

Abstract
The aim of this study was to develop a physiologically based pharmacokinetic (PB-PK) model capable of describing and predicting terbinafine concentrations in plasma and tissues in rats and humans. A PB-PK model consisting of 12 tissue and 2 blood compartments was developed using concentration-time data for tissues from rats (n = 33) after intravenous bolus administration of terbinafine (6 mg/kg of body weight). It was assumed that all tissues except skin and testis tissues were well-stirred compartments with perfusion rate limitations. The uptake of terbinafine into skin and testis tissues was described by a PB-PK model which incorporates a membrane permeability rate limitation. The concentration-time data for terbinafine in human plasma and tissues were predicted by use of a scaled-up PB-PK model, which took oral absorption into consideration. The predictions obtained from the global PB-PK model for the concentration-time profile of terbinafine in human plasma and tissues were in close agreement with the observed concentration data for rats. The scaled-up PB-PK model provided an excellent prediction of published terbinafine concentration-time data obtained after the administration of single and multiple oral doses in humans. The estimated volume of distribution at steady state (Vss) obtained from the PB-PK model agreed with the reported value of 11 liters/kg. The apparent volume of distribution of terbinafine in skin and adipose tissues accounted for 41 and 52%, respectively, of the Vss for humans, indicating that uptake into and redistribution from these tissues dominate the pharmacokinetic profile of terbinafine. The PB-PK model developed in this study was capable of accurately predicting the plasma and tissue terbinafine concentrations in both rats and humans and provides insight into the physiological factors that determine terbinafine disposition.
Terbinafine is an antifungal agent from the allylamine class that inhibits the fungal squalene epoxidase enzyme, leading to the intracellular accumulation of squalene, which causes the rapid death of fungi (36, 37). It has demonstrated activity against most superficial fungal infections, including onychomycosis and dermatomycosis (11, 17), and systemic fungal infections, such as histoplasmosis (1), Pneumocystis carinii infection, (8), and aspergillosis (39). Terbinafine is highly lipophilic and keratophilic, so it is extensively distributed throughout adipose tissue, dermis, epidermis, and nails in humans (12, 19). The apparent volume of distribution in humans is relatively large and has been reported to be in the range of 780 to 2,000 liters (22, 25, 27, 28, 33). The large volume of distribution of terbinafine and its accumulation in peripheral tissue as well as the slow redistribution of the drug into blood are likely to significantly influence the half-life of terbinafine. Previous studies have variously reported the elimination half-life of terbinafine in humans to be 15 h (33), 26 h (27), 290 h (35), and 22 days (33, 35, 43). Terbinafine is extensively metabolized in the liver by oxidation or N demethylation of the three carbon atoms bound to the central nitrogen atom or of the nitrogen atom itself (2), and the metabolites lack the antimycotic activity of the parent drug (34).
Physiologically based pharmacokinetic (PB-PK) models provide the ability to relate the pharmacokinetic behavior of a drug to its physiochemical properties. These models can also predict variations in the drug concentration in particular tissues of interest as a function of whole-body pharmacokinetics. This study aimed to establish a PB-PK model for evaluating terbinafine pharmacokinetic data for rats and humans in an informative way. The PB-PK model presented in this study describes the pharmacokinetics of terbinafine in rats by use of physiological, anatomical, and pharmacokinetic data and enables the prediction of concentration-time data for terbinafine in humans through interspecies scaling.
MATERIALS AND METHODS
Terbinafine concentration data for rats.
We have previously reported the concentration-time data for terbinafine in rat plasma and tissues (19). Briefly, these data were collected from 33 male Sprague-Dawley rats after intravenous (i.v.) bolus administration of 6 mg of terbinafine per kg of body weight. Rats were sacrificed in groups of three at 11 time points (5, 10, 15, 30, and 45 min and 1, 2, 4, 8, 12, and 24 h after terbinafine administration), and their plasma and tissues were sampled, dissected, and stored prior to high-performance liquid chromatography (HPLC) analysis. The terbinafine concentrations in plasma and tissues were determined using a previously described HPLC assay (20). In short, terbinafine and an internal standard (clotrimazole) were extracted from plasma or tissue homogenates at a pH of 9 into hexane and subsequently back extracted from the organic phase into a mixture of 0.5 M sulfuric acid and isopropyl alcohol; an aliquot of this mixture was then directly injected onto the column. The interday and intraday precision levels for terbinafine were between 0.2 and 16%. The limits of quantification of terbinafine for plasma and tissues were 1 ng/ml and 2 ng/g (except for skin tissue [20 ng/g]), respectively, when 200 μl of reconstituted samples was injected into the HPLC system (20).
Terbinafine concentration data for humans.
The pharmacokinetic data for terbinafine in human tissues, including the concentration-time profiles of terbinafine in plasma and skin tissue, were reconstructed from the graphical concentration-time data reported by Kovarik et al. (27), Nedelman et al. (32), and Faergemann et al. (13) and by using the method described by Mayersohn and Tannenbaum (31) for recovering data from the literature. Only those studies with a selective and validated HPLC assay were selected. In those studies, single or multiple oral doses of terbinafine were administered to healthy subjects. In a single-dose study (27), different doses of terbinafine (125, 250, 500, and 750 mg) were administered and blood samples were collected up to 48 h after terbinafine administration. In the multiple-dose studies reported by Faergemann et al. (13) and Nedelman et al. (32), 250 mg of terbinafine was administered to healthy subjects once daily for 14 days. Faergemann et al. (13) collected blood and tissue samples (including stratum corneum, dermis, epidermis, hair, and nail samples) up to 68 days after the cessation of therapy, whereas Nedelman et al. (32) collected only blood samples on days 8 and 15 at specified time points up to 24 h.
