

Abstract
Mycobacteria and other actinomycetes do not produce glutathione but make mycothiol (MSH; AcCys-GlcN-Ins) that has functions similar to those of glutathione and is essential for growth of Mycobacterium tuberculosis. Mycothiol synthase (MshD) catalyzes N acetylation of Cys-GlcN-Ins to produce MSH in Mycobacterium smegmatis mc2155, and Cys-GlcN-Ins is maintained at a low level. The mycothiol synthase mutant, the mshD::Tn5 mutant, produces high levels of Cys-GlcN-Ins along with two novel thiols, N-formyl-Cys-GlcN-Ins and N-succinyl-Cys-GlcN-Ins, and a small amount of MSH. The nonenzymatic reaction of acyl-coenzyme A (CoA) with Cys-GlcN-Ins to produce acyl-Cys-GlcN-Ins is a facile reaction under physiologic conditions, with succinyl-CoA being an order of magnitude more reactive than acetyl-CoA. The uncatalyzed reaction rates are adequate to account for the observed production of N-succinyl-Cys-GlcN-Ins and MSH under physiologic conditions. It was shown that the N-acyl-Cys-GlcN-Ins compounds are maintained in a substantially reduced state in the mutant but that Cys-GlcN-Ins exists in disulfide forms at 5 to 40% at different stages of growth. MSH was able to facilitate reduction of N-succinyl-Cys-GlcN-Ins disulfide through thiol-disulfide exchange, but N-formyl-Cys-GlcN-Ins was ineffective. The oxidized state of Cys-GlcN-Ins in cells appears to result from a high susceptibility to autoxidation and a low capacity of the cell to reduce its disulfide forms. The mutant exhibited no enhanced sensitivity to hydrogen peroxide, tert-butyl hydroperoxide, or cumene hydroperoxide relative to the parent strain, suggesting that the most abundant thiol, N-formyl-Cys-GlcN-Ins, functions as a substitute for MSH.
Mycothiol (MSH; AcCys-GlcN-Ins) (Fig. 1) is the principal thiol in mycobacteria and in most other actinomycetes (12). Mycobacterium smegmatis has proven to be a useful organism for studying the biosynthesis of MSH because it does not require MSH for growth and mutants in MSH biosynthesis are readily isolated and studied (10, 14). Previous studies of such mutants have demonstrated that MSH plays an important role in protecting against various toxins, including peroxides, alkylating agents, and certain antibiotics (4, 16, 21). However, MSH is essential for the growth of Mycobacterium tuberculosis (23), and the enzymes involved in MSH biosynthesis are therefore of potential interest as targets for drugs to treat tuberculosis.
FIG. 1.

Reaction catalyzed by mycothiol synthase (MshD) and the structures for novel thiols produced by the mycothiol synthase mutant mshD::Tn5.
The final step of MSH biosynthesis is catalyzed by mycothiol synthase (MshD; Rv0486 in M. tuberculosis) (10) and involves the acetylation of Cys-GlcN-Ins by acetyl-coenzyme A (CoA) (Fig. 1) (3). MshD is a member of the GNAT (for Gcn5-related N-acetyltransferase) family of acetyltransferases (31), and the crystal structure of the M. tuberculosis protein has been determined (32). The mshD gene was identified by transposon mutagenesis of M. smegmatis to produce the mutant mshD::Tn5 (10). In our initial study, we found that the mshD::Tn5 mutant makes only ∼1% of the normal amount of MSH and accumulates high levels of Cys-GlcN-Ins and significant amounts of an unidentified thiol (10).
In the present study, we have characterized the mshD::Tn5 mutant in greater detail. We have identified two novel thiols in the mutant that are related to MSH and show that the cellular thiol redox status of the mutant is significantly perturbed. In addition, we demonstrate that chemical transacylation of acyl-CoA can serve as a route for production of the novel thiols from Cys-GlcN-Ins. The ability of mycothiol disulfide (mycothione; MSSM) reductase (18) to reduce disulfides of the novel thiol components was characterized. Finally, we show that the ability of the mshD::Tn5 mutant to resist challenge by peroxides is essentially unaltered.
MATERIALS AND METHODS
Materials.
