

Abstract
The 20S proteasome is made up of four stacked heptameric rings, which in eucaryotes assemble from 14 different but related subunits. The rules governing subunit assembly and placement are not understood. We show that a different kind of proteasome forms in yeast when the Pre9/α3 subunit is deleted. Purified pre9Δ proteasomes show a two-fold enrichment for the Pre6/α4 subunit, consistent with the presence of an extra copy of Pre6 in each outer ring. Based on disulfide engineering and structure-guided suppressor analyses, Pre6 takes the position normally occupied by Pre9, a substitution that depends on a network of intersubunit salt bridges. When Arabidopsis PAD1/α4 is expressed in yeast, it complements not only pre6Δ but also pre6Δ pre9Δ mutants; therefore, the plant α4 subunit also can occupy multiple positions in a functional yeast proteasome. Importantly, biogenesis of proteasomes is delayed at an early stage in pre9Δ cells, suggesting an advantage for Pre9 over Pre6 incorporation at the α3 position that facilitates correct assembly.
Keywords: proteasome, protein assembly, proteolysis, ubiquitin
Introduction
Most of the regulated degradation of intracellular proteins in eucaryotes occurs through the ubiquitin–proteasome system (Pickart, 2001; Glickman and Ciechanover, 2002). Substrates are polyubiquitinated, and the tagged proteins are then degraded by the 26S proteasome, which is composed of a proteolytically active 20S proteasome core bound at each end by a 19S regulatory complex (Baumeister et al, 1998; DeMartino and Slaughter, 1999). The latter confers energy- and ubiquitin-dependence on substrate proteolysis.
20S proteasomes are found in all three branches of life, and all eucaryotic species examined have this protease (Baumeister et al, 1998; Kruger et al, 2001). Its subunits assemble into a cylindrical stack of four seven-subunit rings (Figure 1A). β subunits comprising the inner rings bear the active sites, which are exposed to the interior chamber, and substrates enter through a narrow annulus in the outer α rings (Löwe et al, 1995; Arendt and Hochstrasser, 1997; Groll et al, 1997). Whereas the proteasome of the archaeon T. acidophilum is composed of homomeric α and β rings (Löwe et al, 1995), in the yeast Saccharomyces cerevisiae and other eucaryotes there are seven distinct subunits in each α and each β ring (Heinemeyer et al, 1994; Chen and Hochstrasser, 1995; Groll et al, 1997).
Figure 1.
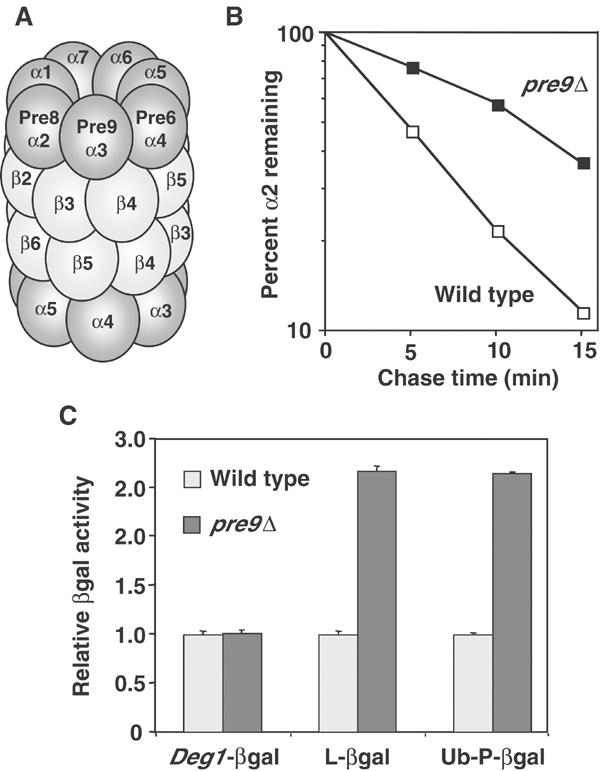
Composition and function of pre9Δ 20S proteasomes. (A) Schematic of subunit positions in wild-type proteasomes. The 28-subunit complex has C2 symmetry. (B) Matα2 repressor degradation rate in pre9Δ (MHY1069) and wild-type (MHY501) cells measured by pulse–chase analysis at 30°C. (C) βgal activity assays for Deg1-βgal, Leu-βgal, and Ub-Pro-βgal from pre9Δ and wild-type cells.
Each subunit of the eucaryotic proteasome is present twice per particle in specific dyad-related positions in the complex. This organization is conserved from yeast to mammals and is thought to be identical in all eucaryotes (Groll et al, 1997; Unno et al, 2002). All subunits have a comparable tertiary fold. Sequence identities among subunits within a species are usually ∼20–40%, but orthologous subunits from different species often have identities in the 55–95% range. Current data suggest that subunit duplication and diversification from simpler homoheptameric ring-forming subunits occurred very early in eucaryotic evolution (Kruger et al, 2001). A limited set of subunit replacements in the β ring of vertebrate proteasomes has also been documented. Specifically, each of the three β subunits that harbor the catalytic centers can be replaced by a γ-interferon-inducible subunit that is ∼60–70% identical. These subunit replacements are important for MHC class I antigen processing (Kruger et al, 2001).
How can multiple, structurally similar polypeptides assemble into a large and stereotypical structure with such high fidelity? This problem is not unique to 20S proteasomes. Other protein complexes have subunits arranged in rings or stacks of rings that are composed of different but related polypeptides. An example is the eucaryotic class II chaperonin, a protein-folding catalyst (Archibald et al, 1999). Often eucaryotic ring complexes are related to similar assemblages in other species that have just one or a few different subunits. Ring structures with related but distinct subunit composition sometimes exist in the same cells, and these alternative rings usually have different functions. Examples include Sm/Lsm RNA-processing oligomers and exosomes, which are complexes of ribonuclease subunits (Pannone and Wolin, 2000; Raijmakers et al, 2002). For all these complexes, the mechanism of assembly in vivo remains an important but unanswered question.
Archaeal 20S proteasomes self-assemble from purified α and β subunits, but eucaryotic proteasome assembly requires both intramolecular and exogenous chaperones (Maurizi, 1998; Kruger et al, 2001). Certain β-subunit propeptides, particularly that of the β5 subunit, promote assembly by mechanisms that are still obscure (Chen and Hochstrasser, 1996; Kruger et al, 2001). A single-turnover chaperone, Ump1, has also been shown to facilitate proteasome biogenesis (Ramos et al, 1998). Assembly of eucaryotic and eubacterial proteasomes appears to take place via a half-proteasome intermediate, which contains one full α ring and a β ring with unprocessed precursors (Yang et al, 1995; Chen and Hochstrasser, 1996; Nandi et al, 1997; Schmidtke et al, 1997; Zühl et al, 1997).
To begin dissecting the rules by which proteasome subunits associate and assemble, we have investigated the molecular basis for an observation made over a decade ago with yeast (Emori et al, 1991). Of the 14 genes encoding the 14 different 20S proteasome subunits in S. cerevisiae, all but one are essential for viability (Emori et al, 1991; Heinemeyer et al, 1994). The lone exception is Pre9/α3, but neither structural nor previous biochemical studies gave any indication as to why this subunit is uniquely dispensable.
We report that proteasomes purified from pre9Δ cells have replaced the missing Pre9 subunit with an additional copy of the Pre6/α4 subunit. Remarkably, a Pre6 ortholog from the plant Arabidopsis thaliana is also able to fill this position. The Pre6 subunit therefore can take two different slots within the hetero-oligomer, and this capacity is evolutionarily conserved. Analysis of proteasome assembly intermediates in pre9Δ cells revealed an accumulation of free proteasome subunits and a strong reduction in the level of half-proteasome intermediates and mature proteasomes. This suggests that correct subunit arrangement is achieved at least in part through more efficient incorporation of Pre9 relative to Pre6 at the α3 position during an early stage in proteasome assembly. These data have implications for our understanding of both proteasome assembly and evolution, and are also probably relevant to other ring-shaped protein assemblies.