Estimation of Kp.
The tissue-to-plasma partition coefficients (Kp) for all tissues following i.v. bolus administration of terbinafine were reported previously (19). This value was determined by comparison of the areas under the concentration-time curves for terbinafine in tissues and arterial plasma, as described previously by Gallo et al. (15). The Kp estimates used in this study are presented in Table 1.
TABLE 1.
Physiological parameters of tissues in a 250-g rat and a 70-kg human and Kp of terbinafine for various rat tissuesg
| Tissue | Ratd
|
Humane
|
Kpf | ||
|---|---|---|---|---|---|
| VT (ml) | QT (ml/min) | VT (liter) | QT (liter/h) | ||
| Adipose | 10.0 | 0.4 | 10.0 | 15.6 | 39.9 |
| Skin | 40.0 | 5.8 | 7.8 | 18.0 | 40.5 |
| Small intestine | 11.4 | 7.5 | 1.7 | 66.0 | 3.1 |
| Stomach | 1.1 | 1.1 | 0.2 | 2.3 | 7.1 |
| Testis | 2.5 | 0.5 | 0.04 | NAc | 2.5 |
| Liver | 10.3 | 11.8a | 1.7 | 98.8a | 1.5 |
| Lung | 2.1 | 43 | 1.2 | 314.4 | 2.6 |
| Heart | 1.2 | 3.9 | 0.3 | 9.0 | 1.8 |
| Kidney | 3.7 | 9.2 | 0.3 | 66.0 | 2.8 |
| Brain | 1.2 | 1.3 | 1.4 | 42.0 | 1.2 |
| Spleen | 0.6 | 0.6 | 0.2 | 4.6 | 1.4 |
| Muscle | 122 | 7.5 | 30.0 | 45.0 | 1.0 |
| Arterial blood | 11.3 | 43 | 1.8 | NA | NA |
| Venous blood | 5.6 | 43 | 3.6 | NA | NA |
| Hepatic artery | NA | 2.0 | NA | 18.0 | NA |
| Portal vein | NA | 9.8b | NA | 80.8b | NA |
Sum of the hepatic-artery and portal-vein blood flow rates.
Sum of the rates of blood flow to the small intestine, spleen, and stomach.
NA, not available.
Data from reference 19.
VT, tissue volume; QT, blood flow rate.
PB-PK model.
The PB-PK model was initially developed using rats. The model consists of 12 tissue and 2 blood compartments, as shown in Fig. 1A. The tissues are connected in parallel between the arterial and venous circulations in this global PB-PK model. The blood passes from the venous pool via the pulmonary artery into the lungs and then out of the lungs via the pulmonary vein into the arterial pool. The lungs in the model close the circulation loop and receive blood at a flow rate equal to that of the cardiac output. Except for the liver, all tissues are supplied from the arterial circulation and blood coming out of the tissues flows directly into the venous circulation. The liver receives its blood supply from the hepatic artery and the portal vein, which itself receives the pooled blood supply from the spleen, stomach, and small intestine. It was assumed in this model that the elimination of terbinafine occurs solely in the liver and that other tissues have no effect on the clearance of terbinafine. Terbinafine was administered by bolus injection into the venous compartment.
FIG. 1.

General structure of the PB-PK model for predicting terbinafine concentrations after i.v. bolus administration (A) and oral administration (B). The arrows show the direction of blood flow. Indicated are the blood flows (Q) to heart (He), brain (Br), kidney (Ki), muscle (Mu), skin (Sk), adipose (Ad), testis (Te), stomach (St), small intestine (SI), and spleen (Sp) tissues and the blood flow to the liver via the hepatic artery (HA) and the hepatic portal vein (HV) as well as the total blood flow out of the liver (H) and the clearance of terbinafine from the liver (CL).
Two structural PB-PK models were investigated and compared in this study. In the first model, all tissues were represented as single well-stirred compartments and the distribution of terbinafine was assumed to be perfusion rate limited (model I). In the second PB-PK model, skin and testis tissues were described as compartments with membrane permeability limitations (19) but distribution through the rest of the tissues was described as being perfusion rate limited (model II). The differential equations describing the PB-PK models are shown in the appendix.
The organ and tissue volumes and blood flow rates for a 250-g rat were obtained from the literature (4, 6, 9) and are listed in Table 1. The blood flow rate was used in this model rather than the plasma flow rate, as it has been shown that in rats and humans, the ratio of the distribution of terbinafine in blood to that in plasma is close to one (21). By use of a hybrid PB-PK model (19), we estimated the permeability surface area products (PS) for rat skin and testis tissues to be 0.016 and 0.053 ml/min, respectively. The value of the terbinafine clearance employed in this study was estimated from the experimental data following administration of an i.v. bolus dose of terbinafine (6 mg/kg) in rats and was found to be 1.7 liters/h/kg (19).
The differential equations listed in the appendix were integrated numerically and simultaneously using a personal computer (Pentium III processor) with the Scientist software program (version 2.0; MicroMath Scientific Software, Salt Lake City, Utah).
Scaled-up results for humans.