M. smegmatis mc2155 was provided by W. R. Jacobs (Albert Einstein College of Medicine, Bronx, N.Y.). M. smegmatis mshD::Tn5 was isolated in this laboratory by using the EZ::TN<Kan2> transposon system (Epicenter) (10). Dithiothreitol (DTT; High Purity) was from Calbiochem, monobromobimane (mBBr) was from Molecular Probes, N-ethylmaleimide (NEM) was from Sigma, and methanesulfonic acid was provided by Fluka. All other chemicals were purchased from Fisher unless otherwise specified.
Cell culture.
M. smegmatis mc2155 was cultured in Middlebrook 7H9 medium (Difco) with 0.05% Tween 80 and 1% glucose; M. smegmatis mshD::Tn5 was grown in the same medium plus 25 μg of kanamycin per ml at 37°C. For the growth curve experiments, duplicate cultures of each strain were inoculated in 4 liters of medium. At each time point, samples of each culture were taken for thiol and disulfide analysis with volumes adequate to generate ∼0.2 g (wet weight) of pellet following centrifugation (4,500 × g) at 4°C. The pellets were analyzed for thiols and disulfides as described below.
Determination of cellular thiols and disulfides.
For thiol analysis, bacterial pellets were extracted in 1 ml of 50% acetonitrile containing 20 mM HEPES (pH 8.0) and 2 mM mBBr at 60°C for 10 min. The thiol samples were acidified with 3 μl of 5 M methanesulfonic acid, cooled on ice, clarified by centrifugation at 13,000 × g for 5 min, and diluted as needed in 10 mM methanesulfonic acid for high-performance liquid chromatography (HPLC).
For disulfide analysis, 5 mM NEM replaced mBBr in the extraction. Pellets were cooled on ice, and the supernatant was clarified by centrifugation at 13,000 × g for 5 min. A 100-μl sample was removed to a separate tube and reacted with 2 mM mBBr for 10 min at 60°C to serve as a control sample to identify fluorescent HPLC peaks not derived from thiols. To the remaining 900 μl, 2-mercaptoethanol was added to a final concentration of 5 mM to scavenge excess NEM, and the sample was incubated at 23°C for 10 min. The sample was concentrated to a volume of 100 μl by using a Savant Speed Vac, and disulfides were reduced with 2 mM DTT (High Purity; Calbiochem) for 15 min at 23°C. The reduced sample was derivatized with mBBr at a final concentration of 9 mM for 15 min at 23°C. The sample was acidified with 2 μl of 5 M methanesulfonic acid and diluted with 10 mM methanesulfonic acid for HPLC. The pellets from the thiol and disulfide extracts were dried in a vacuum oven to a constant weight, and the results are reported as means and ranges of duplicate culture samples in micromoles per gram of residual dry weight of the pellet.
The thiol-bimane derivatives were analyzed by HPLC on a Waters 600 chromatograph equipped with a Farrand Optical Components and Instruments model A-1 filter fluorometer. The HPLC protocol has been previously described (10), and the retention times for the bimane derivatives of Cys-GlcN-Ins, Cys, U18 (fCys-GlcN-Ins), MSH, and U22 (N-succinyl-Cys-GlcN-Ins [succ-Cys-GlcN-Ins]) were 6, 9.8, 18, 20.7, and 21.2 min, respectively. Fluorescence factors for unknown thiols were assumed to be the same as that for CySmB until authentic standards were obtained.
Structure of U18.
