types of lesions, to drive the earliest phosphorylation event in the signaling cascade. The complete absence of Rad14 and Rad2 in the deletion strains could affect the structure of the NER complex. We then tested a point mutant, rad3K48R, defective in the helicase activity that generates the single-stranded bubble in the lesion-containing region. Our results show that when rad3K48R cells are damaged with zeocin the checkpoint is functional, while after 4NQO treatment Ddc2 phosphorylation is not detectable and Rad53 activation is severely diminished. The observed low level of Rad53 phosphorylation is likely due to the residual incision activity of the rad3K48R mutant, supporting the notion that lesion processing is needed for checkpoint activation. Since NER involves the intervention of multiple NEFs, we have tested mutations affecting each of them. The functions of NEF1, NEF2 and NEF3 are necessary for the proper checkpoint response to UV lesions. On the other hand, loss of GGR in a rad7Δ strain or loss of TCR in rad26Δ cells does not affect Rad53 phosphorylation, which is instead completely abolished in the rad7Δ rad26Δ double mutant. These observations suggest that TCR or GGR activities are sufficient for recognizing and processing an adequate amount of lesions for checkpoint activation.
The functional interplay between NER and checkpoint mechanisms is strongly supported by the fact that Rad14 was also isolated in a two-hybrid screen aimed at identifying partners of the Ddc1 subunit of the PCNA-like clamp. This physical interaction was further confirmed by co-immunoprecipitating the endogenous proteins and by GST pull-down experiments, which suggest a direct interaction between Rad14 and Ddc1. This is the first demonstration of a physical interaction between NER and checkpoint proteins.
The possibility that NER factors may be involved in the actual recruitment of checkpoint proteins onto damaged DNA was examined by assessing DNA loading of Ddc1 and Ddc2. The UV damage-dependent association of these two checkpoint complexes to chromosomes was observed by chromatin cosedimentation and chromosome spreading, and is lost in rad14Δ and rad3K48R strains. These findings may suggest that NER factors possibly recruit checkpoint complexes in close proximity of the lesions and that subsequent processing is required to achieve stable DNA binding and kinase activation. While this manuscript was in preparation, a report showing that NER did not have any effect on DNA damage checkpoints was published (Zhang et al, 2003). We have now established that the conclusions presented by Zhang et al stand on a prolonged incubation used to monitor checkpoint activation in response to UV under their experimental conditions. Accumulation of DNA breaks, leading to partial Rad53 phosphorylation, occurs in NER-deficient strains if cells are heavily treated with UV or 4NQO and kept arrested for some time before extract preparation (data not shown, but see Figures 2 and 3). In our hands not only NER mutants do not display modification of Rad53, but also show complete loss of G1 and G2 delays after UV treatment. Moreover, logarithmically growing rad14Δ cells fail to accumulate large-budded cells in response to UV treatment, demonstrating that NER does indeed play a physiological role in obtaining a proper response to UV (data not shown, but see Figure 4).
Altogether, we suggest that NER plays a role in checkpoint function by identifying UV-specific lesions in the genome. The physical interaction detected between Rad14 and Ddc1 could be relevant to allow checkpoint complexes to find damaged DNA within the whole genome and accumulate in its vicinity. Subsequent processing of the primary lesion by NER activity would be required to activate Mec1 kinase, probably through the generation of RPA-coated ssDNA (Zou and Elledge, 2003). This mechanism would explain the sensitivity and rapidity of the checkpoint response and its ability to discriminate between ssDNA intermediates generated after DNA damage and ssDNA generated through normal DNA metabolic processes. This hypothesis will be further investigated by searching for point mutations affecting Ddc1 or Rad14 that disrupt the physical interaction between these factors, while maintaining the functionality in checkpoint signaling and repair, respectively.
Since protein structures and general pathways implicated in checkpoints and repair are evolutionarily conserved between yeast and mammals, it will be interesting to extend the findings reported in this work to cells derived from XP and CS individuals. Most XP patients show a deficiency in both TCR and GGR and could be defective in checkpoint activation, resulting in elevated genomic instability; on the other hand, CS patients, where only TCR is nonfunctional, display less chromosomal instability and could possibly be checkpoint proficient. This might explain the heterogeneity in cancer susceptibility found in CS and XP patients.