The pharmacokinetics of terbinafine in humans were predicted with the PB-PK model after modification of the model structure to allow simulation of the concentration-time profile of terbinafine following oral administration. The modification included the addition of the intestinal lumen compartment to the model as a drug receiver compartment, as described by Carlton et al. (7) (Fig. 1B). This modification required the addition of a differential equation for intestinal lumen and the alteration of the equation for the small intestine, as shown in the appendix. It was assumed that the permeability of cellular membranes in tissues and the structures of organs are conserved among mammals; therefore, the PS of human skin and testis tissues were determined by scaling up the PS obtained for rats with the following allometric equation:
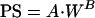 |
(1) |
where W is the body weight and A and B are the coefficient and power function for the allometric relationship, respectively. The coefficient A was estimated from the rat PS values, and B was assumed to be 0.67 (14). By use of this equation, the PS values for human skin and testis tissues were calculated to be 6.98 and 23.12 ml/min, respectively. The Kp values were assumed to be identical among mammals. The organ and tissue volumes and blood flow rates for humans that were used in the global PB-PK model were obtained from the literature (3, 10, 14, 18, 24, 38) and are presented in Table 1.
The estimates of the apparent total clearance of terbinafine that were used in these simulations were obtained from the literature (and were based on the relationship between the area under the concentration-time curve and the dose amount) and ranged from 25 to 40 liters/h (25, 27). For prediction of the pharmacokinetics of terbinafine in humans after oral administration, the bioavailability was assumed to be 80% based on the study by Jensen (25), who reported that the absorbed fraction of terbinafine in the gastrointestinal tract following oral administration of a single 250-mg dose of [14C]terbinafine was about 80%. The hepatic-extraction ratio for terbinafine in humans, calculated as the ratio of the terbinafine clearance to the hepatic blood flow rate, was estimated to be in the range from 0.21 to 0.79. The absorption rate constant (ka) was obtained from the literature and was reported to be in the range from 0.6 to 0.9 h−1 (25). The ka used in the model is a function of both the intestinal permeability and dissolution for terbinafine. The value for the distribution rate constant governing the transfer of terbinafine into the intestinal lumen from the intestinal tissue was assumed to be negligible.
Estimation of Vss.
The volume of distribution at steady state (Vss) was calculated using the data for the plasma terbinafine concentration and was also estimated with the following equation:
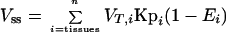 |
(2) |
where VT,i and Kpi are the anatomical tissue volume and tissue-to-plasma partition coefficient, respectively, and Ei is the extraction ratio of the ith tissue. The extraction ratio for noneliminating organs was zero.
RESULTS
Figures 2 and 3 show the predicted concentration-time profiles of terbinafine that were obtained with the global PB-PK model together with the observed concentration-time data from the plasma and tissues of 33 rats after administration of an i.v. bolus dose (6 mg/kg). Generally, there was close agreement between the simulated and observed concentration data over 24 h for tissues and plasma. For plasma and the majority of tissues studied, the accuracy of the PB-PK model simulations was within 3 to 30% of the experimental determinations. The most accurate predictions were those made for the heart, and the model provided the least accurate predictions for the small intestine and testis.
FIG. 2.

Simulated and observed concentration-time profiles for terbinafine in rat plasma and selected tissues after i.v. bolus administration of a 6-mg/kg dose. The solid and dashed lines show the data obtained from simulations with PB-PK models I and II, respectively, and the filled circles and squares indicate the experimental concentration data. The insert plots show the concentration-time profiles in a linear scale up to 10 h.
FIG. 3.

Simulated and observed concentration-time profiles for terbinafine in rat tissues after i.v. bolus administration of a 6-mg/kg dose. The solid and dashed lines show the data obtained from simulations with PB-PK models I and II, respectively, and the filled circles and squares indicate the experimental concentration data. The insert plots show the concentration-time profiles in a linear scale up to 10 h.
In some tissues, such as brain, heart, liver, muscle, and adipose tissues, the model predicted terbinafine concentrations at early times after administration (less than 2 h for adipose tissue and 30 min for other tissues) that were less than the observed terbinafine concentrations. For the small intestine and stomach tissues, the PB-PK model did not provide an adequate description of the experimental observations of the terbinafine concentration over the entire time course of the study.
The terbinafine concentration-time profiles for the testis and skin tissues predicted by the perfusion rate-limited PB-PK model provided a poor description of the experimental data. The use of a PB-PK model with membrane permeability rate limitation provided a significantly improved description of terbinafine uptake into testis tissue.
The concentrations of terbinafine in human plasma after administration of single doses of 125, 250, 500, and 750 mg of terbinafine (27) were compared to those simulated by the PB-PK model that incorporated oral absorption, and the results are presented in Fig. 4. There was excellent agreement between the concentration-time data obtained by observation and those obtained by simulation with the PB-PK model.
FIG. 4.

Simulated and observed concentration-time profiles for terbinafine in human plasma after oral administration of a single dose of 125 (A), 250 (B), 500 (C), or 750 (D) mg of terbinafine. The solid and dashed lines show data obtained from simulations with PB-PK models I and II, respectively, and the filled circles indicate the experimental concentration data, which were obtained from Kovarik et al. (27).
The predicted terbinafine concentrations in various tissues following the administration of a single oral dose of 125, 250, 500, or 750 mg of terbinafine are presented in Fig. 5. In general, the temporal profile of terbinafine concentrations in tissues parallels that observed in plasma. Differences can be observed during the initial distribution, which tends to be slower in some tissues (such as adipose and skin tissues) than in plasma. The maximum concentration of terbinafine in serum (Cmax) was predicted by PB-PK models I and II to be 0.97 and 1.07 μg/ml, respectively. These values are in close agreement with the Cmax reported by Kovarik et al. (1.15 ± 0.66 μg/ml) (27). The Cmaxs based on the model simulations were achieved 1.5 h after administration, while that reported by Kovarik et al. was achieved 1.3 h after administration.
