

Abstract
Phosphoinositides, synthesized from myo-inositol, play a critical role in the development of growth cones and in synaptic activity. As neurons cannot synthesize inositol, they take it up from the extracellular milieu. Here, we demonstrate that, in brain and PC12 cells, the recently identified H+/myo-inositol symporter HMIT is present in intracellular vesicles that are distinct from synaptic and dense-core vesicles. We further show that HMIT can be triggered to appear on the cell surface following cell depolarization, activation of protein kinase C or increased intracellular calcium concentrations. HMIT cell surface expression takes place preferentially in regions of nerve growth and at varicosities and leads to increased myo-inositol uptake. The symporter is then endocytosed in a dynamin-dependent manner and becomes available for a subsequent cycle of stimulated exocytosis. HMIT is thus expressed in a vesicular compartment involved in activity-dependent regulation of myo-inositol uptake in neurons. This may be essential for sustained signaling and vesicular traffic activities in growth cones and at synapses.
Keywords: growth cones, myo-inositol, neurons, SLC2A13, synapses, transporter
Introduction
Phosphatidylinositol and inositol polyphosphates (phosphoinositides) are key regulators of several processes taking place at synapses and growth cones, in particular, vesicle endo- and exocytosis, polymerization of microfilaments, regulation of ion channels and signal transduction by Gq-coupled receptors (Berridge, 1993; Czech, 2000; Sechi and Wehland, 2000; Cremona and De Camilli, 2001; Osborne et al, 2001).
Regulated exocytosis of synaptic vesicles (SVs) and dense-core vesicles (DCVs) is a multistep process in which vesicles are first targeted and docked to the plasma membrane, then undergo an ATP-dependent priming step, which precedes the Ca2+-induced exocytotic event (Chapman, 2002). Ptdins(4,5)P2 synthesis is required for the ATP-dependent priming step of DCV and requires the participation of a DCV-associated PI-4-kinase (Wiedemann et al, 1996) and a plasma membrane-associated PI-5-kinase (Hay et al, 1995; Wenk et al, 2001). The Ca2+-dependent fusion event also requires the participation of PtdIns(4,5)P2, as exocytosis is inhibited by the absence of CAPS, a Ca2+- and PtdIns(4,5)P2-binding protein (Renden et al, 2001). In the case of SV exocytosis, evidence for involvement of PtdIns(4,5)P2 in secretion comes, at least in part, from the recognition that proteins required for exocytosis, such as synaptotagmin, rabphilin, DOC and Munc13, have essential Ca2+- and acidic phospholipid-binding C2 domains. Furthermore, displacement of PtdIns(4,5)P2 binding to the C2B domain of synaptotagmin inhibits exocytosis (Ohara-Imaizumi et al, 1997; Mehrotra et al, 2000).
Endocytosis of clathrin-coated vesicles at the synapse is also dependent on the presence of PtdIns(4,5)P2 and its hydrolysis. Indeed, genetic inactivation of synaptojanin 1, a polyphosphoinositides phosphatase present in clathrin-coated pits of neurons, prevents normal dissociation of the clathrin coat and leads to impaired SV re-endocytosis and to synaptic depression (Gaidarov et al, 1996; Rapoport et al, 1997; Cremona et al, 1999). PtdIns(4,5)P2 is also involved in the regulation of SV recycling through its ability to interact with Rho-GTPase to regulate actin nucleation (Sechi and Wehland, 2000; Cremona and De Camilli, 2001), and probably also participates in the regulation of ion channels (Suh and Hille, 2002). Inositol polyphosphate and pyrophosphate also play a role in controlling endocytosis (Saiardi et al, 2002).
Impaired brain inositol metabolism has been linked to psychiatric diseases, in particular Bipolar disorders. Indeed, current treatments of these mood disorders rely on the use of lithium salt, valproic acid and carbamazepine, drugs whose action may interfere with inositol metabolism. It is well established that one mechanism of action of Li+ is the inhibition of inositol monophosphate phosphatase and polyphosphoinositide 1-phosphate phosphatase (Berridge et al, 1989; Williams and Harwood, 2000), which blocks recycling of inositol phosphate and effectively reduces the availability of inositol for subsequent cycles of intracellular signal transduction. Recently, a common action of Li+, valproate and carbamazepine has been described on stabilization of the extended conformation of growth cones. More importantly, these effects could all be reversed by supplementing the cells with excess inositol (Williams et al, 2002). Thus, inositol and its metabolites not only play an essential role in regulating synaptic activity and growth cone expansion but may also participate in common forms of psychiatric disorders.
Myo–inositol synthesis from glucose in the brain is restricted to the endothelial cells forming the blood–brain barrier (Wong et al, 1987; Zhang and Majerus, 1998; Novak et al, 1999). Its utilization by neurons thus requires the presence of an active uptake system. Recently, we described the molecular and functional characterization of HMIT, an H+/myo-inositol symporter expressed at the highest level in the brain (Uldry et al, 2001). Here, we show that brain HMIT is expressed in neurons. In brain and in PC12 cells, HMIT is located in intracellular vesicles that are distinct from both the DCV and SV. Depolarization and activation of protein kinase C (PKC) leads to translocation of HMIT to the plasma membrane, preferentially at growth cones and varicosities, and surface expression is associated with increased myo-inositol uptake. These data indicate the presence in neurons of an activity-dependent mechanism involved in the regulation of myo-inositol uptake.
Results
HMIT expression in neurons
HMIT expression in neurons and glial cells was first evaluated in aggregating brain cell cultures consisting either of a mixed-cell population, or enriched in glial cells or in neurons. Figure 1A shows a Western blot analysis of HMIT protein in total rat brain membranes and in membranes prepared from the different types of cell aggregates. The symporter was found in each membrane preparation, with the highest level present in the neuron-enriched cultures. After deglycosylation by PNGaseF treatment, HMIT electrophoretic mobility was similarly increased in all membrane preparations but still appeared as multiple bands, suggesting the presence of other post-translational modifications. The enrichment of each type of cell aggregates in glial cells or neurons was confirmed by Western blot analysis of glial fibrillary acidic protein (GFAP) and the neuronal marker synaptophysin (Figure 1B). The glial cell aggregates were devoid of synaptophysin, whereas the neuron-enriched cultures had a high content of synaptophysin. The relative symporter expression level in the different cultured cells was closely correlated to the expression of HMIT mRNA (Figure 1C).
Figure 1.
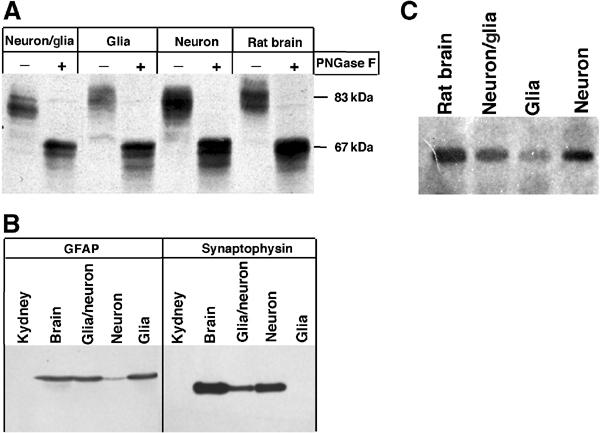
HMIT is strongly expressed in neurons and glial cells. (A) Immunodetection of HMIT in membrane preparations from rat brain or from mixed, glial and neuronal aggregating cell cultures. Samples were subjected (+) or not (−) to a PNGase F treatment. (B) Immunodetection of the glial marker GFAP and the neuronal marker synaptophysin in membrane preparation from mixed, glial and neuronal aggregating cell cultures or from the brain and kidney. (C) Northern blot analysis of HMIT mRNA expression in total RNA from rat brain and mixed, glial and neuronal aggregating cell cultures.