Materials and methods
Strains and plasmids
EGY48 is the host strain for the two-hybrid system (Gyuris et al, 1993). Deletions of MEC3, RAD14, RAD2, RAD4, RAD7 and RAD26 and tagged DDC1, DDC2 and RAD9 were generated using the one-step PCR system (Longtine et al, 1998). All the strains used in this work are derivatives of W303 (K699, MATa ade2–1 trp1–1 can1–100 leu2–3,12 his3–11,15 ura3) and were obtained by classical genetics. Further details are available on request. Ethyl methane sulfonate mutagenesis was performed as previously described (Longhese et al, 1996).
All the plasmids, expressing lexA fusions with checkpoint proteins, were obtained by amplifying the relative coding sequence from genomic DNA and ligating it in pEG202 (kind gift from R Brent). pJG4-5-RAD14TH1 and pJG4-5-RAD14TH2 were isolated from the two-hybrid screen using Ddc1 protein as a bait. pFLB7 contains a GST fusion with MEC3, pFLB10 contains a GST fusion with full-length DDC1, pFLB11 contains a GST fusion with DDC1 (between nucleotides 1 and 1311) and pFLB12 contains a GST fusion with DDC1 (between nucleotides 1317 and 1819). Specific information regarding these constructs is available on request.
GST pull-down
Escherichia coli DH5α cells carrying pFLB7, pFLB10, pFLB11, pFLB12 or pGEX4T3 were grown at 37°C until the optical density was 4–5. The cells were induced with 1 mM of IPTG for 3–4 h and then washed with cold water. Cells were resuspended in PBS 1 × plus complete protease inhibitors (Roche), 1 mM Na3VO4, 1 mM PMSF and 10 mM NaF. Cells were then sonicated and extracts were clarified by centrifugation. Glutathione–sepharose (Amersham) was incubated with the supernatant and washed extensively with PBS. Loaded glutathione–sepharose beads were incubated with 50 μl of the transcription–translation reaction and extensively washed. 35S-labeled Rad14-TH1 was produced with TNT T7 quick PCR DNA (Promega). Proteins that remained bound to the resin were analyzed by autoradiography after SDS–PAGE.
Cell cycle blocks and UV irradiation
Cells were grown in YEPD medium at 28°C to the concentration of 5 × 106 cells/ml and arrested with nocodazole (20 μg/ml) (USB) or α-factor (20 μg/ml). After the arrest, 50 ml cells were spun, resuspended in 500 μl of fresh YEPD and plated on a Petri dish (14 cm diameter). Plates were quickly irradiated with a Stratalinker 2400 (Stratagene) with a 254 nm light source at the indicated UV dosages. Immediately after treatment, cells were resuspended in 20% TCA for extract preparation. No more than 10 min occurred between cell plating and TCA resuspension in all the experiments described. Protein extracts were prepared as described (Muzi-Falconi et al, 1993).
SDS–PAGE and Western blot conditions
Protein extracts were separated by SDS–PAGE in 10% acrylamide gels; for analysis of Rad9, NuPAGE Tris-acetate 3–8% gels (Novex) were used. Western blotting was performed, using standard techniques, with α-Rad53, α-MYC tag (9E10) or α -HA (12CA5) antibodies.
MMS, 4NQO and zeocine treatment
Exponentially growing cells were spun and resuspended in fresh YEPD medium containing 0.02% MMS and incubated for 3 h. Zeocine and 4NQO were used at 200 and 2 μg/ml, respectively, unless otherwise mentioned. Cells were kept in the presence of the drugs for 15 min and then TCA extracts were prepared.
G1/S and G2/M checkpoint assays
Cells were blocked in G1 (2 μg/ml α-factor) or in G2 (2 μg/ml nocodazole) and then irradiated with 40 J/m2 as described above. Cells were released by washing with fresh medium and put back into culture. The percentage of budded cells (G1/S assay) or uninucleated dumbbell cells (G2/M assay) was calculated by microscopic observations at different time points after release.
Co-immunoprecipitation
Extracts from 4NQO- or mock-treated cells were prepared in PBS and incubated with 9E10 α-myc antibodies, crosslinked to protein G–sepharose. After extensive washes, samples were analyzed by Western blotting. Good co-immunoprecipitation was observed when substantial immunodepletion was achieved.
Chromatin binding
The experiments were performed following published procedures (Liang and Stillman, 1997) with slight modifications. Briefly, exponentially growing cells were arrested in G1 with 20 μg/ml α-factor. After genotoxin treatment, cells were washed and resuspended in buffer A: 50 mM HEPES–KOH (pH 7.5), 100 mM NaCl, 0.4 M sorbitol, 50 mM NaF, 60 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM PMSF and protease inhibitors (Complete, Roche). Lysis was performed with zymolyase followed by addition of 0.25% Triton X-100. The chromatin-containing fraction was isolated by centrifugation, extensively washed and analyzed by Western blotting.