Results
Loss of Pre9/α3 has only modest effects on proteasome function
Deletion of PRE9 causes only minor phenotypic abnormalities, in contrast to deletion of any of the remaining 20S subunit genes (Emori et al, 1991; Fu et al, 1998). Cell doubling time in our strain background was 8% slower for pre9Δ cultures in rich medium at 30°C relative to wild type. Haploid pre9Δ cells were more severely impaired for growth when incubated at 37°C or when exposed to the amino-acid analog canavanine, and they were more resistant than wild type to cadmium (not shown, but see Figure 2C). These traits are hallmarks of weak proteasomal mutants (Arendt and Hochstrasser, 1997). We also measured the degradation of several proteasome substrates in pre9Δ cells. Degradation of Matα2, an endogenous substrate, showed only an approximately two-fold slowdown (Figure 1B). When we measured β-galactosidase (βgal) activity levels of several different short-lived βgal-based test proteins, at most very small increases (∼2.5-fold) were seen in pre9Δ compared to wild-type cells (Figure 1C), suggesting very minor changes in the degradation rates of these substrates.
Figure 2.
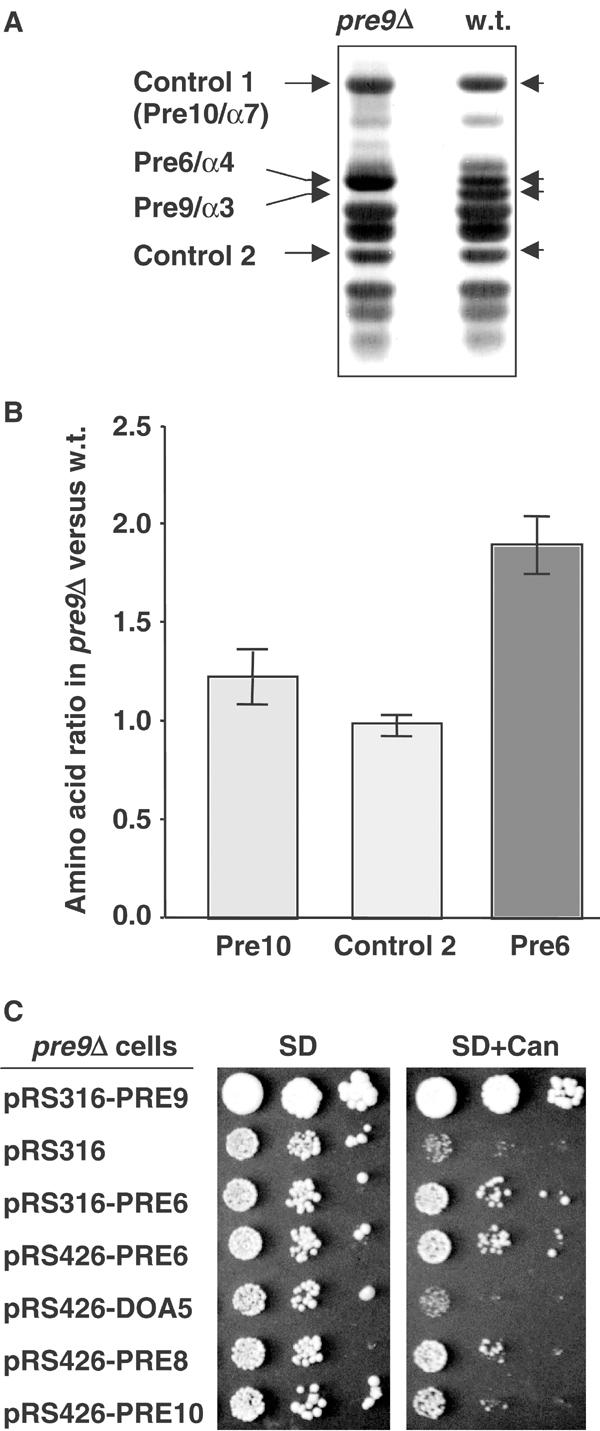
Two extra copies of the Pre6/α4 subunit in each pre9Δ proteasome. (A) Purified 20S proteasomes analyzed by gradient SDS–PAGE and Coomassie blue staining. (B) Average ratio of amino-acid levels in the indicated species (all are 20S proteasome subunits) from panel (A) in pre9Δ relative to wild-type proteasomes. (C) Partial suppression of pre9Δ by increased dosage of PRE6. Cells were spotted in 10-fold serial dilutions on minimal medium (SD) or SD containing 1 μg/ml canavanine.
20S proteasomes from wild-type and pre9Δ strains were partially purified on glycerol gradients. As previously observed (Emori et al, 1991), basal peptidase activities derived from several of the catalytic centers were higher in the mutant particles than in wild type (not shown). Wild-type particles have relatively weak peptidase activity because the proteasome channel in the α ring is predominantly closed (Osmulski and Gaczynska, 2000). The N-terminus of Pre9/α3 provides a key part of the ‘gate' controlling this channel, consistent with the higher apparent activity of pre9Δ proteasomes (Groll et al, 2000).
In summary, changes in proteasome function are observed in vitro and in vivo when the Pre9 subunit is missing, but the alterations are relatively subtle.
Purified pre9Δ proteasomes have two extra copies of Pre6/α4
Several hypotheses could explain the high level of proteasome function in the pre9Δ mutant. The remaining α subunits could arrange themselves into a six-membered ring analogous to the homohexamer formed by the proteasome-related hslV protease in bacteria (Baumeister et al, 1998). Alternatively, a gap could exist in the α ring lacking Pre9, which might be stabilized by contacts among the remaining subunits. Finally, the position normally occupied by Pre9 could be taken by another protein, such as another proteasome subunit.
We purified 20S proteasomes from wild-type and pre9Δ cells and compared their subunit composition (Figure 2A). At least 11 distinct species (of the total of 14 expected) were resolved. One band of the size predicted for Pre9 was missing from the pre9Δ particles, as expected. More interestingly, the band immediately above the Pre9 band consistently stained more intensely in pre9Δ samples than in wild-type ones. This band from an SDS gel-separated pre9Δ proteasome preparation was excised and subjected to matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) mass fingerprinting. The protein was identified as the proteasome subunit Pre6/α4. Pre6 and Pre9 share only ∼33% identity, which is comparable to the similarity of Pre9 to other α subunits.
The increased levels of Pre6 in pre9Δ proteasomes might reflect the ability of the Pre6 subunit to occupy two positions in each α ring, which would predict a two-fold increase in Pre6 subunits per proteasome in the pre9Δ mutant relative to wild type. To quantitate the actual increase, equivalent amounts of purified pre9Δ and wild-type proteasomes were resolved by SDS–PAGE, and subunits were subjected to quantitative amino-acid analysis. The Pre6 bands and two control 20S proteasome bands were analyzed. As shown in Figure 2B, the amount of Pre6 was indeed approximately two-fold higher in the pre9Δ proteasomes relative to wild-type particles. In contrast, for the two control subunits, little or no increase was observed in the mutant. For both wild-type and pre9Δ proteasomes, comparison of the experimentally determined amino-acid content of the Pre6 and Pre10 (control 1) species to the values predicted from their known sequences showed strong agreement. This provided independent evidence for the initial mass spectrometric identification of Pre6, and indicated that there was minimal contamination of these bands by other proteins in the gel (see Materials and methods).
We conclude that in pre9Δ cells, most or all 20S proteasomes contain twice the wild-type number of Pre6 subunits. The Pre6 substitution could explain why only modest proteolytic abnormalities characterize the pre9Δ mutant.
Disulfide engineering indicates that Pre6 occupies the α3 position
The simplest model for subunit arrangement in the pre9Δ proteasome would place the extra Pre6 subunit in the position normally occupied by Pre9 in the wild-type particle, next to Pre8/α2 (Figure 1A). If Pre6 occupation of the α3 position is not favored, then overexpression of Pre6 might partially suppress phenotypic anomalies associated with pre9Δ. In fact, an increased dosage of PRE6 could modestly suppress the poor growth of pre9Δ cells on canavanine plates (Figure 2C). Little or no suppression was seen with high-copy expression of two other α subunits, Doa5/α5 or Pre10/α7. We observed extremely weak but reproducible growth suppression of pre9Δ with high-copy PRE8, suggesting that higher levels of Pre8/α2 might also facilitate slightly the incorporation of its predicted new neighbor, Pre6.