U18mB was generated from exponential-phase cultures of the mshD::Tn5 mutant by the protocol of Newton and Fahey (15) involving thiol affinity chromatography on thiopropyl-Sepharose 6B activated by treatment with 2,2′-dithiodipyridine, conversion of the isolated thiols to the monobromobimane derivatives, and purification of U18mB by preparative HPLC. The purified U18mB was examined by electrospray mass spectrometry (positive-ion mode; The Scripps Research Institute, La Jolla, Calif.). fCySmB-GlcN-Ins was synthesized from CySmB-GlcN-Ins. The latter was generated from 25 g of the mshD::Tn5 mutant by extraction of the cells with 50% acetonitrile (60°C) containing 2 mM mBBr and 20 mM HEPES, pH 8.0. The supernatant was concentrated in a Savant Speed Vac to remove acetonitrile and purified by preparative HPLC chromatography on a Vydac 218TP1022 C18 column in aqueous 0.1% trifluoroacetic acid (TFA) with a linear 0 to 20% methanol gradient over 50 min (5 ml/min). Formylation of CySmB-GlcN-Ins by cyanomethyl formate [(formyloxy)acetonitrile; Aldrich] was performed by a minor modification of the method of Deutsch and Niclas (5). CySmB-GlcN-Ins was dissolved at 1 mM in aqueous 75% DMSO containing 20 mM sodium phosphate, and the pH was adjusted to 7.0 with sodium hydroxide. Cyanomethyl formate was added to a final concentration of 140 mM, and the sample was incubated for 15 h at 23°C in the dark. The synthetic fCySmB-GlcN-Ins was purified by preparative HPLC as described above for U18 with a 72% overall yield. The synthetic fCySmB-GlcN-Ins coeluted with isolated U18mB with HPLC protocol 2 (7) and the HPLC protocol of Koledin et al. (10).
Structure of U22.
U22 was isolated from 10 100-mm plates of Middlebrook 7H9 agar spread with M. smegmatis mshD::Tn5 and cultured for 5 days at 37°C. The plates were scraped, and the cells were labeled with mBBr as described above for thiols. The U22mB produced was purified by preparative HPLC as described above for fCySmB-GlcN-Ins (U18mB). The purified U22mB was analyzed by electrospray mass spectrometry in the positive-ion mode (Micromass ZMD; Waters).
To verify the structure of U22, authentic succ-Cys-GlcN-Ins was synthesized from Cys-GlcN-Ins (thiol form). Cys-GlcN-Ins was obtained from the mshD::Tn5 mutant by a protocol analogous to that used for isolation of MSH from wild-type M. smegmatis (30). Final purification of Cys-GlcN-Ins was on a reversed-phase C18 HPLC column where it could be separated from other thiols but not from unretained salts such as sodium trifluoroacetate and HEPES, with which it coeluted. Unlike MSH (13), Cys-GlcN-Ins oxidizes very readily at neutral pH and must be stored at a mildly acidic pH and −70°C. A 0.5-ml aqueous solution containing 2 mM Cys-GlcN-Ins, 2.4 mM succinyl-CoA (Sigma), 5 mM MgCl2, and 1 mM DTT was adjusted to pH 7 with NaOH and allowed to react for 3 h at 23°C. The loss of Cys-GlcN-Ins was followed by HPLC analysis of samples prepared with and without reduction by additional DTT prior to labeling with mBBr. After 3 h, 15% of the starting Cys-GlcN-Ins still remained but was completely oxidized. An additional 3 mM DTT was added, and the mixture was adjusted to pH 8 and incubated overnight at 23°C. succ-Cys-GlcN-Ins was isolated by preparative HPLC on a Vydac 218TP1022 C18 column with isocratic elution in 0.1% TFA-water (5 ml/min), with the thiol eluting at 23 min. After being labeled with mBBr, this material coeluted with U22mB with the two HPLC protocols described above for U18mB, demonstrating that U22mB is succ-Cys-GlcN-Ins.
Rate of acylation of Cys-GlcN-Ins by acyl-CoA.
Transacylation reactions were run in 150-μl solutions containing 100 mM NaCl, 250 μM Cys-GlcN-Ins (purified as described above), 5 mM MgCl2, 1 mM DTT, and 50 mM HEPES (pH 7.4) at 30°C, conditions chosen to approximate the cellular environment. The reaction was initiated by the addition of 1 mM acetyl-CoA, propionyl-CoA, or succinyl-CoA (all from Sigma). Samples were taken at intervals by mixing a 20-μl aliquot of the reaction mixture with 180 μl of 25 mM HEPES, pH 8.0, containing 2 mM mBBr. The mixture was incubated for 10 min at 30°C, and the derivatization was stopped with the addition of 1 μl of 5 M methanesulfonic acid. After dilution 10 fold into 10 mM methanesulfonic acid, the bimane derivatives were analyzed by HPLC by the method of Koledin et al. (10). Apparent second-order rate constants were calculated from the slope of the linear plot of ln(A0B/B0A) versus t, where A0 and B0 are the initial concentrations of Cys-GlcN-Ins and acyl-CoA, respectively, and A and B are the corresponding concentrations at time t (9). Least-squares fit of the data was obtained using KaleidaGraph 3.6.2.