FIG. 5.

Simulated concentration-time profiles obtained with PB-PK model I for terbinafine in some human tissues after oral administration of a single dose of 125, 250, 500, or 750 mg.
The concentrations of terbinafine in human plasma following multiple oral doses were also investigated using the PB-PK model with membrane permeability rate limitations for skin and testis tissues. The terbinafine distribution in human skin tissue and plasma following oral administration of 250 mg of terbinafine for 14 days was accurately predicted during the course of treatment by use of PB-PK model II data scaled up from data from rats (with repeated oral dosing taken into consideration). A comparison of published concentration-time data for humans following administration of 250 mg of terbinafine for 14 days (13, 32) and the simulated concentration data is presented in Fig. 6. The highest terbinafine concentration that was reported for skin tissue following oral administration of 250 mg of terbinafine for 14 days was 7.6 μg/g (13), and the terbinafine concentration predicted by the PB-PK model was in close agreement (approximately 8.0 μg/g).
FIG. 6.

Simulated and observed concentration-time profiles for terbinafine in human plasma and skin tissue during repeated oral administration of 250 mg of terbinafine once a day for 14 days. Indicated are the data obtained from simulations with PB-PK model II (solid lines) and the observed concentration data for skin tissue (•) and plasma (○) obtained from the studies by Faergemann et al. (13) (A) and Nedelman et al. (32) (B).
The apparent volumes of terbinafine distribution for human tissues are shown in Table 2. The Vss values indicate that at steady state almost all of the terbinafine in the body resides in adipose and skin tissues (collectively accounting for 94% of the total distribution).
TABLE 2.
The apparent volumes of distribution of terbinafine in human tissues, with values scaled up from those in rat tissues
| Tissue | Apparent Vss (liter)a | % of total Vssb |
|---|---|---|
| Adipose | 399.2 | 52.3 |
| Skin | 316.1 | 41.4 |
| Small intestine | 5.1 | 0.7 |
| Stomach | 1.1 | 0.1 |
| Testis | 0.1 | 0.01 |
| Liver | 1.3 | 0.2 |
| Lung | 3.1 | 0.4 |
| Heart | 0.5 | 0.1 |
| Kidney | 0.9 | 0.1 |
| Brain | 1.7 | 0.2 |
| Spleen | 0.3 | 0.04 |
| Muscle | 28.8 | 3.8 |
| Arterial blood | 1.8 | 0.2 |
| Venous blood | 3.6 | 0.5 |
Calculations based on equation 2.
Total Vss, 763.5 liters.
DISCUSSION
For the majority of tissues, the perfusion rate-limited PB-PK model provided better descriptions of the experimentally observed concentrations of terbinafine than the membrane permeability rate-limited PB-PK model. The reason for the underestimation of terbinafine concentrations in some tissues by the PB-PK models is unclear, but it could be due to the increase in the heart rates of the rats (and subsequently the cardiac output) as a result of stress (experienced during handling and dose administration). This increase in heart rate of the rats has the potential to lead to an increase in the regional blood flow and the delivery of more of the drug to the tissues than expected. For the small intestine and stomach tissues, the PB-PK model did not provide an adequate description of experimental observations of the terbinafine concentration over the entire time course of the study. This could be due to the more complicated mechanisms (such as permeability limitation or enterohepatic recycling) involved in the movement of terbinafine in these tissues, especially the small intestine. Investigation of the distribution of terbinafine in the small intestine and stomach with the PB-PK hybrid model has shown that the terbinafine concentration profile in these tissues is best described by a perfusion rate-limited hybrid PB-PK model (20). The use of a PB-PK model with membrane permeability rate limitation for the small intestine or a PB-PK model with a more complicated intratissue distribution involving multiple compartments representing the stomach and small intestine was investigated, but neither model produced higher agreement between the simulated and experimentally observed terbinafine concentration data.
The dominant route of elimination of [14C]terbinafine metabolites in rats has been reported to be biliary excretion; about 80% of the radioactive absorbed dose of [14C]terbinafine is eliminated via this route (2). The same investigators found that about 10% of the metabolites initially eliminated with the bile undergo enterohepatic recycling. Thus, the global PB-PK model was modified to investigate the potential effects of enterohepatic recycling on terbinafine disposition and on the concentration-time profile of terbinafine in the stomach and small intestine. In this modified PB-PK model, a fraction of terbinafine was assumed to be secreted via the bile into the intestinal lumen, from where it could be reabsorbed into the systemic bloodstream. It was assumed that the entire amount of secreted drug is subsequently reabsorbed and not excreted with the feces (the differential equations describing this modified PB-PK model for the intestinal lumen, small intestine, and liver are shown in the appendix). However, despite the inclusion of enterohepatic recirculation, this model did not provide a better description of terbinafine concentrations in the small intestine or stomach (data not shown).