To obtain direct physical evidence for juxtaposition of Pre6 and Pre8 in the pre9Δ proteasome, we attempted to crosslink cysteine residues that were introduced into the two subunits at positions predicted to be in close proximity (Figure 3 cartoon; Figure 4A). [Attempts to crystallize pre9Δ proteasomes have so far failed (M Groll and M Hochstrasser, unpublished).] The feasibility of subunit crosslinking was first tested by engineering a disulfide bond between Pre8 and its normal neighbor, Pre9. From the yeast proteasome structure (PDB entry 1RYP), we identified Pre9 residues that contacted Pre8 and, by sequence alignment, were conserved in Pre6. The β carbons of Pre8-Lys160 and Pre9-Leu56 are within 4.9 Å of each other, close to β-carbon separations seen in natural disulfide bonds (Skiba et al, 1999), so these residues were changed to cysteines.
Figure 3.
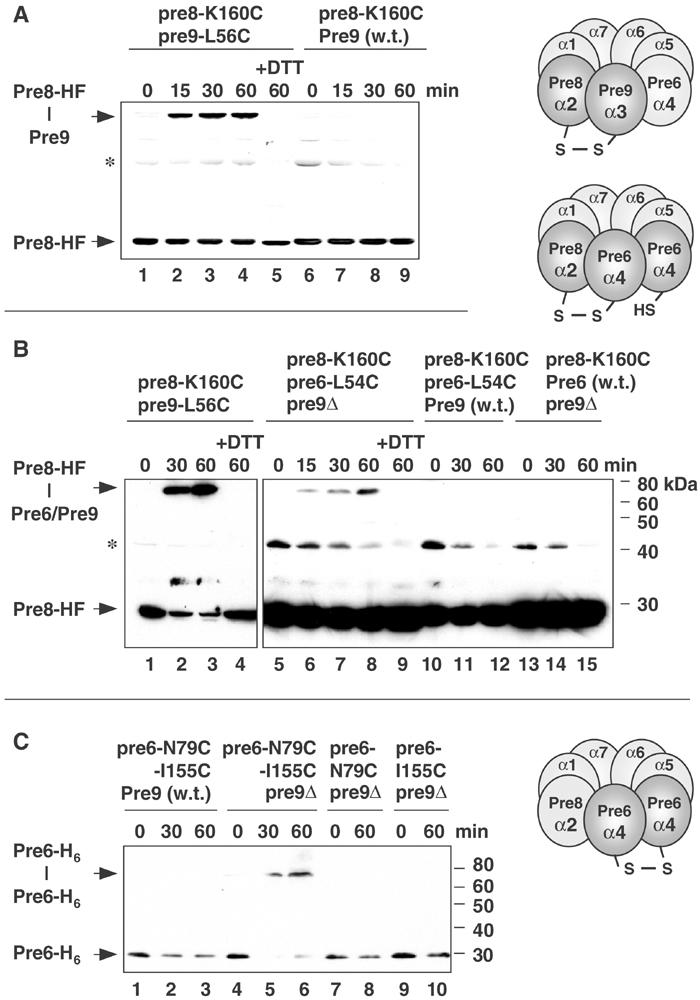
Juxtaposition of Pre6 and Pre8 in pre9Δ proteasomes revealed by disulfide engineering. (A) Control disulfide crosslinking in aqueous iodine of Pre8-HF(His/Flag)–Pre9-T7 for wild-type proteasomes analyzed by anti-Flag immunoblot. +DTT, reducing agent added prior to electrophoresis. (B) Anti-Flag immunoblot of CuCl2-induced crosslinking of Pre8-HF to Pre6 in extracts from strains MHY2863 (lanes 5–9), MHY2865 (lanes 10–12), and MHY2867 (lanes 13–15). A parallel Pre8-HF–Pre9 (MHY1839; lanes 1–4) crosslinking reaction allowed direct comparison of crosslinking efficiency. Asterisk, an unknown cross-reacting band. (C) Crosslinking (CuCl2) of two engineered His6-tagged Pre6 subunits in pre9Δ proteasomes visualized by anti-His immunoblotting. Strains used were MHY2900 (lanes 1–3), MHY2901 (lanes 5–6), MHY2896 (lanes 7–8), and MHY2897 (lanes 9–10).
Figure 4.
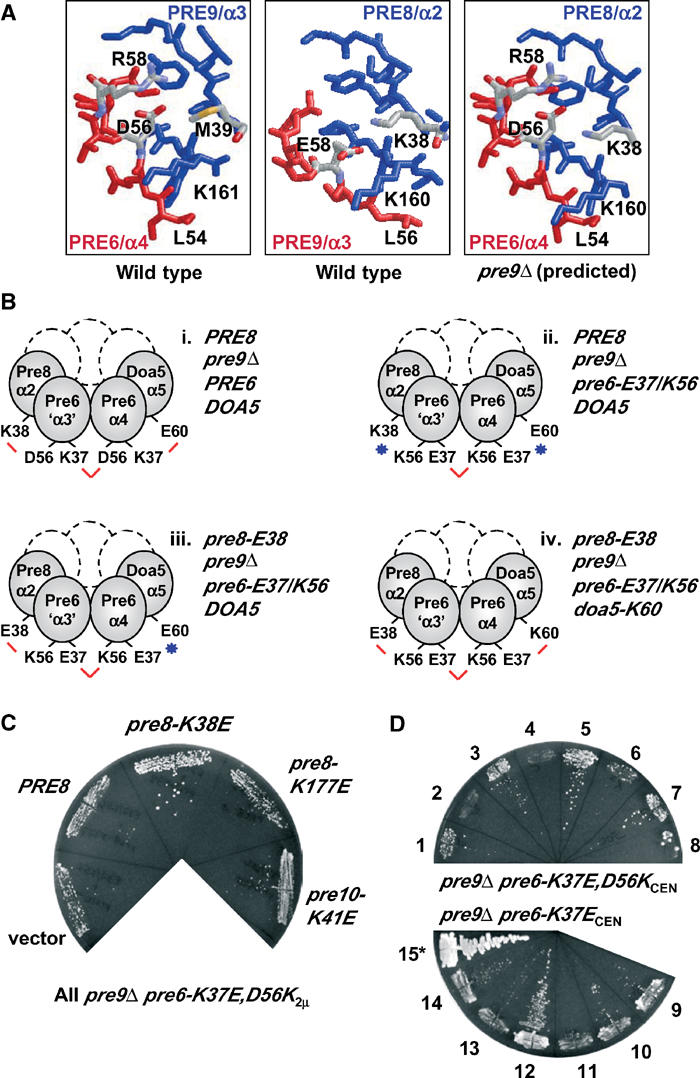
A network of salt bridges important for Pre6 subunit occupation of the α3 position in pre9Δ proteasomes. (A) RasMol figures (PDB 1RYP) showing subunit interfaces. For the predicted interface between Pre8 and Pre6 in the pre9Δ proteasome (right panel), the Cα backbone of Pre6 was fitted onto the Pre9 structure. (B) Summary of key suppression tests with engineered complementary side-chain changes at Pre6 interfaces in the pre9Δ proteasome. Salt bridges are indicated by red lines and charge clashes by blue stars. (C) Suppression of the growth defect associated with pre9Δ pre6-K37E,D56K by pre8-K38E (see Figure 4Biii). The pre6 allele was on a high-copy (2μ) plasmid. MHY1603 cells expressing the indicated pRS425 plasmid-borne alleles were grown on FOA medium (30°C, 5 d) to evict the PRE6/URA3 plasmid originally present. (D) Suppressor analysis on FOA of pre9Δ pre6-K37E,D56KCEN and pre9Δ pre6-K37ECEN with high-copy (2μ) pre8-K38E and/or doa5-E60K. High-copy plasmid-borne alleles present (empty plasmids not listed): 1, pre8-K38E; 2, PRE8; 3, doa5-E60K; 4, DOA5; 5, pre8-K38E+doa5-E60K; 6, pre8-K38E+DOA5; 7, PRE8+doa5-E60K; 8, PRE8+DOA5; 9, none; 10, pre8-K38E; 11, PRE8; 12, doa5-E60K; 13, DOA5; 14, pre8-K38E+doa5-E60K; 15, none (*but carries PRE6, not pre6-K37E). 1–8, 30°C for 7 d; 9–15, 30°C for 4 d.