Preparation of disulfides.
MSSM was produced from MSH purified from M. smegmatis mc2155 as described by Unson et al. (30). A 1.4-ml aqueous solution containing 2.8 mM MSH was adjusted to pH 9 with NH4OH, and CuSO4 was added to a final concentration of 5 μM. Autoxidation was allowed to continue unstirred in air overnight at 23°C. The thiol level was checked by titration with 5,5′-dithiobis(2-nitrobenzoic acid) (Sigma) as described by Ellman (6) and found to be completely oxidized. The pH was adjusted to 2 to 3 with TFA, and the MSSM was purified by preparative HPLC on a Vydac 218TP1022 C18 column. Isocratic elution with 0.1% TFA-water (5 ml/min) was monitored at 220 nm, and the disulfide was eluted at 18 min. The pH was adjusted to 7 with NH4OH, and the solvent was removed on a Savant Speed Vac.
The disulfide of fCys-GlcN-Ins was prepared in analogous fashion from the thiol which was purified from the mshD::Tn5 mutant as described above. A 2.8-ml aqueous solution containing 2.6 mM fCys-GlcN-Ins was oxidized as above; upon HPLC purification, the disulfide eluted at 16.5 min. It was neutralized and dried as for MSSM above.
Oxidation of succ-Cys-GlcN-Ins, prepared as described above, to the disulfide was performed with a 1-ml solution of 0.4 mM succ-Cys-GlcN-Ins as described for MSSM. The succ-Cys-GlcN-Ins disulfide was purified by preparative HPLC on a Vydac 218TP1022 C18 column with a 2 to 15% methanol gradient in 0.1% aqueous TFA over 50 min. The disulfide eluted at 43 min; it was neutralized and dried as for MSSM above.
Substrate specificity of M. tuberculosis Mtr.
His6-tagged M. tuberculosis mycothione reductase (Mtr) was isolated from M. smegmatis mc2155 transformed with the plasmid pMV261-MycRHis6 (18), kindly provided by John S. Blanchard (Albert Einstein College of Medicine), using a modified version of the published protocols (17, 18). A 50-ml column of nickel-nitrilotriacetic acid, His Bind resin (Novagen) was employed for purification; repeated chromatography on this column was required to achieve >95% purity as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. An assay of steady-state activities with the various disulfides was performed at 23°C by monitoring A340 with a Beckman DU 640 spectrophotometer using a 100-μl microcell with a 1-cm path length 1-cm-path-length. Assay solutions contained 100 mM NaCl, 0.1 mM EDTA, 100 μM NADPH (Sigma), and 50 mM HEPES (pH 7.4).
Autoxidation of Cys-GlcN-Ins and MSH.
Autoxidation reactions were conducted at 30°C with 50 mM HEPES, pH 7.3, containing 100 or 200 μM concentrations of specified thiols in a total volume of 150 μl. A zero time sample was taken prior to the addition of CuSO4 to a final concentration of 0.3 μM. At various times, 20-μl samples were mixed with 20 μl of 10 mM mBBr in aqueous 25 mM HEPES (pH 8.0) and incubated for 10 min in the dark. After dilution with 360 μl of 10 mM aqueous methanesulfonic acid, the samples were analyzed by HPLC as described above.
Sensitivity to peroxides.
Sensitivity to hydrogen peroxide (Fisher), tert-butyl hydroperoxide (Sigma), and cumene hydroperoxide (Aldrich) was determined by the broth dilution method. Exponential-phase mc2155 and mshD::Tn5 mutant cells cultured in 7H9 Middlebrook medium containing 1% glucose were harvested at A600 of <1.0 and diluted to A600 = 0.1 in the same medium. The cells were aliquoted (5 ml) into 14-ml polypropylene snaptop culture tubes (Falcon 302059) containing various amounts of peroxide: 0 to 1.0 mM in 0.1 mM increments for tert-butyl and cumene hydroperoxide and 0 to 10 mM in 1 mM increments for hydrogen peroxide. Cells were cultured 2 to 5 days with shaking (250 rpm) at 37°C, and the MIC was defined as the minimum concentration of peroxide that resulted in no increase in the A600 value.