The use of a PB-PK model with membrane permeability rate limitation provided a significantly improved description of terbinafine uptake into testis tissue, suggesting that the uptake of terbinafine into this tissue is limited by the transport across the capillary membrane. The capillary-membrane rate limitation of drug transport in rat testis tissue has already been demonstrated (5, 30). For example, the distributions of tenoxicam (30) and a series of barbiturates (5) in rat testis tissue were best described by a permeability rate-limited PB-PK model. Although the existence of a permeability barrier in testis tissue is pharmacokinetically interesting, it is of little clinical interest with respect to terbinafine as testis tissue is not a potential target tissue for this drug. For skin tissue, however, although the perfusion rate-limited PB-PK model did not describe terbinafine distribution at later times (greater than 2 h) after terbinafine administration, it provided good predictions of the drug uptake at early times (up to 2 h). The simulated concentration data for skin from the global PB-PK model with membrane permeability rate limitation for skin tissue showed better agreement with the observed concentration data for skin at times later than 1 h after terbinafine administration and over the remaining time course of the study. The distribution of terbinafine in skin soon after administration (less than 1 h), however, cannot be adequately described with this model. The simulated concentration data for skin from the perfusion rate-limited PB-PK model showed a faster redistribution of terbinafine from skin than did the membrane permeability rate-limited PB-PK model.
Terbinafine has a high affinity for skin and adipose tissues, and these tissues act as a major depot for terbinafine (19). In contrast, terbinafine uptake in muscle, spleen, and brain tissues is significantly less (Kp = 1.0 to 1.4). The slow uptake and efflux of terbinafine in skin and adipose tissues, which lead to the long elimination half-life of terbinafine, made it logistically and ethically impossible to administer a constant-rate infusion of the drug for a sufficiently long time to achieve steady state or to collect samples over a sufficiently long time to fully characterize the pharmacokinetics in peripheral tissues. Therefore, for the purpose of the development of a PB-PK model, the Kp for different tissues were estimated in this study according to the area method described by Gallo et al. (15) and published earlier (19).
To adequately predict the concentrations of terbinafine in plasma following high doses of terbinafine (i.e., 500 and 750 mg), it was necessary to adjust the ka value to 0.3 h−1 to account for the change in the dissolution of terbinafine at the higher doses. The resulting PB-PK models were able to accurately predict the concentrations of terbinafine in plasma following these dosing regimens. According to the PB-PK models, the maximum concentrations of terbinafine at 1.5 h after the administration of single doses of 500 and 750 mg were 1.2 and 1.6 μg/ml, respectively. These values were in close agreement with the previously reported values of 1.1 and 1.3 μg/ml after single doses of 500 and 750 mg, respectively (27).
The terbinafine concentrations in various human tissues following administration of a single oral dose were also predicted. Analysis of the predicted events in tissues clearly showed that the slow and substantial accumulation of terbinafine in skin and adipose tissues has a great influence on the temporal profile of terbinafine concentrations after a single oral dose. There are no data available in the literature regarding the concentrations of terbinafine in visceral and peripheral human tissues following a single oral dose. Thus, the predictions of the PB-PK model could not be directly compared to observed concentration data for these tissues. However, it can be concluded from the prediction of the terbinafine concentration in plasma that the model is able to validly provide a description and prediction of terbinafine pharmacokinetics after a single oral dose.
The concentration-time data for terbinafine in plasma after 7 and 15 days of treatment that were described in the study by Nedelman et al. (32) showed good agreement with the terbinafine concentrations in plasma predicted by the global PB-PK model proposed in this study. The Cmax after the seventh dose of terbinafine was found to be 1.3 μg/ml, which was achieved within 2 h of terbinafine administration. The predicted value from the global PB-PK model was 1.5 μg/ml, which was achieved within 1.5 h of administration. These terbinafine concentrations are also in close agreement with the data presented by Villars and Jones (42) that were obtained after administration of multiple doses of terbinafine.
The estimated Vss for terbinafine in human tissues that was scaled up from that in rat tissues is in close agreement with the Vss in human tissues reported in the literature. The Vss for terbinafine has been reported to be in the range of 11 (33) to 28 (25) liters/kg. Kovarik et al. (27), Humbert et al. (23), and Nejjam et al. (33), however, reported an estimated Vss of approximately 11 liters/kg, which agrees closely with the estimated Vss in human tissues of the present study (10.9 liters/kg).
In spite of the relatively high clearance of terbinafine from plasma, the rapid distribution of terbinafine into tissues (especially adipose and skin tissues) and its slow efflux result in a long elimination half-life. These data indicate that distribution of terbinafine into skin (the target tissue) and adipose tissues dominate the pharmacokinetic profile of this drug.
Terbinafine has a broad spectrum of activity in vitro and in vivo against pathogenic fungi responsible for superficial fungal infections (such as dermatophytosis and onychomycosis) and systemic fungal infections (such as histoplasmosis, aspergillosis, and Pneumocystis carinii infection) (1, 8, 39, 40). The data presented in this study clearly show that terbinafine achieves high and sustained concentrations in skin, which is the target tissue for this drug. The lipophilic and keratophilic nature of terbinafine leads to the accumulation of this drug in adipose and keratinous tissues such as nail, hair, and particularly skin. These characteristics of terbinafine make it the drug of first choice in the treatment of dermatophytosis and onychomycosis (11, 17). Furthermore, it has been shown that systemic treatment with terbinafine is more efficient than topical administration in the treatment of onychomycosis (41).
The rapid and extensive distribution of terbinafine in tissues dominates the pharmacokinetic characteristics of this drug in the body. The slow redistribution of terbinafine from tissues is responsible for the long elimination half-life observed for this drug. Based on these pharmacokinetic characteristics, terbinafine can be expected to provide sustained protection from a relapse of fungal infections following therapy (26); these pharmacokinetic characteristics also provide a rationale for a potentially shorter treatment time (13, 28, 43), which may be more convenient for patients.