Incubation of extracts from pre8-K160C pre9-L56C yeast cells under oxidative conditions resulted in the time-dependent formation of a novel larger species that was detected when the samples were run on nonreducing SDS gels and subjected to immunoblot analysis with an antibody against Flag epitope-tagged Pre8 (Figure 3A, lanes 1–4). Consistent with the inference that this is a disulfide-linked Pre9–Pre8 species, the low mobility band disappeared when the reducing agent dithiothreitol (DTT) was added to a crosslinked sample prior to electrophoresis (lane 5). Although Pre9 was tagged with a T7 epitope, it reacted very poorly with anti-T7 epitope antibodies. To verify the presence of Pre9 in the crosslinked species, we carried out an identical oxidative time course using a Pre9 allele lacking the Cys substitution (Figure 3A, lanes 6–9). The low mobility band was no longer observed. Crosslinking also required the K160C substitution in Pre8 (not shown). These data indicate that intersubunit contacts can be monitored by disulfide engineering.
We tested whether crosslinking could be detected between Pre8 and Pre6 specifically when proteasomes lacked Pre9 (Figure 3B). In pre9Δ proteasomes, Pre6-Leu54 should be in a position similar to that of Pre9-Leu56 in wild-type particles if Pre6 occupied the α3 position (Figure 4A). For the pre9Δ proteasome with Pre8-Lys160 and Pre6-Leu54 mutated to Cys, we could not know exactly how close the mutant residues would be to one another, so crosslinking might not occur as readily as in the control experiments (Figure 3A and B). Nevertheless, a time-dependent accumulation of a DTT-sensitive crosslinked species was observed in pre6-L54C pre8-K160C pre9Δ cell extracts (Figure 3B, lanes 5–9). In Pre9+ proteasomes, the Pre6–Pre8 crosslinked species was no longer detected (lanes 10–12), and it was also lost if Pre6 lacked Cys54 (lanes 13–15) or Pre8 lacked the Cys160 mutation (not shown).
A second prediction for Pre6 substitution at the Pre9 position in pre9Δ proteasomes would be that two Pre6 subunits would abut one another (Figure 3C). We tested this by substituting cysteines at a distinct set of Pre6 residues, Asn79 and Ile155, which should be in close proximity across the predicted Pre6–Pre6 interface. As shown in Figure 3C, efficient Pre6–Pre6 crosslinking occurred in pre9Δ (lanes 4–6) but not PRE9 cells (lanes 1–3). Crosslinking did not occur in pre9Δ cells unless Asn79 and Ile155 of Pre6 were both replaced with Cys (lanes 7–10).
We conclude that the extra copies of Pre6 in the pre9Δ proteasome can take the positions normally occupied by Pre9 in the α rings and that the new Pre6 interfaces with surrounding subunits are very similar to those used by Pre9. This conclusion was confirmed by the genetic data in the next section.
A network of salt bridges is important for Pre6 positioning in pre9Δ proteasomes
We previously developed structure-guided pseudoreversion strategies to probe the functional significance of specific subunit interactions within the proteasome (Chen and Hochstrasser, 1996; Arendt and Hochstrasser, 1997). An analogous approach was designed to investigate interactions of the Pre6 subunits in pre9Δ cells. Several Pre9 residues that contact the adjacent Pre8 subunit in wild-type proteasomes are conserved in Pre6. When Pre6 is located in the α3 position in the pre9Δ proteasome, these Pre6/‘α3' residues could make similar contacts with Pre8. We focused on a salt bridge between Pre9-Glu58 and Pre8-Lys38; Pre6 has an Asp residue (D56) at the position corresponding to Pre9-E58 (Figure 4A). In pre9Δ cells, where Pre6 is in both the α3 and α4 slots, D56 of Pre6/‘α3' could salt bridge with Pre8-K38 while D56 of Pre6/α4 might pair with K37 of the Pre6/‘α3' subunit (Figure 4Bi).
We reasoned that mutations in the Pre6 subunit that affect the putative Pre8-K38–Pre6-D56 salt bridge might be deleterious, which in turn might be alleviated by a compensatory mutation in the adjacent Pre8 subunit that might restore the salt bridge. However, mutation of Pre6-Asp56 to Asn or Lys had little if any effect on growth (i.e., on essential proteasome functions) in either a PRE9 or pre9Δ background (not shown). In contrast, the pre6-K37E mutation was strongly deleterious but only when PRE9 was deleted (Figure 4D and not shown). This suggested that loss of Pre9 sensitized the proteasome to perturbations of certain neighbor-interacting residues of the duplicated Pre6 subunit. Specifically, the pre6-K37E mutation might cause charge clashes with both Pre6-D56 in the α4 position and with Doa5-E60 at the α5 position.
To alleviate the predicted clash of Pre6-E37 in the α3 position with Pre6-D56 at α4, we engineered a D56K substitution into pre6-K37E, creating the pre6-K37E,D56K double mutant (Figure 4Bii). However, the double mutant grew worse, not better, than pre6-K37E when combined with pre9Δ. When on a low-copy (CEN) plasmid, pre6-K37E allowed very slow growth but pre6-K37E,D56K was lethal (Figure 4D); weak growth was only seen if pre6-K37E,D56K was overproduced (Figure 4C, ‘vector').
Poor growth could reflect the fact that in pre6-K37E,D56K pre9Δ proteasomes, the mutant K56 of Pre6/‘α3' should appose Pre8-K38 (Figure 4Bii). We therefore introduced a high-copy pre8-K38E allele into pre6-K37E,D56K pre9Δ cells to see if growth would be enhanced (Figure 4B iii). Indeed, significant suppression of the pre6-K37E,D56K pre9Δ growth defect was observed at 30°C (Figure 4C). Importantly, the suppression effects were both allele- and gene-specific (Figure 4C). A pre8 allele encoding a Lys-to-Glu mutation at residue 177 (which affects the surface facing the 19S cap) did not suppress, nor did wild-type PRE8. A Lys-to-Glu mutation of a residue equivalent to pre8-K38 in another α subunit, Pre10-K41, also failed to suppress.
While the pre8-K38E mutation restored Pre8/α2–Pre6/‘α3' intersubunit contact, the Pre6/α4–Doa5/α5 interface would still be defective in the pre8-K38E pre6-K37E,D56K pre9Δ mutant (Figure 4Biii). We therefore asked if simultaneous introduction of high-copy pre8-K38E and doa5-E60K alleles into a pre6-E37,K56CENpre9Δ strain could suppress the original lethality. This should effectively restore all the targeted salt bridges linked to the Pre6 subunits (Figure 4Biv), although the polarity is reversed and impaired expression/folding of the multiple mutated subunits might occur. In fact, more vigorous growth was observed with the two suppressors than with pre8-K38E alone (Figure 4D, strain 5 versus 6). This suppression was largely dependent on doa5-E60K (strains 3,7). Tellingly, however, when suppression of the pre6-K37E single mutant was evaluated (Figure 4D, 9–14), enhanced growth was only seen with high-copy doa5-E60K and not with both pre8-K38E and doa5-E60K (strain 12 versus 14), consistent with enhanced interaction of doa5-K60–pre6-E37 (at α4) but a deleterious interaction between pre8-E38 and the normal pre6-D56 residue (at α3).
These structure-based pseudoreversion analyses strongly support the conclusion that the extra Pre6 subunits in the pre9Δ proteasome sit between Pre8/α2 and Pre6/α4 in each α ring. More importantly, they make a compelling case for a network of salt-bridging interactions between neighboring α subunits making substantial contributions to proper proteasome subunit arrangement, at least for the alternative pre9Δ proteasome.
Evolutionary conservation of Pre9/α3 replacement by α4
Previously, we had found that several yeast 20S proteasome subunits, including the α subunits Doa5/α5 and Pre9/α3, could be replaced by their orthologs from A. thaliana (Fu et al, 1998). In Figure 5 (top left), it can be seen that the plant α4 subunit, PAD1, could also replace its yeast counterpart, Pre6. We then tested whether PAD1 shared the ability of Pre6 to replace the α3 subunit in the yeast pre9Δ proteasome. Indeed, PAD1 supported the growth of a pre6Δ pre9Δ strain as well (Figure 5). Complementing activity was specific to the PAD1 gene. This finding was especially remarkable given that pre9Δ cells were extremely sensitive to mutations in PRE6 (Figure 4 and not shown). Pre6 and PAD1 are ∼58% identical over 249 residues. Thus, even though 42% of the residues between the orthologous subunits are different, the plasticity of α4 subunit placement has been conserved, at least in the context of the yeast proteasome. Pre6 residues, which when mutated lead to synthetic growth defects with pre9Δ, for example Lys37, are similar or identical in PAD1 and α4 orthologs from most other species.