Subcellular fractionation of mouse brain HMIT compartment
Our initial characterization of HMIT (Uldry et al, 2001) revealed that it was located in an intracellular compartment and could reach the plasma membrane only after mutation of internalization signals. Thus, here, we first aimed at better characterizing the intracellular HMIT-containing vesicles. This was achieved by subcellular fractionation experiments, as our antibodies directed either against the C-terminal tail or intracellular middle loop of HMIT failed to recognize the symporter in immunomicroscopic analysis. Brain homogenates were separated by centrifugation on sucrose density gradients and each fraction was analyzed for the presence of HMIT and for markers of different membrane compartments. Figure 2A shows that HMIT was present in fractions of the gradients which were distinct from those containing the endosomal marker EEA1 and the lysosomal marker Lamp1. There was partial overlap with the distribution of the Na+/K+-ATPase, which, however, showed a much broader distribution on the gradient. The endoplasmic reticulum marker calreticulin showed a bimodal distribution and HMIT comigrated with the light-density calreticulin-positive vesicles. A partial comigration with the synaptophysin-containing vesicles was also observed.
Figure 2.
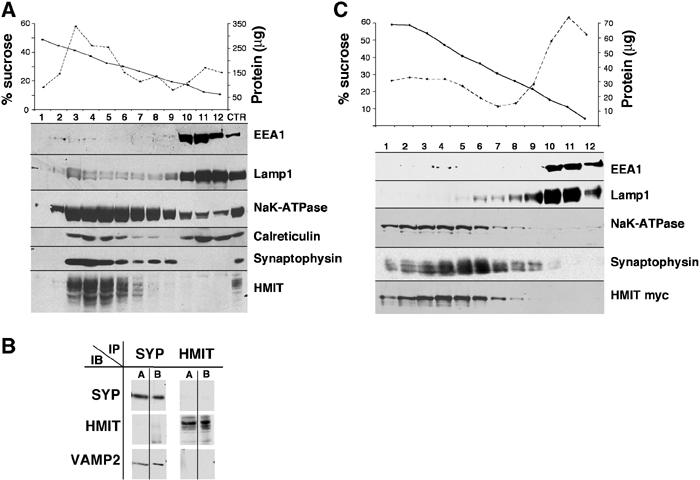
Subcellular fractionation of brain HMIT compartment. (A) Fractionation of mouse brain membrane preparation on a sucrose density gradient (10–50%). Fractions were analyzed for protein and sucrose concentration (top). HMIT and several membrane fraction markers were detected by Western blot. (B) Immunoprecipitation of HMIT- and synaptophysin-containing vesicles. Magnetic beads coupled with antibodies against HMIT or synaptophysin were incubated in the presence of mouse brain membrane proteins. The immunoprecipitated vesicles were first solubilized with Triton X-100 and the proteins remaining attached to the beads were removed by an SDS-containing buffer. Both the Triton X-100-solubilized proteins (lanes A) and the SDS-elution proteins (lanes B) were analyzed by Western blot for the detection of HMIT, synaptophysin or Vamp2. (C) Fractionation on sucrose density gradient of homogenates of HMIT-myc-expressing PC12, as described in (A).
To determine directly whether HMIT and synaptophysin were present in the same membrane compartment, we immunoprecipitated HMIT- and synaptophysin-containing vesicles from whole-brain homogenates and performed Western blot analysis (Figure 2B). Immunoprecipitated vesicles were first solubilized in Triton X-100 and the proteins remaining associated with the immunoprecipitation beads were then released in the presence of SDS-containing buffer. Both the Triton X-100-solubilized fraction (Figure 2B, lanes A) and SDS-released proteins (Figure 2B, lanes B) were analyzed for the presence of HMIT, synaptophysin and VAMP2. The data show that vesicles immunopurified with synaptophysin antibodies contained synaptophysin and VAMP2, but were devoid of HMIT. Inversely, HMIT vesicles contained the symporter but did not contain synaptophysin or VAMP2. Together, the above data show that brain HMIT is present in a compartment distinct from SVs, early endosomes and lysosomes.
HMIT subcellular localization in PC12 cells
To study HMIT trafficking, we stably transfected PC12 cells with a cDNA construct encoding HMIT, tagged with a myc epitope inserted in the extracellular loop present between transmembrane domains 9 and 10. Subcellular localization of HMIT-myc was first evaluated by centrifugation of cell homogenates on sucrose density gradients. As shown in Figure 2C, HMIT was present mostly in fractions 2–6, a distribution similar to that of Na/KATPase. The HMIT-containing fractions were clearly distinct from those containing Lamp1 and EEA1 and partially overlapped the synaptophysin-containing fractions. These results show that the subcellular distribution of HMIT is very similar in PC12 and brain. By confocal immunofluorescence microscopy, HMIT showed a broad intracellular distribution (Figure 3), both in the cell body and in growth cones, but no cell surface expression as assessed with anti-myc antibodies in the absence of cell permeabilization (see Figures 4 and 5). Costaining of HMIT and synaptophysin or chromogranin A (Figure 3) showed a mostly distinct localization of the symporter and the SV or dense-core granule markers. No colocalization with calreticulin, EEA1 or Lamp1 could be observed. Thus when expressed in PC12 cells, HMIT was also associated with an intracellular compartment distinct from SVs and large dense-core granules.
Figure 3.
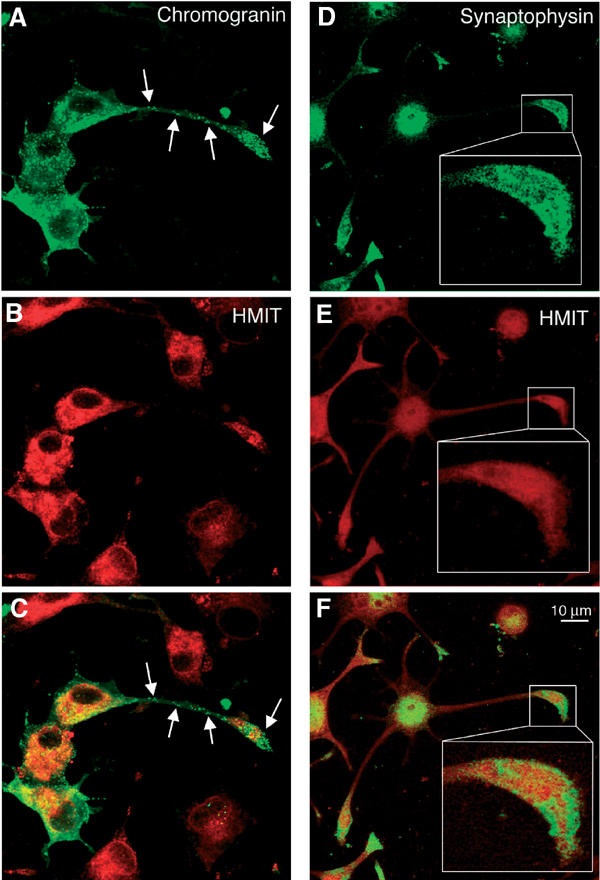
HMIT does not colocalize with chromogranin or synaptophysin in PC12 cells. (A–C) Immunofluorescence microscopy detection of HMIT, chromogranin and merged picture. Arrows point to distinct chromogranin vesiclesthat are devoid of HMIT staining. (D–F) Immunofluorescence microscopy detection of HMIT, synaptophysin and merged picture. The inset shows a growth cone region where HMIT and synaptophysin localization is in distinct structures. Scale bar: 10 μm.
Figure 4.