A PB-PK model for terbinafine has been developed and validated for rats and successfully scaled up for use with humans. This model was capable of accurately predicting the concentration-time data of terbinafine in both rats and humans. The PB-PK model developed in this study can be used for the evaluation of dose schedules by predicting the concentration of terbinafine in target tissues.
Acknowledgments
M.H.-Y. was supported by a Pharmacy Research Trust Scholarship.
We are grateful to Novartis Pharmaceuticals Australia for the gift of terbinafine and thank Richard Upton for insightful advice on model development.
APPENDIX
Terbinafine concentrations in tissues with blood flow-limited uptake.
(i) Noneliminating tissues.The following differential equation describes the mass balance of terbinafine concentrations in noneliminating tissues (such as muscle, adipose, brain, spleen, kidney, heart, stomach, and small intestine tissues):
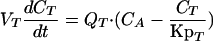 |
(A1) |
where QT and VT represent the blood flow rate and the volume of the tissue, respectively; CA and CT are the terbinafine concentrations in the arterial plasma and tissue, respectively; and KpT is the tissue-to-plasma partition coefficient.
(ii) Liver.For the liver, the equation is
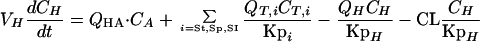 |
(A2) |
where QHA is the hepatic-artery blood flow rate; i represents the stomach (St), spleen (Sp), and small intestine (SI); QH is the sum of the hepatic-artery and portal-vein blood flow rates; CL is the clearance of terbinafine; Kpi is the stomach, spleen, or small intestine tissue-to-plasma partition coefficient; and KpH is the liver tissue-to-plasma partition coefficient.
(ii) Lungs.For the lungs, the equation is
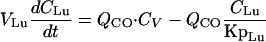 |
(A3) |
where QCO is the cardiac output (sum of all blood flow rates) and CV is the terbinafine concentration in venous plasma.
Terbinafine concentrations in tissues with permeability rate-limited uptake.
(i) Noneliminating tissues.The terbinafine concentrations in noneliminating tissues with permeability rate-limited uptake, such as skin and testis tissues, were determined with the following differential equations:
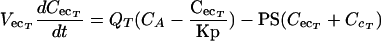 |
(A4) |
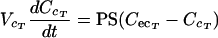 |
(A5) |
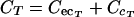 |
(A6) |
where CA and CT are the terbinafine concentrations in the arterial plasma and noneliminating tissues, respectively; VT is tissue volume; PS is the permeability surface area coefficient (in milliliters per minute); CecT and CcT are the extracellular and cellular terbinafine concentrations, respectively; and VecT and VcT represent the volumes of the extracellular and cellular spaces, respectively, of the tissue.
(ii) Venous plasma.For venous plasma, the equation is
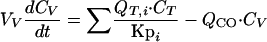 |
(A7) |
where i represents tissues such as brain, heart, kidney, muscle, testis, skin, adipose, and liver tissues; Vv is the volume of the venous blood pool; Cv is the terbinafine concentration in mixed venous plasma; and QCO is the cardiac output.
(iii) Arterial plasma.For arterial plasma, the equation is
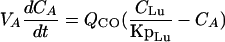 |
(A8) |
where VA is the volume of the arterial blood pool and CLu and CA are the terbinafine concentrations in the lungs and arterial plasma, respectively.
Terbinafine concentrations following oral administration.The following differential equations describe the mass balance of terbinafine concentrations in the intestinal lumen and small intestine following oral administration of terbinafine. (i) For the intestinal lumen, the equation is
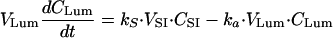 |
(A9) |
(ii) For the small intestine, the equation is
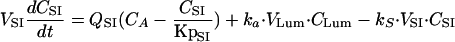 |
(A10) |
where ka and ks are the absorption and reabsorption rate constants, respectively; CLum and CSI are the concentrations of terbinafine in the intestinal lumen and small intestine, respectively; VLum and VSI are the volumes of the intestinal lumen (VLum = 1 liter [7]) and small intestine, respectively; and KpSI is the small intestine tissue-to-plasma partition coefficient.
Consideration of enterohepatic recirculation.The modified differential equations describing the terbinafine concentrations in the liver, small intestine, and intestinal lumen, with enterohepatic recirculation taken into consideration, are as follows. (i) For the liver, the equation is
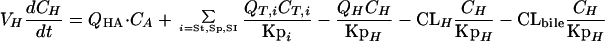 |
(A11) |
(ii) For the intestinal lumen, the equation is
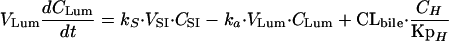 |
(A12) |
(iii) For the small intestine, the equation is
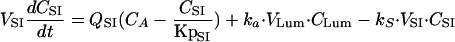 |
(A13) |
where CLbile and CLH are the biliary and hepatic clearances, respectively. For the purpose of these simulations, it was assumed that the biliary clearance is equal to the bile flow rate in rats (0.02 ml/min [29]). The hepatic clearance (7.98 ml/min in a 250-g rat) was equal to the difference between the total clearance (8.0 ml/min in a 250-g rat [19]) and the biliary clearance.
REFERENCES
- 1.Bankole Sanni, R., C. Denoulet, B. Coulibaly, R. Nandiolo, E. Kassi, M. Honde, and M. L. Mobiot. 1998. Apropos of 1 Ivoirian case of osseus and cutaneous histoplasmosis by Histoplasma capsulatum var. duboisii. Bull. Soc. Pathol. Exot. 91:151-153. (In French.) [PubMed] [Google Scholar]
- 2.Battig, F., M. Nefzger, and G. Schulz. 1987. Major biotransformation routes of some allylamine antimycotics, p. 479-495. In R. Fromtling (ed.), Recent trends in the discovery, development and evaluation of antifungal agents. Prous Science Publishers, Barcelona, Spain.