Figure 5.
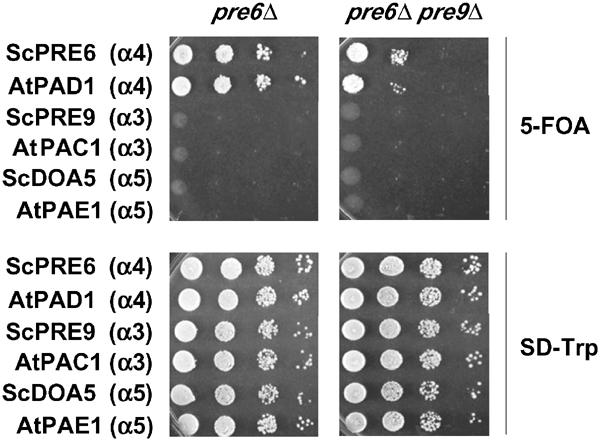
The A. thaliana Pre6/α4 ortholog PAD1 can replace Pre6 in both pre6Δ and pre6Δ pre9Δ cells. Genes encoding the indicated proteins were expressed from TRP1 plasmids in MHY1600 (left) and MHY1603 (right), which both originally also carried a PRE6/URA3 plasmid. Cells were spotted in 10-fold serial dilutions and incubated at 23°C; growth on FOA requires loss of the PRE6/URA3 plasmid. AtPAC1 and AtPAE1 were shown previously to complement deletion of their yeast orthologs, Pre9/α3 and Doa5/α5 (Fu et al, 1998).
Mutant pre9Δ cells have a defect early in proteasome assembly
The ability of the Pre6/α4 subunit to assume the α3 position raises the question of why Pre6 is not observed in this position in wild-type cells in at least a fraction of proteasomes. We reasoned that Pre9 might have some advantage over Pre6 in incorporating stably into the assembling particle during proteasome biogenesis. A prediction of this hypothesis is that assembly would be delayed in cells lacking Pre9. We evaluated this by gel filtration, which allows the resolution of 26S and 20S proteasomes from unincorporated subunits and precursor subparticles such as the 15S intermediate (‘half-proteasome') that is bound to the Ump1 chaperone (Ramos et al, 1998). Congenic wild-type and pre9Δ strains were generated that expressed Ump1 and Pup1/β2 proteins as HA2-epitope-tagged derivatives. Size-fractionated extracts from these cells were analyzed by immunoblotting (Figure 6).
Figure 6.
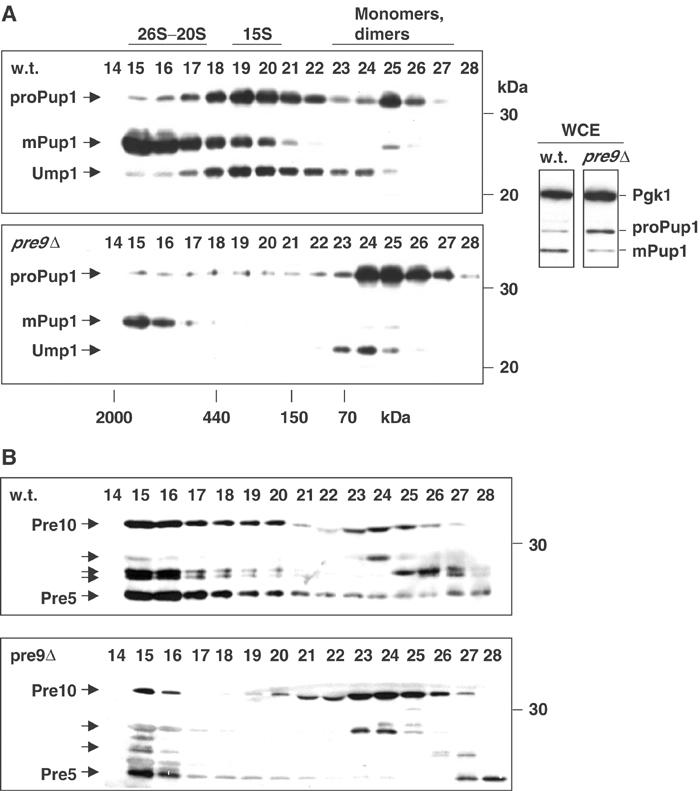
An early proteasome assembly defect in pre9Δ cells. (A) Anti-HA immunoblots of Superose 12 column fractions from extracts of wild-type and congenic pre9Δ cells. Both the Pup1 and Ump1 proteins have C-terminal HA epitope tags. Right, whole-cell extract (WCE). Pgk1 was used as a loading control. (B) Immunoblot analysis of the same fractions run on separate gels using the anti-α subunit monoclonal antibody MCP231. The antibody recognizes a common epitope in multiple α subunits (arrows), reacting most strongly with Pre10/α7 and Pre5/α6.
In wild-type cells, the Pup1 precursor, proPup1, was detected primarily in the 15S intermediate and to a lesser extent as free subunit (Figure 6A, top panel). Pup1 is proteolytically processed at a late stage in proteasome assembly to its mature form (mPup1). The Ump1 chaperone was also primarily detected in the 15S intermediate, as expected (Ramos et al, 1998). Strikingly, when Pre9 was absent from cells, a very different profile was observed (Figure 6A, lower panel). Most of the Pup1 precursor and Ump1 were found as smaller species, and significantly less mature Pup1 accumulated in 20S/26S proteasomes. Reduced levels of mature proteasomes were also seen in whole-cell extracts as measured by relative levels of mature to precursor Pup1 even though total Pup1 subunit levels are comparable in the two strains (Figure 6A, right). Analysis of a tagged Doa3/β5 subunit showed a similar difference in distribution between these two strains (not shown). Instability of pre9Δ proteasomes is unlikely to account for these data because very little processed Pup1 (or Doa3) appeared free or in small oligomers, which would have been expected if mature proteasomes were falling apart more readily in pre9Δ cells or in extracts (Figure 6A).
These results indicated that in the pre9Δ mutant, 15S assembly intermediates and mature proteasomes were not forming at normal efficiency. We verified this by examining the same samples used for Figure 6A with an antibody that recognizes several α subunits (Hendil et al, 1995). Relative to wild type, a much larger fraction of the detected subunits accumulated as monomers (or at most small oligomers) in pre9Δ cells (Figure 6B). This result suggests that deletion of Pre9 slows down assembly (or reduces the stability) of a full heteromeric α ring, which is thought to form prior to β subunit addition (Nandi et al, 1997). Within the sensitivity of the gel filtration assay, overexpression of Pre6, although able to suppress weakly the growth defect of pre9Δ cells, did not detectably affect proteasome subunit distributions (not shown).
Thus, it appears that Pre9 normally has an advantage over Pre6 in incorporating stably at the α3 position in early proteasome precursors. Despite their reduced levels of mature proteasomes, pre9Δ cells suffer relatively minor disabilities, suggesting that wild-type cells express proteasomes at levels well above what is necessary for viability under optimal growth conditions.
Discussion
Comparative analysis of purified 20S proteasomes from wild-type and pre9Δ cells revealed that the mutant proteasomes can replace the missing Pre9/α3 subunits with additional copies of Pre6/α4. The extra Pre6 subunit assumes the position normally occupied by Pre9. The ability of the α4 subunit to occupy simultaneously the α3 and α4 positions is evolutionarily conserved, suggesting that this plasticity in subunit organization reflects an important feature of proteasome assembly or function. Suppressor analysis revealed the importance of specific electrostatic interactions between subunits for proper subunit arrangement in the particle. These approaches provide a framework for structure–function analysis of proteasome structure and assembly that can be extended to other large protein complexes. Finally, our results indicate that incorporation of the Pre9 subunit into early proteasome precursor particles enhances the assembly of proteasomes, which suggests a general mechanism by which subunits in heteromeric ring complexes achieve their correct arrangement.