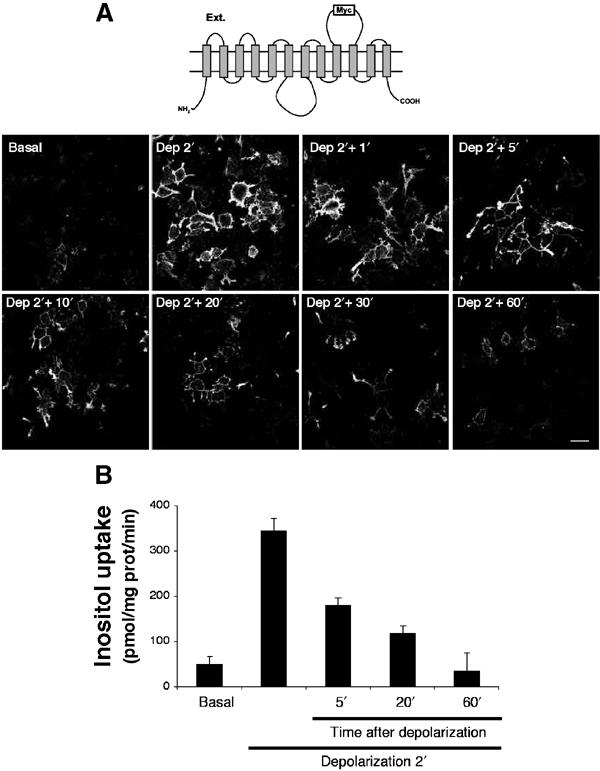
Regulated cell surface expression of HMIT in PC12 cells. (A) Scheme of the structure of HMIT-myc (top). Lower part: Immunofluorescence microscopy detection of cell surface-expressed HMIT-myc on nonpermeabilized cells. HMIT was not detectable on the plasma membrane under basal condition, but was strongly expressed after 2 min of depolarization (Dep 2′). Following transfer of the cells back into the basal medium for the indicated periods of time (min), there is a progressive disappearance of immunostaining, which returns to basal levels at 60 min. Scale bar: 30 μm. (B) Myo-inositol uptake was measured in infected PC12 cells in basal solution, after 2 min of depolarization and quickly washing the cells in basal solution, and after 5, 20 or 60 min of incubation in basal solution following depolarization. The transient increase in myo-inositol uptake parallels the pattern of immunostaining.
Figure 5.
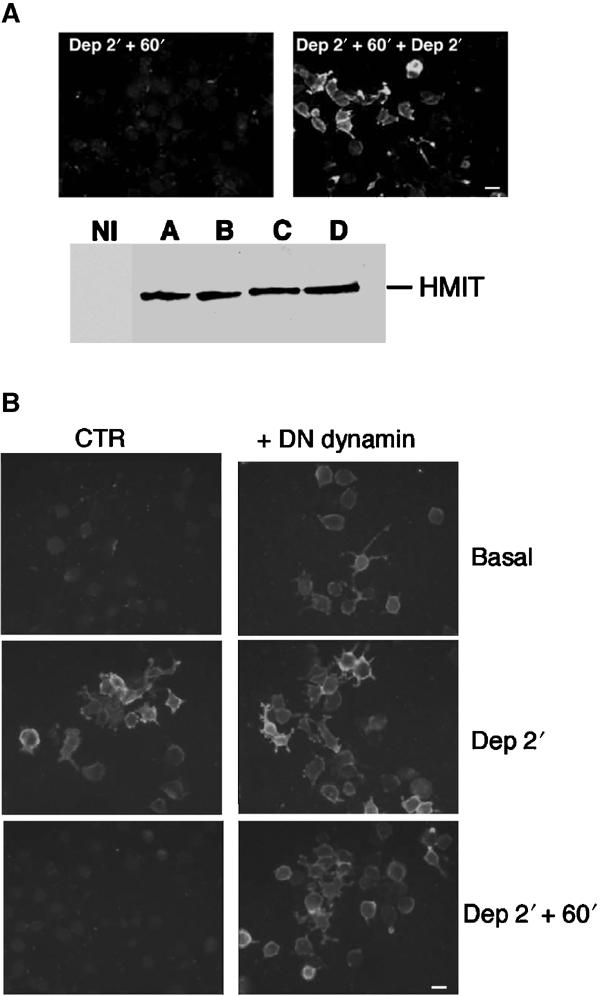
Following surface expression, HMIT is endocytosed in a dynamin-dependent manner and becomes available for a subsequent round of stimulated exocytosis. (A) Upper part: Immunodetection of HMIT on the surface of: (left) cells depolarized for 2 min and returned to basal medium for 60 min or (right) cells exposed to a subsequent 2 min depolarization period. Lower part: HMIT protein content assessed by Western blot in noninfected cells (NI) or in infected cells in basal solution (A), after 2 min of depolarization (B), after 2 min of depolarization followed by 60 min in basal medium (C) and after a subsequent 2 min depolarization. Cellular expression of HMIT is constant over the time course of the experiment. Scale bar: 25 μm. (B) HMIT-expressing PC12 cells were infected or not with a recombinant adenovirus for overexpression of a dominant-negative form of dynamin, under conditions in which 100% of the cells were infected. Under basal conditions, surface expression of HMIT was clearly detectable in the presence of dynaminK44A. Depolarization increased HMIT surface expression in both controlled and dynaminK44A-infected cells. Returning the cells for 60 min to a basal medium was associated with HMIT endocytosis in control cells but not dynaminK44A-expressing cells. These observations indicate a dynamin-dependent mechanism for HMIT endocytosis and suggest that HMIT constitutively recycles between its intracellular storage compartment and the cell surface. Scale bar: 25 μm.
HMIT-stimulated translocation to the cell surface
Next, we evaluated whether HMIT cell surface expression could be triggered by different stimuli. HMIT-expressing PC12 cells were first exposed to 80 mM extracellular potassium and appearance of HMIT on the plasma membrane was revealed by immunostaining of nonpermeabilized cells with anti-myc antibodies (Figure 4A). Under basal conditions, no cell surface staining was detected. At 2 min after initiation of treatment, a strong plasma membrane immunostaining was observed (Figure 4A). Maximal cell surface expression was already seen after less than 10 s of exposure to the high potassium medium, indicating an extremely fast exocytotic process (not shown). Replacing the cells in basal solution after the depolarization period led to progressive disappearance of HMIT-myc surface staining, a process that was complete after 60 min (Figure 4A).
To determine whether surface expression was correlated with an increase in myo-inositol transport, HMIT-expressing cells were depolarized for 2 min or depolarized for 2 min and replaced in basal medium for different periods of time. At each time point, myo-inositol uptake was assayed over a period of 60 s. As shown in Figure 4B, depolarization of the cells induced an increase in myo-inositol uptake from 70 to 350 pmol/mg prot/min. No such increase could be observed in nontransfected PC12 cells (not shown). Myo-inositol uptake measured at different times after replacing the cells in basal medium showed a time-dependent decrease that returned to the basal level after 60 min. This kinetics of myo-inositol uptake paralleled that of HMIT surface expression determined by immunofluorescence microscopy.
We next determined whether disappearance of HMIT from the cell surface 60 min after depolarization was due to symporter internalization and whether HMIT was still available for a subsequent round of exocytosis. HMIT-expressing PC12 cells were thus depolarized for 2 min; following a 60 min incubation in basal medium, the cells were again depolarized for 2 min. Figure 5A shows that a second depolarization very efficiently triggered a new round of HMIT cell surface expression. The total cellular content of HMIT, assessed by Western blot analysis, was unchanged over the time course of the experiment (Figure 5A).