- 3.Bell, G. H., D. Emslie-Smith, and C. R. Paterson. 1976. Textbook of physiology and biochemistry, 9th ed. Churchill Livingstone, Edinburgh, Scotland.
- 4.Bernareggi, A., and M. Rowland. 1991. Physiologic modeling of cyclosporin kinetics in rat and man. J. Pharmacokinet. Biopharm. 19:21-50. [DOI] [PubMed] [Google Scholar]
- 5.Blakey, G. E., I. A. Nestorov, P. A. Arundel, L. J. Aarons, and M. Rowland. 1997. Quantitative structure-pharmacokinetics relationships. I. Development of a whole-body physiologically based model to characterize changes in pharmacokinetics across a homologous series of barbiturates in the rat. J. Pharmacokinet. Biopharm. 25:277-312. [DOI] [PubMed] [Google Scholar]
- 6.Brown, R. P., M. D. Delp, S. L. Lindstedt, L. R. Rhomberg, and R. P. Beliles. 1997. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 13:407-484. [DOI] [PubMed] [Google Scholar]
- 7.Carlton, L. D., G. M. Pollack, and K. L. Brouwer. 1996. Physiologic pharmacokinetic modeling of gastrointestinal blood flow as a rate-limiting step in the oral absorption of digoxin: implications for patients with congestive heart failure receiving epoprostenol. J. Pharm. Sci. 85:473-477. [DOI] [PubMed] [Google Scholar]
- 8.Contini, C., D. Colombo, R. Cultrera, E. Prini, T. Sechi, E. Angelici, and R. Canipari. 1996. Employment of terbinafine against Pneumocystis carinii infection in rat models. Br. J. Dermatol. 134:30-32. [DOI] [PubMed] [Google Scholar]
- 9.Davies, B., and T. Morris. 1993. Physiological parameters in laboratory animals and humans. Pharm. Res. 10:1093-1095. [DOI] [PubMed] [Google Scholar]
- 10.Dedrick, R., D. Forrester, and D. Ho. 1972. In vitro-in vivo correlation of drug metabolism--deamination of 1-beta-d-arabinofuranosylcytosine. Biochem. Pharmacol. 21:1-16. [DOI] [PubMed] [Google Scholar]
- 11.Evans, E. G., and B. Sigurgeirsson. 1999. Double blind, randomised study of continuous terbinafine compared with intermittent itraconazole in treatment of toenail onychomycosis. Br. Med. J. 318:1031-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faergemann, J., H. Zehender, J. Denouel, and L. Millerioux. 1993. Levels of terbinafine in plasma, stratum corneum, dermis-epidermis (without stratum corneum), sebum, hair and nails during and after 250 mg terbinafine orally once per day for four weeks. Acta Dermato-Venereol. 73:305-309. [DOI] [PubMed] [Google Scholar]
- 13.Faergemann, J., H. Zehender, and L. Millerioux. 1994. Levels of terbinafine in plasma, stratum corneum, dermis-epidermis, sebum, hair and nails during and after 250 mg terbinafine orally once daily for 7 and 14 days. Clin. Exp. Dermatol. 19:121-126. [DOI] [PubMed] [Google Scholar]
- 14.Fiserova-Bergerova, V. (ed.). 1983. Modeling of inhalation exposure to vapors, vol. I. CRC Press, Boca Raton, Fla.
- 15.Gallo, J. M., F. C. Lam, and D. G. Perrier. 1987. Area method for the estimation of partition coefficients for physiological pharmacokinetic models. J. Pharmacokinet. Biopharm. 15:271-280. [DOI] [PubMed] [Google Scholar]
- 16.Gallo, J. M., F. C. Lam, and D. G. Perrier. 1991. Moment method for the estimation of mass transfer coefficients for physiological pharmacokinetic models. Biopharm. Drug Dispos. 12:127-137. [DOI] [PubMed] [Google Scholar]
- 17.Gupta, A., and N. Shear. 1997. Terbinafine: an update. J. Am. Acad. Dermatol. 37:979-988. [DOI] [PubMed] [Google Scholar]
- 18.Guyton, A. C., and J. E. Hall. 1996. Textbook of medical physiology, 9th ed. W. B. Saunders, Philadelphia, Pa.