Proteasome assembly
Multisubunit protein assemblies often involve extensive subunit interfaces in which single point mutations have little if any detectable effect on the function of the complex. A similar impediment often makes it a challenge to design small molecule inhibitors that interfere with protein–protein interactions. For the α–α ring interactions examined here, we have found striking synthetic effects when combining mutations in Pre6 neighbor-contacting residues with the pre9Δ allele (Figure 4 and not shown). Use of synthetic growth effects provides a general tack for assessing the importance of particular intersubunit contacts by suppressor analysis when single mutations are of little consequence. This approach should be useful for the study of other large complexes with multivalent interactions. [Analyses of interactions between pre6 mutations and pre9ΔN showed no synthetic interactions (not shown). The latter is an allele that removes just the N-terminal tail of Pre9 and causes a constitutively open α-ring channel but leaves Pre9 in its normal position (Groll et al, 2000). Hence, the strong synthetic growth defects associated with pre9Δ are not a result of an open channel but are consistent with there being a duplicate Pre6 subunit at the α3 position.]
Until now, there has been virtually no analysis of the basis for the stereospecific placement of proteasome subunits in vivo. Analysis of the Pre9–Pre6 replacement provides a starting point for addressing the molecular basis for subunit arrangement in this complex machine. Specifically, replacement of Pre9 by Pre6 in pre9Δ cells appears to be due to a combination of favorable electrostatic and van der Waals interactions between Pre6 at the α3 position and its subunit neighbors there, together with the absence of strong steric clashes. If we consider just the prominent intersubunit interaction involving Pre6-Asp56 and Pre6-Lys37 that occurs when Pre6 is at both the α3 and α4 positions, only one other subunit, Scl1/α1, would have charge complementarity at this position with both neighboring α subunits. In addition, by fitting the backbones of all seven α subunits into the α3 position, we found that several would have strong steric overlaps with neighboring subunits (not shown). Scl1 is one that would suffer substantial steric conflict. Although undoubtedly oversimplified, these simple criteria can in principle account in large measure for the apparently unique ability of Pre6 to take both the α3 and α4 positions within the proteasome.
Specific α–β subunit contacts are also expected to be important for proteasome assembly. In pre9Δ cells, Pre6 must be able to interact productively with β subunits that normally contact Pre9 in wild-type cells. We have found that pre6 mutations at these predicted β interfaces can also cause synthetic growth effects with pre9Δ (unpublished data). Therefore, inter-ring contacts impose additional constraints on the positioning of specific subunits.
An important question is why Pre9 is invariably at the α3 position when both Pre9 and Pre6 are available. The detailed shape and electrostatic complementarities between Pre9 and adjoining subunits are evidently more favorable than when Pre6 is at this position. Interaction surfaces between subunits may have evolved to the point where their incorporation into the proteasome complex is strongly codependent. An assembly advantage for Pre9 over Pre6 at the α3 position can be inferred from the gel filtration data in Figure 6. For other proteasome subunits, the bias toward a single specific subunit in a particular slot is evidently even more acute, with little or no possible incorporation of any of the other subunits at noncanonical locations.
Evolution and assembly of heteromeric ring complexes
Proteasomes share a number of structural and evolutionary features with other large but functionally distinct protein complexes. Like the eucaryotic 20S proteasome, eucaryotic class II chaperonins, the Sm core complex of snRNP complexes, the Mcm DNA replication-licensing complex, and probably Lsm complexes and exosomes assemble into precisely ordered heteromeric rings or stacks of rings (Archibald et al, 1999; Kambach et al, 1999; Prokhorova and Blow, 2000; Raijmakers et al, 2002). The six ATPases of the eucaryotic 19S proteasomal subcomplex are also believed to form a heterohexameric ring (DeMartino and Slaughter, 1999). In most of these examples, related proteins can be identified from eubacterial or archaeal species that comprise simpler, usually homopolymeric ring structures, e.g. the archaeal class II chaperonins and an archaeal Sm/Lsm-related ring.
Similar to Pre9+ and pre9Δ proteasomes, alternative heteroheptameric Lsm rings that differ in composition by a single subunit can assemble within eucaryotic cells. These different Lsm rings have distinct functions (Pannone and Wolin, 2000). It is possible that there are growth conditions or points in the yeast life cycle that require or favor the alternative 20S proteasome structure that we have described. In metazoans, particular tissues or cell types can harbor specific variants of the proteasome (Yuan et al, 1996), and such variation might extend to duplication of certain subunits within a ring. Our data indicate that a plant α4 subunit, PAD1 of Arabidopsis thaliana, retains the capacity for assuming both the α3 and α4 positions in the α ring (Figure 5). In general, the evolution of heteromultimeric forms of protein rings allows for alternative but architecturally similar assemblies that can assume divergent functions. Our analysis of pre9Δ proteasome assembly and subunit interactions gives a first clue as to how such structural diversity can be maintained.
The ability of α4/PAD1 to assume two positions in the yeast proteasome does not necessarily mean that the α3 subunit in plants (or other organisms) will also be dispensable, as it is in yeast. The assembly defect in cells lacking α3 (Figure 6) might be sufficiently severe to reduce proteasome production below that needed for viability. Proteasome subunit knockdown by RNA interference in Drosophila cells, for example, suggests that α3 depletion causes a loss of 20S proteasomes without the efficient formation of an alternative 20S species (Wojcik and DeMartino, 2002). This does not exclude a role for positional plasticity of subunits under certain growth conditions or as a part of the proteasome assembly mechanism.
During evolution, position-specific incorporation of new subunits derived from gene duplication would be expected to depend on coadaptation of residues at subunit interfaces that favor interactions between particular subunits. There are examples of what appears to be incipient diversification of proteasome subunits in certain prokaryotes where two different subunits with over 80% identity can assume any position in the ring and in all possible ratios (Zühl et al, 1997). Among human α subunits, the C8/α7 subunit can form homoheptamers when expressed on its own, and coexpression with certain other α subunits in Escherichia coli results in mixed rings with all varieties of stoichiometries (Gerards et al, 1998). In vivo, there must be some means to ensure that specific subunit arrangements are favored. Our data indicate that Pre6 subunits can interact with one another in 20S proteasome particles in vivo, yet the Pre9–Pre6 pairing predominates. This arrangement is dictated at a very early stage of proteasome assembly.
We suggest that it is also during early stages of assembly that particular arrangements of subunits will usually be established for other ring complexes because removal and replacement of subunits from a full ring would be expected to be more energetically costly. Where alternative rings coexist in the same cell, it is necessary either to invoke specific chaperone activities that direct assembly through different pathways or to assume that interactions between certain subunits are sufficiently similar that alternative complexes can form simply by mass action. It will be important to establish which of these mechanisms predominate for the many different ring assemblies found in eucaryotic cells.
Materials and methods
Yeast and bacterial media and methods
Yeast rich (YPD) and minimal (SD) plates were prepared as described, and standard methods were used for genetic manipulation of yeast (Guthrie and Fink, 1991). Standard techniques were used for recombinant DNA work.
Plasmid and mutant allele construction
The PRE8, PRE9, and PRE10 genes were isolated by PCR amplification from genomic DNA and subcloned into yeast-E. coli shuttle vectors. Their functional integrity was verified by complementation of yeast null mutants. DOA5 cloning was described previously (Chen and Hochstrasser, 1995). A PRE6 plasmid was obtained from Heinemeyer et al (1994). Mutations and epitope-coding segments were introduced either by a two-step PCR method or by Quikchange (Stratagene). DNA sequences were verified by DNA sequencing. To make the YIp′PRE8-HF plasmid, pJD416 (Ramos et al, 1998) was digested with Asp718I and SacI to remove the PRE1 insert, and the vector fragment was ligated to a PCR-amplified Asp718I-SacI PRE8 fragment encoding a C-terminal segment of the protein. Ligation fused the PRE8 3′ sequence in-frame to sequences encoding the Flag and His6 epitope tags. YIp′PRE8-HF was integrated into the PRE8 locus, resulting in a full-length, fully functional epitope-tagged version of PRE8. Correct integration was verified by colony PCR. YIp-′pre8-K160C-HF was integrated into yeast by the same procedure following mutagenesis. Other proteasome gene mutants were carried on CEN or 2μ plasmids. To clone the A. thaliana PAD1 gene into yeast expression vectors, PAD1 was first amplified from a λ ZAPII/pBluescript cDNA clone (Fu et al, 1998). The PCR product was cloned into p414-PGPD and p424-PGPD (Mumberg et al, 1995).