In order to investigate whether HMIT internalization was dependent on dynamin-mediated endocytosis, we infected HMIT-expressing cells with the GTPase-deficient dominant-negative form of dynamin (dynaminK44A) (Ceresa et al, 1998; Kao et al, 1998). Under basal conditions, cell surface expression of HMIT could be clearly detected in cells expressing dynaminK44A but not in control cells. This suggested that HMIT traffics between the cell surface and its intracellular compartment in a dynamin-dependent manner even in the absence of stimuli. After cell depolarization, HMIT plasma membrane expression was markedly increased in PC12 cells expressing or not dynaminK44A. However, whereas the HMIT staining disappeared from the cell surface in control cells, those expressing the dynamin mutant remained strongly positive. These results show that after depolarization, HMIT is internalized by a dynamin-dependent pathway.
To identify the intracellular signals that could trigger exocytosis of HMIT, we next evaluated whether phorbol myristate acetate (PMA), an activator of PKC and also of Munc13 (Brose and Rosenmund, 2002), could induce its cell surface expression. Exposure of the cells to PMA for 10 min produced a very strong increase in HMIT cell surface expression, which was obvious over the entire surface of the cell (Figure 6A). Increasing intracellular Ca2+ by exposure of the cells to the calcium ionophore A23187 also led to a strong cell surface expression of the symporter, which was, however, preferentially associated with growth cones (Figure 6A). Combined treatment of the cells with PMA and the calcium ionophore displayed an additive effect on translocation. In contrast to the above treatments, exposure of the cells to dibutyryl cAMP in the presence of isobutyl-methyl-xanthine (IBMX) did not trigger HMIT translocation nor enhanced PMA- or calcium ionophore-induced HMIT translocation (Figure 6A and not shown).
Figure 6.
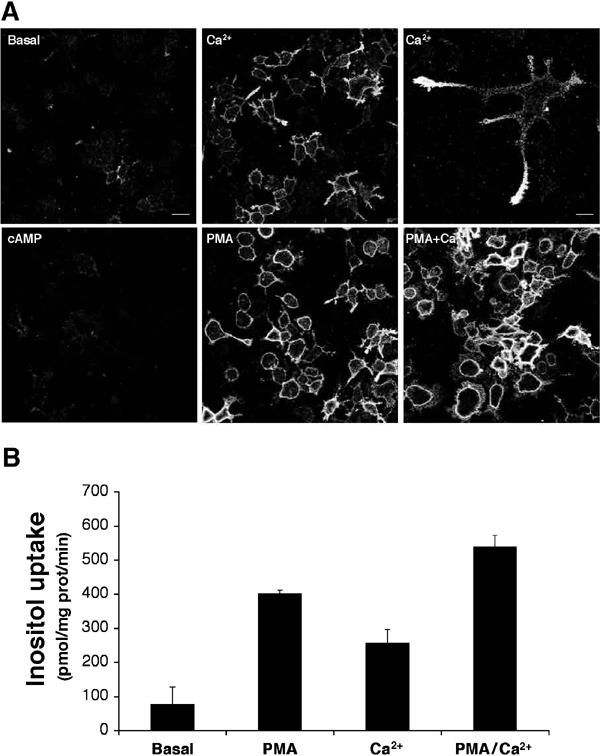
HMIT cell surface expression is triggered by activation of PKC or by influx of Ca2+. (A) Plasma membrane expression of HMIT was assessed under basal conditions (basal), after treatment with the Ca2+ ionophore A23187 (Ca2+), after addition of dbcAMP in the presence of IBMX (cAMP), after activation of PKC by PMA (PMA) or after treatment with both PMA and calcium ionophore (PMA+Ca2+). Calcium ionophore induced plasma membrane expression of HMIT preferentially in growth cones of PC12 cells, as shown at higher magnification in the upper right panel, whereas PMA induced a more homogeneous expression over the entire surface of the cells. Scale bar left 30 μm; high power picture (top right) 10 μm. (B) Myo-inositol uptake was measured under the same condition as that described in (A). A good correlation between cell surface expression and myo-inositol uptake is observed.
Myo-inositol uptake experiments performed under the same stimulatory conditions showed that PMA treatment induced a five-fold increase in myo-inositol uptake over basal conditions (Figure 6B). Calcium ionophore treatment induced a relatively lower increase in myo-inositol uptake, and a maximal uptake rate was obtained after combined treatment with PMA and the Ca2+ ionophore. Thus, increased cell surface expression observed by immunofluorescence microscopy correlated with an increase in myo-inositol uptake.
HMIT translocation in hippocampal neurons and aggregating brain cell cultures
To evaluate whether HMIT translocation could also be evidenced in neurons, we studied translocation in primary culture of neuronal cells using two approaches. Firstly, we measured increased myo-inositol uptake by the endogenous HMIT expressed in primary cultures of brain cells and, secondly, we transiently transfected HMIT-myc in primary culture of hippocampal neurons and studied its expression by immunofluorescence microscopy.
Myo-inositol uptake experiments were performed using neuronal or mixed glial/neuron aggregates. These experiments were carried out in a sodium-free medium and at pH 6, conditions under which transport activity catalyzed by the sodium-myo-inositol transporter (SMIT) could not be detected. Under these conditions, myo-inositol uptake by unstimulated neuronal aggregates was approximately 40 pmol/mg prot/min (Figure 7). After a 10 min treatment with PMA and calcium ionophore, this value was increased to 200 pmol/mg prot/min, indicating HMIT translocation to the plasma membrane. Similar results were obtained when using mixed glial/neuronal aggregates. Furthermore, we could also measure induction of myo-inositol uptake in those cells after depolarization (Figure 7). These data therefore suggested that the endogenous form of HMIT also undergoes stimulation-dependent translocation to the cell surface.
Figure 7.
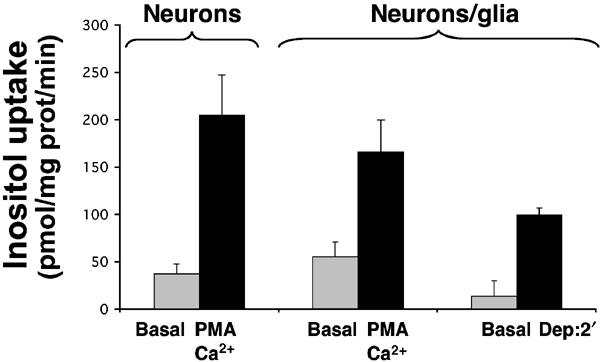
Myo-inositol uptake in aggregating brain cell cultures. Transport of myo-inositol was evaluated under basal condition or after a 10 min of treatment with both PMA and Ca2+ ionophore in neuronal aggregating cell cultures. Uptake experiments were also performed on mixed (neuronal and glial) aggregating cell culture. In addition, in those latter cells, myo-inositol uptake was measured after 2 min of depolarization. Both PMA and Ca2+ ionophore treatments (10 min) or depolarization by high K+ (2 min) stimulated myo-inositol uptake.
HMIT-myc was then transfected in primary cultures of hippocampal cells. Cell surface expression of HMIT-myc was barely detectable at the plasma membrane when evaluated in the basal culture medium (Figure 8A). However, following a 2 min treatment with the calcium ionophore, a strong cell surface expression was observed (Figure 8B). Higher magnification confocal microscopy revealed that HMIT cell surface expression was not over the entire surface of the neuron, but was strikingly restricted to varicosities (Figure 8C and inset).
Figure 8.