RESULTS
Novel thiols of M. smegmatis mshD::Tn5.
In our initial study, we found that the mshD::Tn5 mutant accumulated a high level of Cys-GlcN-Ins (10). We observed that the mutant produced a major unidentified thiol designated U18mB, based on the HPLC elution time of its monobromobimane derivative. Upon closer scrutiny, a second bimane derivative, designated U22mB, was detected at much lower levels. To establish the structures of these novel thiols, they were isolated from cell extracts and purified as the bimane derivatives by reversed-phase preparative HPLC.
U18 was purified from extracts of the mshD::Tn5 mutant by thiol affinity chromatography. It was then converted to the bimane derivative, U18mB, and isolated in pure form by preparative HPLC. When analyzed by positive-ion electrospray mass spectrometry, purified U18mB generated a molecular ion with a mass of 663 Da and a prominent fragment with a mass of 483 corresponding to the loss of an inositol residue, a fragmentation pattern observed previously with mycothiol bimane (MSmB) (13). The molecular ion of 663 Da was 14 mass units less than the bimane derivative of mycothiol, suggesting that U18mB was N-formyl-CySmB-GlcN-Ins (fCySmB-GlcN-Ins). Formylation of CySmB-GlcN-Ins by cyanomethyl formate produced authentic fCySmB-GlcN-Ins; analysis by mass spectrometry as above showed a molecular ion of 663 Da with a prominent 483-Da fragment, as observed for the U18mB isolated from the mshD::Tn5 mutant. Synthetic fCySmB-GlcN-Ins coeluted with isolated U18mB on two different HPLC protocols, establishing that U18 is fCys-GlcN-Ins. The structure of fCys-GlcN-Ins is given in Fig. 1.
The second unknown thiol, U22, was present in small amounts in the mshD::Tn5 mutant but was produced at a fivefold-higher level by cells cultured on agar plates. Cells scraped from plates were extracted and labeled with mBBr, and the U22mB was purified by preparative HPLC. Positive-ion electrospray mass spectrometry of purified U22mB produced a molecular ion of 735 Da and a prominent fragment of 555 Da, corresponding to the loss of the inositol residue, as found above for U18mB. This suggested that U22mB was a relative of MSmB with an additional 59 Da of mass. One possible N-acyl derivative of Cys-GlcN-Ins with a molecular ion of 735 Da is succ-Cys-GlcN-Ins. The presence of succinyl-CoA has been reported in extracts of M. smegmatis as an intermediate in CO2 fixation (28). succ-Cys-GlcN-Ins was synthesized by transacylation of Cys-GlcN-Ins with succinyl-CoA and purified in the thiol form by preparative HPLC. After conversion to the bimane derivative, succ-CySmB-GlcN-Ins was analyzed by positive-ion electrospray mass spectrometry to yield a molecular ion of 735 with a major fragment of 555, as observed for U22mB isolated from the mshD::Tn5 mutant. Synthetic succ-CySmB-GlcN-Ins coeluted with U22mB isolated from mshD::Tn5 on two separate HPLC protocols, establishing that U22 is succ-Cys-GlcN-Ins (Fig. 1).
Thiol-disulfide composition of mshD::Tn5 compared to the parent strain mc2155.
With the structures of the novel thiols established, we next determined the thiol composition of the mutant and parent strains during exponential and stationary growth phases (Fig. 2). The analysis protocol involved chilling the cells on ice and centrifugation prior to extraction of the cell pellet for analysis; it is assumed that this produces little change in the thiol and disulfide content. The parent strain, mc2155, produced MSH as the dominant thiol with little fluctuation in level during growth (Fig. 2A). CoA was the next most prevalent thiol, followed by Cys, and their levels showed significant fluctuations as a function of growth phase. Cys-GlcN-Ins, fCys-GlcN-Ins, and succ-Cys-GlcN-Ins were not detected (<0.01 μmol per g [dry weight]) in the parent strain.
FIG. 2.