Yeast strains
Yeast strains used in this study are listed in Table I. MHY500, MHY501, MHY606, and MHY1069 were described previously (Chen and Hochstrasser, 1995; Fu et al, 1998). A pre6 null mutant was created by transforming MHY606 with a pre6Δ::HIS3 allele (Heinemeyer et al, 1994). The resulting heterozygous diploid was transformed with YCplac33PRE6, and MHY1600 was isolated by sporulation and tetrad dissection of the transformant. MHY1603 was a segregrant from a cross between MHY1600 and MHY1069. MHY2550 and MHY2552 were made by the introduction of UMP1-HA2 and PUP1-HA2 alleles (Ramos et al, 1998) into strains that previously had the doa3Δ::HIS3 and/or pre9Δ::HIS3 alleles (Chen and Hochstrasser, 1995; Fu et al, 1998); growth rates were identical to congenic strains lacking the tagged subunits. Other strains were made by standard yeast genetic techniques.
Table 1.
Yeast strains used in the current study
| MHY500 | a his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 |
| MHY501 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 |
| MHY606 | a/α (MHY500 × MHY501) |
| MHY1069 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre9Δ::HIS3 |
| MHY1600 | a his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 [YCplac33PRE6] |
| MHY1603 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre9Δ::HIS3 [YCplac33PRE6] |
| MHY1831 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre8-K160C-HF pre9Δ::HIS3 |
| MHY1838 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre8-K160C-HF pre9Δ::HIS3 [pRS315PRE9-T7] |
| MHY1839 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre8-K160C-HF pre9Δ::HIS3 [pRS315pre9-L56C-T7] |
| MHY1850 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 PRE8-HF pre9Δ::HIS3 [pRS315pre9-L56C-T7] |
| MHY2475 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre8-K160C-HF pre9Δ::HIS3 [pRS424PRE6-T7] |
| MHY2550 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 doa3Δ::HIS3 ump1::pRS305-UMP1-HA2 pup1::YIplac211-PUP1-HA2 pre9::HIS3 [YCplac22Doa3-His6] |
| MHY2552 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 doa3Δ::HIS3 ump1::pRS305-UMP1-HA2 pup1::YIplac211-PUP1-HA2 [YCplac22Doa3-His6] |
| MHY2863 | a his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre8-K160C-HF pre9Δ::HIS3 [pRS424pre6-L54C] |
| MHY2865 | a his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre8-K160C-HF [pRS424pre6-L54C] |
| MHY2867 | a his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre8-K160C-HF pre9Δ::HIS3 [pRS424PRE6] |
| MHY2896 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre9Δ::HIS3 [pRS317pre6-N79C-H6] |
| MHY2897 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre9Δ::HIS3 [pRS317pre6-I155C-H6] |
| MHY2900 | a his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 [pRS317pre6-N79C,I155C-H6] |
| MHY2901 | α his3-Δ200 leu2-3,112 ura3-52 lys2-801 trp1-1 pre6Δ::HIS3 pre9Δ::HIS3 [pRS317pre6-N79C,I155C-H6] |
Purification of proteasomes
Yeast 20S proteasomes were purified from MHY501 and MHY1069 as described (Chen and Hochstrasser, 1995) except that after the DEAE-Sepharose column, the pooled active fractions were loaded onto a Superose-6 FPLC gel filtration column. Proteins were eluted in 20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 150 mM NaCl, and 10% glycerol. Fractions (0.5 ml) were collected and the peak fractions, as judged by Coomassie blue staining of SDS gels and peptide hydrolysis assays, were used for further analysis. Protein concentration was determined by the Bradford method.
Protein identification by gradient PAGE and MALDI-MS
Equivalent amounts of purified wild-type and pre9Δ proteasomes were loaded on a 10–15% polyacrylamide gel, electrophoresed overnight, transferred to a Millipore Immobilon-P membrane overnight at low voltage, stained with Coomassie blue, destained thoroughly, and washed with deionized water. The single band that stained more intensely in the pre9Δ preparation was excised from the membrane (∼100 pmol recovered) and sent to the Keck Foundation Biotechnology Resource Laboratory (Yale) for protein identification. The sample was digested with trypsin and analyzed by MALDI-MS. Both the OWL database (using ProFound) and the EMBL/nonredundant database (using PeptideSearch) were searched for matches with the detected peptide masses. The yeast proteasome subunit Pre6 was identified with 44% coverage of its sequence.
Amino-acid analysis
Purified proteasomes were separated by gradient SDS–PAGE (10 μg of each preparation). The gel was stained with Coomassie blue, destained, and washed thoroughly in deionized water. Bands were excised using razor blades. The band identified as Pre6 by MALDI-MS and several additional reference bands were excised. Amino-acid analysis of the proteins was performed by the Keck Laboratory on a Beckman Model 6300 ion-exchange instrument following a 16 h hydrolysis at 115°C in 100 μl of 6 N HCl, 0.2% phenol that also contained 2 nmol norleucine (an internal standard). After hydrolysis, HCl was removed in a Speedvac, and the resulting amino-acid mixture was dissolved in 100 μl of sample buffer that contained 2 nmol homoserine (an internal standard). Data analysis was carried out with Perkin-Elmer/Nelson data acquisition software. The amino acids Asx (Asp and Asn), Thr, Ser, Glx (Glu and Gln), Ala, Val, Ile, Leu, Tyr, and Phe were used in our analyses. For each amino acid, the ratio of the amount (in nanomoles) present in the pre9Δ proteasome band to the amount in the wild-type band was calculated. These ratios were then averaged.
By comparing the experimentally determined amino-acid content of the Pre6 and Pre10 species from both wild-type and pre9Δ proteasomes (Figure 2) to the values expected from their known sequences, we found strong agreement in all cases. The deviation from the theoretical values was less than 10% in all four measurements (Pre6: 8.7±4.9% for mutant and 9.9±6.9% for wild type; Pre10: 7.6±5.9% and 8.3±7.7%). For comparison, when the experimental Pre6 values were compared to the predicted fractional amino-acid content of Pre9, the discrepancies were far higher (42±34% and 41±31%).
Disulfide crosslinking of engineered proteasome subunits
Yeast expressing proteasome subunits with engineered Cys residues were grown to mid-log phase, and 10–15 OD600 equivalents were harvested by centrifugation. Cells were converted to spheroplasts with Zymolyase 100T (ICN), and the spheroplasts were lysed in 0.15 ml of ice-cold lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% Triton X-100) to which a protease inhibitor cocktail had been added (5 μg/ml each of pepstatin, leupeptin, chymostatin, aprotinin, and antipain). Lysis was achieved by vortexing 2–3 times for 30 s with a minute on ice in between. The supernatant after pelleting of cell debris was saved and a 20 μl aliquot was removed and added to 2 μl of 10 × stop buffer [10 mM sodium iodoacetate and 50 mM N-ethylmaleimide (NEM)].
Disulfide crosslinking was induced with either 0.5 mM aqueous iodine or 0.2 mM CuCl2 at room temperature. The latter was slightly more efficient. Reactions were stopped at different times by removing aliquots and adding to tubes on ice that contained 10 × stop buffer. Neither Pre8 nor Pre9 has any Cys residues in its normal sequence, but Pre6 has four, although none is at a subunit interface.
Immunological procedures and β-galactosidase activity assays
Western immunoblot and pulse–chase analyses were performed as described (Chen and Hochstrasser, 1995). Antibodies used were an anti-α2 rabbit polyclonal antibody (Hochstrasser and Varshavsky, 1990), the anti-HA mouse monoclonal antibody 16B12 (Covance), the anti-Flag M2 monoclonal antibody (Sigma), and the MCP231 monoclonal antibody that recognizes several different α subunits (Hendil et al, 1995). Proteins were visualized with horseradish peroxidase-coupled secondary antibodies and ECL detection reagents (Amersham).