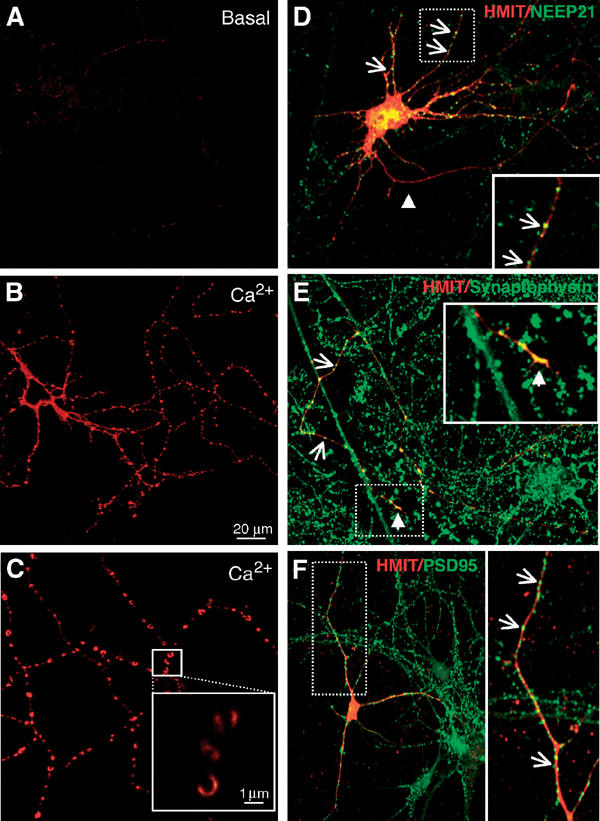
Translocation of HMIT transiently transfected in hippocampal neurons. (A) Very low HMIT staining was detected on the surface of transfected neurons under basal conditions. (B) Ca2+ ionophore treatment strongly increased cell surface expression of HMIT, which was preferentially associated with varicosities (C and inset). Scale bar: 20 μm. (D–F) HMIT is expressed both in the axons and the somatodendritic domain of transfected hippocampal neurons. (D) Both HMIT and NEEP21, a somatodendritic protein, are present in the somatodendritic region and appear as dot-like structures (arrows). Dendrites are colabeled with HMIT and NEEP21, indicating the expression of HMIT in these structures. The inset shows a high magnification of the selected dendrite region (broken line). The arrowhead points to the axon, which is HMIT positive but NEEP21 negative. HMIT is thus present in both the axon and dendrites. (E) Costaining of HMIT and synaptophysin. The figure shows the staining of HMIT in an axon and its association with synaptophysin-positive structures along the axon (arrows) and at the growth cone (arrowhead). The region of the growth cone (broken line) is enlarged in the inset. HMIT is thus present both with varicosities and growth cones. (F) Colocalization of HMIT and PSD95, a marker of the postsynaptic densities. PSD95 is localized to the dendrites of the hippocampal neurons where HMIT is also present. HMIT localization is, however, broader than that of PSD95. Scale bar: 20 μm.
Finally, to evaluate whether HMIT was present in pre- or postsynaptic regions, we performed colocalization studies in transiently transfected hippocampal neurons. Figure 8D shows costaining for HMIT-myc and NEEP21, a somatodendritic marker (Steiner et al, 2002). HMIT was present in the dendrites as revealed by colocalization with NEEP21 (Figure 8D and inset) and in the axon, which is NEEP21 negative (Figure 8D, arrowhead). HMIT was also present along the axon at sites where synaptophysin is detected (Figure 8E, arrows) and at the growth cone (Figure 8E, arrowhead and insert). Thus, HMIT is present both in varicosities and growth cones in addition to a more extensive distribution along the length of the axon. Figure 8F shows that HMIT localization in the dendrites overlaps with sites of PSD95 (a marker of postsynaptic density) staining (Figure 8F, inset), indicating that it is also present in the postsynaptic area.
Discussion
The present data demonstrate that in neurons and in PC12 cells, HMIT resides in an intracellular compartment, distinct from SV and DCV, that can be rapidly translocated to the cell surface. This translocation occurs preferentially at growth cones and varicosities and leads to increased uptake of myo-inositol. These observations provide important new information about the complexity of vesicular traffic in neuronal cells, and uncover a novel mechanism in the regulation of myo-inositol metabolism in neurons.
HMIT subcellular localization and regulated exocytosis
Subcellular fractionation of brain homogenates on sucrose density gradients revealed that HMIT vesicles are clearly distinct from Lamp1-positive lysosomes and EEA1-positive endocytic vesicles. Vesicle immunoprecipitation and Western blot analysis further showed that HMIT was in a compartment distinct from synaptophysin-containing vesicles. A similar distribution of HMIT was found in transfected PC12 cells where immunofluorescence confocal microscopy further confirmed that HMIT was not associated with synaptophysin and chromogranin A. Thus, both in the brain and in PC12 cells, HMIT is located in an intracellular compartment distinct from synaptic-like vesicles and dense-core granules.
The intracellular vesicles in which HMIT is located can be triggered to fuse with the plasma membrane to increase cell surface expression of the symporter and activate myo-inositol uptake. This was demonstrated first in PC12 cells, where depolarization of the cells by high extracellular K+ induced a rapid expression of HMIT-myc to the cell surface. Upon repolarization of the plasma membrane, HMIT was progressively re-internalized by a dynamin-dependent process, which indicates the involvement of either clathrin-coated pits or of caveolae or raft-dependent endocytotic pathways (Nabi and Le, 2003). In the presence of dynaminK44A, HMIT was also expressed at the plasma membrane in the absence of stimuli, suggesting a basal rate of HMIT trafficking between the plasma membrane and its intracellular storage compartment. This is in agreement with the presence of internalization signals in the amino- and carboxy-terminal tails of the symporter, which, when mutated, led to surface expression of HMIT (Uldry et al, 2001). Finally, after endocytosis, HMIT is directed to a compartment from which it can be readily mobilized for a novel translocation cycle.
Surface expression of HMIT was also triggered efficiently by PMA and by increasing the intracellular Ca2+ concentrations with the calcium ionophore A23187. Phorbol esters activate PKC, but can also directly activate exocytosis by interaction with Munc13 (Brose and Rosenmund, 2002). Our present data cannot distinguish between these two mechanisms. Nevertheless, the pattern of appearance of HMIT at the cell surface upon depolarization and Ca2+ stimulation was different from that stimulated by PMA with a more pronounced localization at growth cones with Ca2+ and a more homogeneous distribution over the cell surface with PMA. The translocation of HMIT to the cell surface at growth cones corresponds to the observed preferential increase in [Ca2+]i in these cellular regions following K+ depolarization of PC12 cells (Lorenzon et al, 1995). However, because treatment of the cells with Ca2+ ionophore, which is expected to increase similarly [Ca2+]i over the entire cell area, also stimulates a preferential translocation at growth cones, it is possible that translocation requires Ca2+-sensitive exocytotic proteins preferentially located in these regions.
Translocation of HMIT and increase in myo-inositol uptake could also be demonstrated to occur in primary cultures of neurons. Indeed, translocation to the cell surface of HMIT-myc transiently transfected in hippocampal neurons could be induced by the same stimuli used in PC12 cells. In addition, these experiments demonstrated a strong preferential translocation of HMIT to restricted cell surface areas that could be associated with both pre- and postsynaptic regions, as revealed by the presence of HMIT in regions of the axons, where synaptophysin is detected, and in the dendrites, where PSD95 is found. These translocation events were shown, using primary neuron or mixed glial cells/neuron aggregates, to lead to increased myo-inositol uptake. Together, these data indicate that HMIT expressed in neuronal cells can be triggered to reach the cell surface where it catalyzes myo-inositol uptake and support the hypothesis that its stimulus-dependent translocation may play a role in some aspects of synaptic activity.
Importance of myo-inositol uptake in the control of inositol metabolism
Cellular uptake of myo-inositol represents the only source of inositol for neurons besides recycling from inositol phosphates. Inositol uptake has previously been reported to occur mostly through the Na+-dependent inositol symporter SMIT1 and, more recently, SMIT2 (Coady et al, 2002). Furthermore, expression of SMIT1 is upregulated by osmolarity and its role is thought to be, in large part, to stimulate the uptake of inositol, which acts as an osmolyte, during osmotic shock. However, recently, genetic inactivation of SMIT1 by gene targeting in the mouse has been shown to lead to early death associated with a reduction in brain inositol content and apnea, possibly caused by dysfunction of the respiration center of the brainstem (Berry et al, 2003). HMIT, which has no sequence similarity with SMIT1 and SMIT2, presents the unique functional characteristic of being an H+-dependent symporter activated by decreasing extracellular pH, whereas SMIT1 is inhibited by low pH. Furthermore, HMIT surface expression is dependent on cell depolarization, influx of Ca2+ and PKC activation, whereas cell surface expression of SMIT is constitutive.