- 19.Hosseini-Yeganeh, M., and A. J. McLachlan. 2001. Tissue distribution of terbinafine in rats. J. Pharm. Sci. 90:1817-1828. [DOI] [PubMed] [Google Scholar]
- 20.Hosseini-Yeganeh, M., and A. J. McLachlan. 2000. Determination of terbinafine in tissues. Biomed. Chromatogr. 14:261-268. [DOI] [PubMed] [Google Scholar]
- 21.Hosseini-Yeganeh, M., and A. J. McLachlan. 2002. In vitro distribution of terbinafine in rat and human blood. J. Pharm. Pharmacol. 46:702-707. [DOI] [PubMed] [Google Scholar]
- 22.Humbert, H., M. Cabiac, J. Denouel, and S. Kirkesseli. 1995. Pharmacokinetics of terbinafine and of its five main metabolites in plasma and urine, following a single oral dose in healthy subjects. Biopharm. Drug Dispos. 16:685-694. [DOI] [PubMed] [Google Scholar]
- 23.Humbert, H., J. Denouel, M. D. Cabiac, H. Lakhdar, and A. Sioufi. 1998. Pharmacokinetics of terbinafine and five known metabolites in children, after oral administration. Biopharm. Drug Dispos. 19:417-423. [DOI] [PubMed] [Google Scholar]
- 24.Igari, Y., Y. Sugiyama, Y. Sawada, T. Iga, and M. Hanano. 1983. Prediction of diazepam disposition in the rat and man by a physiologically based pharmacokinetic model. J. Pharmacokinet. Biopharm. 11:577-593. [DOI] [PubMed] [Google Scholar]
- 25.Jensen, J. 1989. Clinical pharmacokinetics of terbinafine. Clin. Exp. Dermatol. 14:110-113. [DOI] [PubMed] [Google Scholar]
- 26.Jones, T., and V. Villars. 1989. Lamisil, a new antifungal drug. Clin. Res. Bull. 6:9-27. [Google Scholar]
- 27.Kovarik, J., S. Kirkesseli, H. Humbert, P. Grass, and K. Kutz. 1992. Dose proportional pharmacokinetics of terbinafine and its N-demethylated metabolite in healthy volunteers. Br. J. Dermatol. 126:8-13. [DOI] [PubMed] [Google Scholar]
- 28.Kovarik, J., E. Mueller, H. Zehender, J. Denouel, H. Caplain, and L. Millerioux. 1995. Multiple-dose pharmacokinetics and distribution in tissue of terbinafine and metabolites. Antimicrob. Agents Chemother. 39:2738-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuipers, F., R. Havinga, H. Bosschieter, G. P. Toorop, F. R. Hindriks, and R. J. Vonk. 1985. Enterohepatic circulation in the rat. Gastroenterology 88:403-411. [DOI] [PubMed] [Google Scholar]
- 30.Kwon, K., and D. W. Bourne. 1987. Physiological pharmacokinetic model for the distribution and elimination of tenoxicam. Int. J. Pharm. 37:219-226. [Google Scholar]
- 31.Mayersohn, M., and S. Tannenbaum. 1998. On reclaiming data from the literature: literature data “R and R”(recovery and reanalysis). Am. J. Pharm. Educ. 62:363-371. [Google Scholar]
- 32.Nedelman, J., J. Cramer, B. Robbins, E. Gibiansky, C. Chang, S. Gareffa, A. Cohen, and J. Meligeni. 1997. The effect of food on the pharmacokinetics of multiple dose terbinafine in young and elderly healthy subjects. Biopharm. Drug Dispos. 18:127-138. [DOI] [PubMed] [Google Scholar]
- 33.Nejjam, F., M. Zagula, M. Cabiac, N. Guessous, H. Humbert, and H. Lakhdar. 1995. Pilot study of terbinafine in children suffering from tinea capitis: evaluation of efficacy, safety and pharmacokinetics. Br. J. Dermatol. 132:98-105. [DOI] [PubMed] [Google Scholar]
- 34.Nussbaumer, P., I. Leitner, K. Mraz, and A. Stutz. 1995. Synthesis and structure activity relationships of side chain substituted analogues of the allylamine antimycotic terbinafine lacking the central amino function. J. Med. Chem. 38:1831-1836. [DOI] [PubMed] [Google Scholar]
- 35.Robbins, B., C. Chang, J. Cramer, S. Garreffa, B. Hafkin, T. Hunt, and J. Meligeni. 1996. Safe coadministration of terbinafine and terfenadine: a placebo controlled crossover study of pharmacokinetic interaction in healthy volunteers. Clin. Pharmacol. Ther. 59:275-283. [DOI] [PubMed] [Google Scholar]
- 36.Ryder, N. 1985. Effect of allylamine antimycotic agents on fungal sterol biosynthesis measured by sterol side chain methylation. J. Gen. Microbiol. 131:1595-1602. [DOI] [PubMed] [Google Scholar]
- 37.Ryder, N. 1985. Specific inhibition of fungal sterol biosynthesis by SF 86-327, a new allylamine antimycotic agent. Antimicrob. Agents Chemother. 27:252-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaeffer, J. (ed.). 1953. Human anatomy, 11th ed. Blakiston, London, England.
- 39.Schiraldi, G., and D. Colombo. 1997. Potential use of terbinafine in the treatment of aspergillosis. Rev. Contem. Pharmacother. 8:349-356. [Google Scholar]
- 40.Shadomy, S., A. Espinel-Ingroff, and R. Gebhart. 1985. In vitro studies with SF-86-327, a new orally active allylamine derivative. J. Med. Vet. Mycol. 23:123-132. [DOI] [PubMed] [Google Scholar]
- 41.Torok, I., G. Simon, A. Dobozy, B. Farkas, C. Meszaros, L. Nebenfuhrer, E. Szepes, and E. Toth. 1998. Long-term post-treatment follow-up of onychomycosis treated with terbinafine: a multicentre trial. Mycoses 41:63-65. [DOI] [PubMed] [Google Scholar]
- 42.Villars, V., and T. Jones. 1990. Present status of the efficacy and tolerability of terbinafine used systemically in the treatment of dermatomycoses of skin and nails. J. Dermatol. Treatment 1:33-38. [Google Scholar]
- 43.Zehender, H., M. Cabiac, J. Denouel, J. Faergemann, P. Donatsch, K. Kutz, and H. Humbert. 1994. Elimination kinetics of terbinafine from human plasma and tissues following multiple dose administration and comparison with 3 main metabolites. Drug Investig. 8:203-210. [Google Scholar]