To measure βgal activities for various βgal test substrates, wild-type and pre9Δ cells were transformed with 2μ-based plasmids bearing the corresponding genes. βgal activity was measured for at least three independent transformants as described (Hochstrasser and Varshavsky, 1990).
Gel filtration analysis of proteasomes
Fractionation of cell extracts by gel filtration was performed as described (Ramos et al, 1998), except that Superose 12 was used instead of Superose 6 to enhance the resolution of lower molecular mass complexes. Yeast cells (0.25 l) were grown at 30°C in synthetic medium to an OD600 of 1.4, harvested, washed once with ice-cold water, and then frozen in liquid nitrogen. Cell pellets were ground to a fine powder in a mortar in the presence of liquid nitrogen. Cell powder was then resuspended in 1 ml of sample buffer (50 mM Tris–HCl, pH 7.4, 15% glycerol, 2 mM ATP, 1 mM DTT, 5 mM MgCl2, 1 mM PMSF, 20 μM pepstatin A, 10 μM leupeptin, 10 μM chymostatin, and 10 μM aprotinin). Cell debris was removed by centrifugation at 30 000 rpm for 10 min at 2°C in a TLA 120.2 rotor in an Optima TLX ultracentrifuge. Cleared lysate was centrifuged again at 41 000 rpm for 30 min at 2°C in the same rotor. Using an FPLC system (Amersham-Pharmacia), 0.2 ml of supernatant (5 mg/ml) was fractionated at 4°C on a Superose 12 column equilibrated in sample buffer. Fractions (0.6 ml) were eluted at a 0.3 ml/min flow rate. For each fraction, a 100 μl aliquot was TCA-precipitated, washed with ethanol, resuspended in gel loading buffer, and subjected to immunoblot analysis.
Acknowledgments
We thank H Fu for the Arabidopsis PAD1 cDNA, RJ Dohmen and W Heinemeyer for plasmids, N Luscombe for the computational α subunit superimposition onto Pre9, and M Deng for comments on the manuscript. This work was supported by NIH Grant GM46904 to MH.
References
- Archibald JM, Logsdon JM, Doolittle WF (1999) Recurrent paralogy in the evolution of archaeal chaperonins. Curr Biol 9: 1053–1056 [DOI] [PubMed] [Google Scholar]
- Arendt CS, Hochstrasser M (1997) Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation. Proc Natl Acad Sci USA 94: 7156–7161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister W, Walz J, Zuhl F, Seemuller E (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92: 367–380 [DOI] [PubMed] [Google Scholar]
- Chen P, Hochstrasser M (1995) Biogenesis, structure, and function of the yeast 20S proteasome. EMBO J 14: 2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Hochstrasser M (1996) Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell 86: 961–972 [DOI] [PubMed] [Google Scholar]
- DeMartino GN, Slaughter CA (1999) The proteasome, a novel protease regulated by multiple mechanisms. J Biol Chem 274: 22123–22126 [DOI] [PubMed] [Google Scholar]
- Emori Y, Tsukahara T, Kawasaki H, Ishiura S, Sugita H, Suzuki K (1991) Molecular cloning and functional analysis of three subunits of yeast proteasome. Mol Cell Biol 11: 344–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, Doelling JH, Arendt CS, Hochstrasser M, Vierstra RD (1998) Molecular organization of the 20S proteasome gene family from Arabidopsis thaliana. Genetics 149: 677–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerards WL, de Jong WW, Bloemendal H, Boelens W (1998) The human proteasomal subunit HsC8 induces ring formation of other alpha-type subunits. J Mol Biol 275: 113–121 [DOI] [PubMed] [Google Scholar]
- Glickman MH, Ciechanover A (2002) The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82: 373–428 [DOI] [PubMed] [Google Scholar]
- Groll M, Bajorek M, Kohler A, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D (2000) A gated channel into the proteasome core particle. Nat Struct Biol 7: 1062–1067 [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R (1997) Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature 386: 463–471 [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR (1991) Guide to Yeast Genetics and Molecular Biology. San Diego, CA: Academic Press [Google Scholar]
- Heinemeyer W, Tröndle N, Albrecht G, Wolf DH (1994) PRE5 and PRE6, the last missing genes encoding 20S proteasome subunits from yeast? Indication for a set of 14 different subunits in the eukaryotic proteasome core. Biochemistry 33: 12229–12237 [DOI] [PubMed] [Google Scholar]
- Hendil KB, Kristensen P, Uerkvitz W (1995) Human proteasomes analysed with monoclonal antibodies. Biochem J 305: 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M, Varshavsky A (1990) In vivo degradation of a transcriptional regulator: the yeast α2 repressor. Cell 61: 697–708 [DOI] [PubMed] [Google Scholar]
- Kambach C, Walke S, Young R, Avis JM, de la Fortelle E, Raker VA, Luhrmann R, Li J, Nagai K (1999) Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs. Cell 96: 375–387 [DOI] [PubMed] [Google Scholar]
- Kruger E, Kloetzel PM, Enenkel C (2001) 20S proteasome biogenesis. Biochimie 83: 289–293 [DOI] [PubMed] [Google Scholar]
- Löwe J, Stock D, Jap B, Zwickl P, Baumeister W, Huber R (1995) Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 Å resolution. Science 268: 533–539 [DOI] [PubMed] [Google Scholar]
- Maurizi MR (1998) Proteasome assembly: biting the hand. Curr Biol 8: R453–R456 [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122 [DOI] [PubMed] [Google Scholar]
- Nandi D, Woodward E, Ginsburg DB, Monaco JJ (1997) Intermediates in the formation of mouse 20S proteasomes: implications for the assembly of precursor beta subunits. EMBO J 16: 5363–5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmulski PA, Gaczynska M (2000) Atomic force microscopy reveals two conformations of the 20S proteasome from fission yeast. J Biol Chem 275: 13171–13174 [DOI] [PubMed] [Google Scholar]
- Pannone BK, Wolin SL (2000) Sm-like proteins wRING the neck of mRNA. Curr Biol 10: R478–R481 [DOI] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Ann Rev Biochem 70: 503–533 [DOI] [PubMed] [Google Scholar]
- Prokhorova TA, Blow JJ (2000) Sequential MCM/P1 subcomplex assembly is required to form a heterohexamer with replication licensing activity. J Biol Chem 275: 2491–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raijmakers R, Egberts WV, van Venrooij WJ, Pruijn GJ (2002) Protein–protein interactions between human exosome components support the assembly of RNase PH-type subunits into a six-membered PNPase-like ring. J Mol Biol 323: 653–663 [DOI] [PubMed] [Google Scholar]
- Ramos PC, Hockendorff J, Johnson ES, Varshavsky A, Dohmen RJ (1998) Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell 92: 489–499 [DOI] [PubMed] [Google Scholar]
- Schmidtke G, Schmidt M, Kloetzel PM (1997) Maturation of mammalian 20S proteasome: purification and characterization of 13S and 16S proteasome precursor complexes. J Mol Biol 268: 95–106 [DOI] [PubMed] [Google Scholar]
- Skiba MC, Logan KM, Knight KL (1999) Intersubunit proximity of residues in the RecA protein as shown by engineered disulfide cross-links. Biochemistry 38: 11933–11941 [DOI] [PubMed] [Google Scholar]
- Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, Yasuoka N, Tsukihara T (2002) The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 10: 609–618 [DOI] [PubMed] [Google Scholar]
- Wojcik C, DeMartino GN (2002) Analysis of Drosophila 26 S proteasome using RNA interference. J Biol Chem 277: 6188–6197 [DOI] [PubMed] [Google Scholar]
- Yang Y, Früh K, Ahn K, Peterson PA (1995) In vivo assembly of the proteasomal complexes, implications for antigen presentation. J Biol Chem 270: 27687–27694 [DOI] [PubMed] [Google Scholar]
- Yuan X, Miller M, Belote JM (1996) Duplicated proteasome subunit genes in Drosophila melanogaster encoding testes-specific isoforms. Genetics 144: 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zühl F, Tamura T, Dolenc I, Cejka Z, Nagy I, De Mot R, Baumeister W (1997) Subunit topology of the Rhodococcus proteasome. FEBS Lett 400: 83–90 [DOI] [PubMed] [Google Scholar]