The regulation of substrate uptake by stimulus-dependent translocation of transporters to the plasma membrane has been described in other instances. For example, water reabsorption in the kidney collecting duct is stimulated by vasopressin-triggered translocation of aquaporin to the apical membrane of specialized epithelial cells (Nielsen et al, 1995), and also the increase in glucose uptake by adipose and muscle cells upon insulin stimulation, which is controlled by the translocation of the GLUT4 glucose transporter from an intracellular storage site to the plasma membrane (Watson and Pessin, 2001; Bryant et al, 2002). Numerous studies, in particular based on GLUT4 gene inactivation or overexpression, have demonstrated the critical importance of controlled surface expression of GLUT4 in the regulation of not only adipose and muscle glucose uptake and metabolism but also of whole-body glucose homeostasis (Galuska et al, 1998). By analogy with these previous examples, the present description of an activity-dependent translocation of HMIT to the cell surface of specialized regions of neurons suggests an important physiological role for this mechanism. This may be linked to the very high rates of membrane exo- and endocytosis in growth cones and at synapses, which require active PdtIns synthesis and degradation. Under these particular conditions, recycling of inositol phosphates may not provide sufficient inositol for sustained rates of phosphatidylinositol synthesis; consequently, uptake of inositol from the external medium may be required. As compared to aquaporin and GLUT4, however, a further point of control is exerted on HMIT activity, since uptake is strongly stimulated by decreases in extracellular pH. This means that HMIT exposed to the plasma membrane will be mostly active at sites of extracellular medium acidification, such as those associated with synaptic activity (Chesler and Kaila, 1992).
In summary, we have demonstrated that HMIT is present in a vesicular compartment, distinct from SV and DCV, which undergoes activity-dependent translocation to, and fusion with, the plasma membrane. Increased surface expression of the symporter enhances inositol uptake activity. As this activity is pH-dependent, uptake preferentially increases at the site of low extracellular pH such as generated by synaptic activity or in the vicinity of metabolically active glial cells. Finally, as inositol metabolism and production of phosphoinositides is required for the control of vesicle endo- and exocytosis, microfilament dynamics, ion channel regulation and signal transduction, the regulated surface expression of HMIT may represent an important control step in these mechanisms.
Materials and methods
Primary cultures of brain cells
Aggregates. Sprague–Dawley rats (OFA strain) were obtained from Charles River Laboratories, IFFA CREDO, France. The chemicals used were of the highest purity available. The culture medium used was Dulbecco's modified Eagle's medium, prepared from powdered media (Gibco-BRL, cat. no. 52100-039) containing D-glucose (4.5 g/l) but no pyruvate. The culture medium and buffers used for cell culture were controlled for pH (pH 7.3–7.4) and osmolarity (330–340 mOsm). The culture medium was supplemented with 0.8 μM insulin (Sigma, St Louis, MO, USA), 30 nM triiodothyronine (Sigma), 20 nM hydrocortisone-21-phosphate (Sigma), 1 μg/ml transferrin (Sigma), 4 μM biotin (Life Technologies), 1 μM vitamin B12 (Fluka AG, Buchs, Switzerland), 10 μM linoleate (Sigma), 1 μM lipoic acid (Sigma), 10 μM L-carnitine (Fluka), trace elements (Honegger et al, 1979) and 25 μg/ml gentamicin sulfate (Sigma). Aggregating cell cultures were prepared from mechanically dissociated telencephalon of 16-day rat embryos as described in detail elsewhere (Honegger et al, 1979). All cultures were initiated and grown in a serum-free, chemically defined medium under constant giratory agitation (80 rpm) at 37°C and in an atmosphere of 10% CO2/90% humidified air. Neuron-enriched aggregate cultures were obtained by the treatment of mixed-cell cultures at day 1 with the glial mitogen EGF (20 ng/ml) and subsequently, at days 1 and 2, with cytosine arabinoside (0.4 μM) to eliminate the proliferating glioblasts. These cultures contained >90% neurons (Honegger and Pardo, 1999). Glial cell-enriched aggregate cultures were derived from mixed-cell cultures by the treatment at day 7 with cholera toxin (100 nM, from Vibrio cholerae type Inaba 569B, Gibco-BRL, dialyzed against 0.9% NaCl buffered with 0.5 mM HEPES pH 7.4).
Preparation and transient transfection of hippocampal neurons
Hippocampal neurons were prepared from P0 rats. Hippocampi without dentate gyri were dissociated with papain and triturated using a glass pipette. After centrifugation at 400 g for 2 min, cells were plated at 150 000 cells per 35-mm dish containing poly-D-lysin/laminin-coated borosilicate coverslips in DME/10% FCS. Medium was changed after 3 h to Neurobasal/B27 medium. Neurons were transfected after day in culture 9 according to Xia et al (1996) by the calcium phosphate technique. Immunohistochemical analysis was performed 2 days later as described previously (Steiner et al, 2002), except that fixation in the case of HMIT/PSD-95 colabeling was carried out using ice-cold methanol. Antibodies against the following proteins were applied: myc-tag (monoclonal clone 9E10; 1:100), myc-tag (polyclonal; 1:500, Covance, Berkeley, CA), NEEP21 (polyclonal; 1:300; Steiner et al, 2002), synaptophysin (polyclonal; 1:200; Santa Cruz Biotechnology, Santa Cruz, CA), PSD-95 (monoclonal; 1:200; Affinity BioReagents, Golden, CO). Images were taken on a Leica SP2 AOBS confocal microscope, and processed with Photoshop.
Northern blot
Total RNA extracted (Chomczynski and Sacchi, 1987) from aggregated cultured brain cells were run on a 1% denaturating formaldehyde agarose gel and blotted onto nylon membranes (Genescreen, NEN Life Science). Membranes were hybridized with random-primed 32P-labeled probes for HMIT.
Subcellular fractionation
Sucrose density gradients. One mouse brain was homogenized by eight strokes of a Dounce homogenizer in 5 ml of a solution consisting of 0.25 M sucrose, 1 mM EDTA, 10 mM Tris–HCl pH 7.5 and 1 mM PMSF (homogenization buffer). The homogenate was then centrifuged at 1500 rpm (270 g), the pellet was discarded and the supernatant protein concentration was measured by a BCA assay (Pierce). In all, 2.5 mg of protein was loaded on top of a continuous 12.5–50% sucrose gradient containing 1 mM EDTA, 10 mM Tris–HCl pH 7.5, and centrifuged at 4°C for 5 h at 31 000 rpm (165 000 g) in an SW40Ti rotor (Beckman). In total, 12 fractions (1 ml) were collected and the protein and sucrose concentrations were measured. An equal volume of 80 μl of each fraction was then separated by electrophoresis on 10% SDS–polyacrylamide gels and the proteins were transferred onto nitrocellulose membranes for immunodetection using different antibodies as described (Widmann et al, 1995).