Chromosome spreading
Cells were arrested in G1 and G2 and irradiated, as described. Samples were immediately processed for spreading. Mitotic chromosomes were spread as described by Michaelis et al (1997). Immunofluorescence was performed with 9E10 α-MYC, Cy3-goat α-mouse and Cy3-donkey α-goat antibodies.
Acknowledgments
We thank C Santocanale for Rad53 antibodies, F Lippi-Boncambi for the pFLB plasmids and M Clerici for helping with the chromosome spreading experiments. L Prakash kindly provided a plasmid with the original rad3K48R allele. We thank all the members of the lab for critical discussions. This work was supported by grants from AIRC, MIUR (5%) Biomolecole per la Salute Umana, Progetto FIRB-MIUR ‘Genomica e proteomica nello studio di funzioni cellulari complesse', MIUR ‘Genomica Funzionale', Ministero della Salute Ricerca Finalizzata 2002 to PP and MM-F and from AIRC to MPL. The financial support of Telethon—Italy (grant nos GGP030406 to MM-F and E.1247 to MPL) is gratefully acknowledged.
References
- Bang DD, Verhage R, Goosen N, Brouwer J, van de Putte P (1992) Molecular cloning of RAD16, a gene involved in differential repair in Saccharomyces cerevisiae. Nucleic Acids Res 20: 3925–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AM (2002) DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair 1: 983–994 [DOI] [PubMed] [Google Scholar]
- D'Amours D, Jackson SP (2001) The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev 15: 2238–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ (1996) Cell cycle checkpoints: preventing an identity crisis. Science 274: 1664–1672 [DOI] [PubMed] [Google Scholar]
- Emili A (1998) MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell 2: 183–189 [DOI] [PubMed] [Google Scholar]
- Foiani M, Pellicioli A, Lopes M, Lucca C, Ferrari M, Liberi G, Muzi Falconi M, Plevani P (2000) DNA damage checkpoints and DNA replication controls in Saccharomyces cerevisiae. Mutat Res 451: 187–196 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W (1995) DNA Repair and Mutagenesis. Washington, DC: ASM Press [Google Scholar]
- Giannattasio M, Sommariva E, Vercillo R, Lippi-Boncambi F, Liberi G, Foiani M, Plevani P, Muzi-Falconi M (2002) A dominant-negative MEC3 mutant uncovers new functions for the Rad17 complex and Tel1. Proc Natl Acad Sci USA 99: 12997–13002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenon M, Gilbert C, Lowndes NF (2001) Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat Cell Biol 3: 844–847 [DOI] [PubMed] [Google Scholar]
- Guzder SN, Sung P, Prakash L, Prakash S (1996) Nucleotide excision repair in yeast is mediated by sequential assembly of repair factors and not by a pre-assembled repairosome. J Biol Chem 271: 8903–8910 [DOI] [PubMed] [Google Scholar]
- Gyuris J, Golemis E, Chertkov H, Brent R (1993) Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell 75: 791–803 [DOI] [PubMed] [Google Scholar]
- Hartwell L (1992) Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 71: 543–546 [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246: 629–634 [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411: 366–374 [DOI] [PubMed] [Google Scholar]
- Jansen R, Tollervey D, Hurt EC (1993) A U3 snoRNP protein with homology to splicing factor PRP4 and G beta domains is required for ribosomal RNA processing. EMBO J 12: 2549–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Matsumoto K, Sugimoto K (1999) Role of a complex containing Rad17, Mec3, and Ddc1 in the yeast DNA damage checkpoint pathway. Mol Cell Biol 19: 1136–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K (2001) Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science 294: 867–870 [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE (1998) Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 94: 399–409 [DOI] [PubMed] [Google Scholar]
- Liang C, Stillman B (1997) Persistent initiation of DNA replication and chromatin-bound MCM proteins during the cell cycle in cdc6 mutants. Genes Dev 11: 3375–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Foiani M, Muzi-Falconi M, Lucchini G, Plevani P (1998) DNA damage checkpoint in budding yeast. EMBO J 17: 5525–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhese MP, Fraschini R, Plevani P, Lucchini G (1996) Yeast pip3/mec3 mutants fail to delay entry into S phase and to slow DNA replication in response to DNA damage, and they define a functional link between Mec3 and DNA primase. Mol Cell Biol 16: 3235–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lowndes NF, Murguia JR (2000) Sensing and responding to DNA damage. Curr Opin Genet Dev 10: 17–25 [DOI] [PubMed] [Google Scholar]
- Lydall D, Weinert T (1995) Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science 270: 1488–1491 [DOI] [PubMed] [Google Scholar]