Vesicle immunoprecipitation
For each condition, 30 μl of Dynabeads M-280 Sheep anti-rabbit IgG (Dynal) was blocked with 200 μl of 5% BSA in Buffer A (1 mM EGTA, 150 mM NaCl, 0.1 mM MgCl2, 10 mM HEPES, pH 7.4) for 1 h with rotation at room temperature. Beads were then washed three times with 200 μl of Buffer A, and then incubated with 150 μl of Buffer A containing 2 μg of rabbit affinity-purified antibody against the cytoplasmic C-terminal tail of HMIT (Uldry et al, 2001) or 2 μg of rabbit anti-synaptophysin antibody (Santa Cruz Biotechnology) for 12 h at 4°C. Excess antibody solution was then removed and the beads were washed three times with Buffer A. Brain homogenates prepared as described above were adjusted to a protein concentration of 1 mg/ml, and 200 μg was incubated with 30 μl of antibody-coated Dynabeads M-280 for 4 h at 4°C with rotation. The beads were then washed three times with Buffer A and incubated with Buffer A containing 1% Triton X-100 for 1 h at 4°C. The Triton X-100 supernatant was retained and the beads were further washed once with Triton X-100 prior to elution of the remaining antigens with 150 μl of gel electrophoresis 1 × sample buffer (5% SDS, 280 mM β-mercaptoethanol, 50 mM Tris pH 6.8, 12% glycerol) under rotation for 30 min. The first Triton X-100 eluate and the final sample buffer eluate were then analyzed by Western blot analysis as described above.
Expression of HMIT-myc in PC12 cells
A human GLUT4 cDNA containing seven c-Myc epitope tags (GLUT4-myc) in the first exofacial loop was kindly provided by Jonathan S Bogan and Harvey F Lodish. A fragment of 174 pb containing coding sequence for four c-Myc epitopes (position 193–399 of GLUT4-myc cDNA) in tandem was amplified by polymerase chain reaction (PCR) from this plasmid by using specific primers containing additional 5′ and 3′ extension corresponding to positions 1308–1319 and 1371–1382 of HMIT cDNA sequence. A second fragment (position 1100–1319 of HMIT cDNA) was amplified by PCR with specific primers containing additional 3′ extension corresponding to position 193–209 of GLUT4-myc cDNA. Finally, a third fragment (position 1371–1778) of HMIT cDNA containing a 5′ extension corresponding to position 391–399 of GLUT4-myc cDNA was obtained by PCR. A mix of 30 ng of each fragment was used as a template for a fourth PCR reaction to generate a fragment of 823 pb corresponding to position 1100–1778 of HMIT cDNA with four c-Myc epitopes inserted into the exofacial loop between transmembrane domains 9 and 10. This PCR digested product with KpnI and SacII was subcloned in pcDNA3 vector (Invitrogen) containing the wild type and the triple mutant form of HMIT digested with the same enzymes.
The cDNAs for rat HMIT-myc (Uldry et al, 2001), containing a FLAG epitope at its C-terminal end, was subcloned into a lentiviral vector (SIN-18-PGK-WHV) containing a neomycin resistance gene downstream of an internal ribosome entry site (Dr N Déglon, University Hospital, Lausanne, Switzerland). Stocks of lentiviral vectors were prepared by transient transfection of 293T cells and concentrated by centrifugation for 90 min at 19 000 rpm (100 000 g) in an SW28 rotor (Beckman). To obtain stable expression of HMIT-myc, PC12 cells were infected at a multiplicity of infection of 20 for 48 h. Following selection with neomycin, pools of cells demonstrating 100% of expressing cells were selected by immunofluorescence microscopy for further studies.
Myo-inositol uptake
PC12 cells (7 × 105) were plated on 35 mm dish and treated with 50 ng/ml of NGF 2.5S (Alomone labs) for 48 h. Cells were washed twice with K5 solution (5 mM KCl, 127 mM NMDG, 10 mM D-glucose, 1 mM MgCl2 20 mM HEPES, 2.7 mM CaCl2, pH 7.4). For depolarization-induced myo-inositol uptake, cells were incubated for 2 min in the presence of 2 ml of K80-NMDG (80 mM KCl, 53 mM NMDG, 10 mM D-glucose, 20 mM HEPES, 1 mM MgCl2, 2.7 mM CaCl2, pH 7.4) and then subjected to myo-inositol uptake experiment immediately or after 5, 20, 30 or 60 min of incubation in K5-NMDG. For phorbol ester- or Ca2+ influx-induced myo-inositol uptake, PC12 cells were treated with K5-NMDG containing 0.5 μM PMA (Sigma) or 10 μM A23187 (Calbiochem) or both compounds for 10 min before uptake assay. Radioactive myo-inositol uptake experiments were performed in 1 ml K5-NMDG, pH 6.0, containing 10 μCi of myo-[2-3H]inositol (NEN Life Science) and cold myo-inositol at a final concentration of 200 μM for 90 s at 37°C. Cells were then washed four times with 2 ml of PBS containing 1 mM HgCl2 at 4°C and lysed in 1 ml of 5% SDS. A measure of 20 μl of the lysate was kept for protein concentration determination and the rest was mixed with 10 ml of scintillation liquid and counted. Uptake experiments with cultured aggregates were performed as with PC12 cells for 5 min at 37°C. After washing, cells were lysed in 4 M guanidin isothiocynate, 25 mM sodium citrate and 0.5% lauryl sarcosin pH 7.4. A measure of 20 μl of the lysate solution was kept for protein concentration determination and the rest was mixed with 10 ml of scintillation liquid and counted.
Immunocytochemistry
Detection of HMIT-myc on nonpermeabilized cells was performed after incubation of the cells in K5 solution for 10 min at 37°C or after pretreatment for 2 min with K80 at 37°C followed or not by different periods of time in K5 solution. Cells were then immediately incubated in PBS containing 1% BSA at 4°C. Other pretreatments were performed in K5 containing either 0.5 μM PMA, 10 μM A23187, or 1 μM dibutyryl cyclic-AMP (dbcAMP) and 0.5 mM IBMX. Immunodetection of the myc epitope was carried out for 1 h at 4°C with the first antibody (mouse monoclonal anti-myc antibody, 10 μg/ml (Roche)) and for 45 min with CY3-conjugated goat anti-mouse antibody (Jackson ImmunoResearch). Cells were washed three times for 3 min in PBS at 4°C and fixed with 4% paraformaldehyde in PBS for 15 min. Cells were then mounted in 60% glycerol, 2% n-propyl gallate, 0.2 M Tris–HCl pH 8.0 for immunofluorescence microscopy observation either with a Zeiss Axiophot immunofluorescence microscope or with a Leica TCS NT confocal microscope. Images acquired by confocal microscopy were with identical conditions of optical section thickness and other parameters such as laser intensity.
For immunodetection of intracellular antigens, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 in PBS for 2 min. Detection of antigens was then performed for 1 h at 4°C followed by a 45 min incubation with the secondary antibodies, which were CY3-conjugated goat anti-mouse and fluorescein-conjugated goat anti-rabbit second antibodies. Two separate confocal images of the stained cells for the red and green channels were acquired, and then superimposed by using Adobe Photoshop software.
Acknowledgments
We thank Karin Pierre for her help with handling of the confocal microscope, Michel Dallaporta for his very precious advice and Jeffrey Pessin for the dynaminK44A adenovirus. This work was supported by Swiss National Foundation grant 3100-065219-01 to Bernard Thorens.
References
- Berridge MJ (1993) Inositol trisphosphate and calcium signalling. Nature 361: 315–325 [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Downes CP, Hanley MR (1989) Neural and developmental actions of lithium: a unifying hypothesis. Cell 59: 411–419 [DOI] [PubMed] [Google Scholar]
- Berry GT, Wu S, Buccafusca R, Ren J, Gonzales LW, Ballard PL, Golden JA, Stevens MJ, Greer JJ (2003) Loss of murine Na+/myo-inositol cotransporter leads to brain myo-inositol depletion and central apnea. J Biol Chem 278: 18297–18302 [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C (2002) Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci 115: 4399–4411 [DOI] [PubMed] [Google Scholar]
- Bryant NJ, Govers R, James DE (2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3: 267–277 [DOI] [PubMed] [Google Scholar]
- Ceresa BP, Kao AW, Santeler SR, Pessin JE (1998) Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways. Mol Cell Biol 18: 3862–3870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER (2002) Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat Rev Mol Cell Biol 3: 498–508 [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K (1992) Modulation of pH by neuronal activity. Trends Neurosci 15: 396–402 [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal Biochem 162: 156–159 [DOI] [PubMed] [Google Scholar]
- Coady MJ, Wallendorff B, Gagnon DG, Lapointe JY (2002) Identification of a novel Na+/myo-inositol cotransporter. J Biol Chem 277: 35219–35224 [DOI] [PubMed] [Google Scholar]
- Cremona O, De Camilli P (2001) Phosphoinositides in membrane traffic at the synapse. J Cell Sci 114: 1041–1052 [DOI] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, McCormick DA, De Camilli P (1999) Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell 99: 179–188 [DOI] [PubMed] [Google Scholar]
- Czech MP (2000. PIP2 and PIP3: complex roles at the cell surface. Cell 100: 603–606 [DOI] [PubMed] [Google Scholar]
- Gaidarov I, Chen Q, Falck JR, Reddy KK, Keen JH (1996) A functional phosphatidylinositol 3,4,5-trisphosphate/phosphoinositide binding domain in the clathrin adaptor AP-2 alpha subunit. Implications for the endocytic pathway. J Biol Chem 271: 20922–20929 [DOI] [PubMed] [Google Scholar]
- Galuska D, Ryder J, Kawano Y, Charron MJ, Zierath JR (1998) Insulin signaling and glucose transport in insulin resistant skeletal muscle. Special reference to GLUT4 transgenic and GLUT4 knockout mice. Adv Exp Med Biol 441: 73–85 [DOI] [PubMed] [Google Scholar]
- Hay JC, Fisette PL, Jenkins GH, Fukami K, Takenawa T, Anderson RA, Martin TF (1995) ATP-dependent inositide phosphorylation required for Ca(2+)-activated secretion. Nature 374: 173–177 [DOI] [PubMed] [Google Scholar]
- Honegger P, Lenoir D, Favrod P (1979) Growth and differentiation of aggregating fetal brain cells in a serum-free defined medium. Nature 282: 305–308 [DOI] [PubMed] [Google Scholar]
- Honegger P, Pardo B (1999) Separate neuronal and glial Na+, K+-ATPase isoforms regulate glucose utilization in response to membrane depolarization and elevated extracellular potassium. J Cereb Blood Flow Metab 19: 1051–1059 [DOI] [PubMed] [Google Scholar]
- Kao AW, Ceresa BP, Santeler SR, Pessin JE (1998) Expression of a dominant interfering dynamin mutant in 3T3L1 adipocytes inhibits GLUT4 endocytosis without affecting insulin signaling. J Biol Chem 273: 25450–25457 [DOI] [PubMed] [Google Scholar]
- Lorenzon P, Zacchetti D, Codazzi F, Fumagalli G, Meldolesi J, Grohovaz F (1995) Ca2+ waves in PC12 neurites: a bidirectional, receptor-oriented form of Ca2+ signaling. J Cell Biol 129: 797–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra B, Myszka DG, Prestwich GD (2000) Binding kinetics and ligand specificity for the interactions of the C2B domain of synaptotagmin II with inositol polyphosphates and phosphoinositides. Biochemistry 39: 9679–9686 [DOI] [PubMed] [Google Scholar]
- Nabi IR, Le PU (2003) Caveolae/raft-dependent endocytosis. J Cell Biol 161: 673–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S, Chou CL, Marples D, Christensen EI, Kishor BK, Knepper MA (1995) Vasopressin increases water permeability of kidney collecting duct by inducing translocation of aquaporin-CD water channels to plasma membrane. Proc Natl Acad Sci USA 92: 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JE, Turner RS, Agranoff BW, Fisher SK (1999) Differentiated human NT2-N neurons possess a high intracellular content of myo-inositol. J Neurochem 72: 1431–1440 [DOI] [PubMed] [Google Scholar]
- Ohara-Imaizumi M, Fukuda M, Niinobe M, Misonou H, Ikeda K, Murakami T, Kawasaki M, Mikoshiba K, Kumakura K (1997) Distinct roles of C2A and C2B domains of synaptotagmin in the regulation of exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci USA 94: 287–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne SL, Meunier FA, Schiavo G (2001) Phosphoinositides as key regulators of synaptic function. Neuron 32: 9–12 [DOI] [PubMed] [Google Scholar]
- Rapoport I, Miyazaki M, Boll W, Duckworth B, Cantley LC, Shoelson S, Kirchhausen T (1997) Regulatory interactions in the recognition of endocytic sorting signals by AP-2 complexes. EMBO J 16: 2240–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renden R, Berwin B, Davis W, Ann K, Chin CT, Kreber R, Ganetzky B, Martin TF, Broadie K (2001) Drosophila CAPS is an essential gene that regulates dense-core vesicle release and synaptic vesicle fusion. Neuron 31: 421–437 [DOI] [PubMed] [Google Scholar]
- Saiardi A, Sciambi C, McCaffery JM, Wendland B, Snyder SH (2002) Inositol pyrophosphates regulate endocytic trafficking. Proc Natl Acad Sci USA 99: 14206–14211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi AS, Wehland J (2000) The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J Cell Sci 113: 3685–3695 [DOI] [PubMed] [Google Scholar]
- Steiner P, Sarria JC, Glauser L, Magnin S, Catsicas S, Hirling H (2002) Modulation of receptor cycling by neuron-enriched endosomal protein of 21 kD. J Cell Biol 157: 1197–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Hille B (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35: 507–520 [DOI] [PubMed] [Google Scholar]
- Uldry M, Ibberson M, Horisberger JD, Chatton JY, Riederer BM, Thorens B (2001) Identification of a mammalian H(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J 20: 4467–4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RT, Pessin JE (2001) Subcellular compartmentalization and trafficking of the insulin-responsive glucose transporter, GLUT4. Exp Cell Res 271: 75–83 [DOI] [PubMed] [Google Scholar]
- Wenk MR, Pellegrini L, Klenchin VA, Di Paolo G, Chang S, Daniell L, Arioka M, Martin TF, De Camilli P (2001) PIP kinase Igamma is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron 32: 79–88 [DOI] [PubMed] [Google Scholar]
- Widmann C, Dolci W, Thorens B (1995) Agonist-induced internalization and recycling of the glucagon-like peptide-1 receptor in transfected fibroblasts and in insulinomas. Biochem J 310: 203–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann C, Schafer T, Burger MM (1996) Chromaffin granule-associated phosphatidylinositol 4-kinase activity is required for stimulated secretion. EMBO J 15: 2094–2101 [PMC free article] [PubMed] [Google Scholar]
- Williams RS, Cheng L, Mudge AW, Harwood AJ (2002) A common mechanism of action for three mood-stabilizing drugs. Nature 417: 292–295 [DOI] [PubMed] [Google Scholar]
- Williams RS, Harwood AJ (2000) Lithium therapy and signal transduction. Trends Pharmacol Sci 21: 61–64 [DOI] [PubMed] [Google Scholar]
- Wong YH, Kalmbach SJ, Hartman BK, Sherman WR (1987) Immunohistochemical staining and enzyme activity measurements show myo-inositol-1-phosphate synthase to be localized in the vasculature of brain. J Neurochem 48: 1434–1442 [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME (1996) Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci 16: 5425–5436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Majerus PW (1998) Phosphatidylinositol signalling reactions. Semin Cell Dev Biol 9: 153–160 [DOI] [PubMed] [Google Scholar]