- Majka J, Burgers PM (2003) Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc Natl Acad Sci USA 100: 2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo J, Toczyski D (2002) A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 14: 237–245 [DOI] [PubMed] [Google Scholar]
- Melo JA, Cohen J, Toczyski DP (2001) Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15: 2809–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35–45 [DOI] [PubMed] [Google Scholar]
- Muzi-Falconi M, Piseri A, Ferrari M, Lucchini G, Plevani P, Foiani M (1993) De novo synthesis of budding yeast DNA polymerase alpha and POL1 transcription at the G1/S boundary are not required for entrance into S phase. Proc Natl Acad Sci USA 90: 10519–10523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neecke H, Lucchini G, Longhese MP (1999) Cell cycle progression in the presence of irreparable DNA damage is controlled by a Mec1- and Rad53-dependent checkpoint in budding yeast. EMBO J 18: 4485–4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciotti V, Clerici M, Lucchini G, Longhese MP (2000) The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev 14: 2046–2059 [PMC free article] [PubMed] [Google Scholar]
- Paciotti V, Lucchini G, Plevani P, Longhese MP (1998) Mec1p is essential for phosphorylation of the yeast DNA damage checkpoint protein Ddc1p, which physically interacts with Mec3p. EMBO J 17: 4199–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Hartwell LH (1995) A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82: 841–847 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE (2001) Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 7: 293–300 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Prakash L (2000) Nucleotide excision repair in yeast. Mutat Res 451: 13–24 [DOI] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2000) LCD1: an essential gene involved in checkpoint control and regulation of the MEC1 signalling pathway in Saccharomyces cerevisiae. EMBO J 19: 5801–5812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297: 547–551 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357–360 [DOI] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF (1996) Spk1/Rad53 is regulated by Mec1-dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev 10: 395–406 [DOI] [PubMed] [Google Scholar]
- Sun ZX, Hsiao J, Fay DS, Stern DF (1998) Rad53 Fha domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281: 272–274 [DOI] [PubMed] [Google Scholar]
- Sung P, Guzder SN, Prakash L, Prakash S (1996) Reconstitution of TFIIH and requirement of its DNA helicase subunits, Rad3 and Rad25, in the incision step of nucleotide excision repair. J Biol Chem 271: 10821–10826 [DOI] [PubMed] [Google Scholar]
- Sung P, Higgins D, Prakash L, Prakash S (1988) Mutation of lysine-48 to arginine in the yeast RAD3 protein abolishes its ATPase and DNA helicase activities but not the ability to bind ATP. EMBO J 7: 3263–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svejstrup JQ, Wang Z, Feaver WJ, Wu X, Bushnell DA, Donahue TF, Friedberg EC, Kornberg RD (1995) Different forms of TFIIH for transcription and DNA repair: holo-TFIIH and a nucleotide excision repairosome. Cell 80: 21–28 [DOI] [PubMed] [Google Scholar]
- Thelen MP, Venclovas C, Fidelis K (1999) A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins. Cell 96: 769–770 [DOI] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH (2001) A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 7: 1255–1266 [DOI] [PubMed] [Google Scholar]
- van Gool AJ, Verhage R, Swagemakers SM, van de Putte P, Brouwer J, Troelstra C, Bootsma D, Hoeijmakers JH (1994) RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J 13: 5361–5369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialard JE, Gilbert CS, Green CM, Lowndes NF (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J 17: 5679–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakayama T, Kondo T, Ando S, Matsumoto K, Sugimoto K (2001) Pie1, a protein interacting with Mec1, controls cell growth and checkpoint responses in Saccharomyces cerevisiae. Mol Cell Biol 21: 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert T (1998) DNA damage checkpoints update—getting molecular. Curr Opin Genet Dev 8: 185–193 [DOI] [PubMed] [Google Scholar]
- Zhang H, Taylor J, Siede W (2003) Checkpoint arrest signaling in response to UV damage is independent of nucleotide excision repair in Saccharomyces cerevisiae. J Biol Chem 278: 9382–9387 [DOI] [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ (2002) Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev 16: 198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]