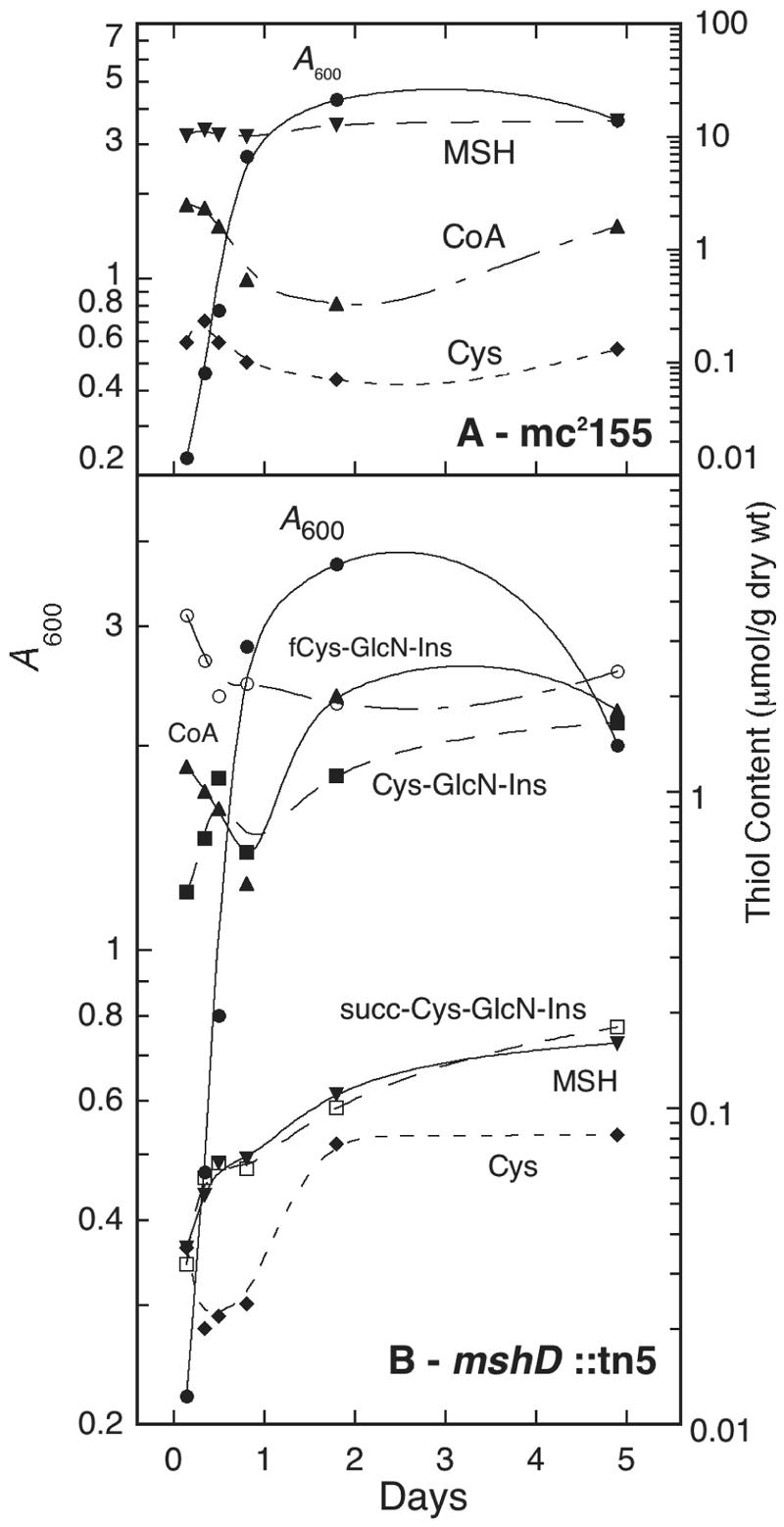
Cellular thiol composition during growth of M. smegmatis mc2155 (A) and M. smegmatis mshD::Tn5 (B). •, A600; ▾, MSH; ▴, CoA; ♦, Cys; ○, fCys-GlcN-Ins; ▪, Cys-GlcN-Ins; □, succ-Cys-GlcN-Ins. Results from two independent cultures are shown; average deviation from mean was ≤20%, except for Cys-GlcN-Ins (where the average deviation was 30%).
The mshD::Tn5 mutant produced low levels of MSH, ∼100 to 200-fold lower than found in the parent strain (Fig. 2B). The major thiol in the mutant was fCys-GlcN-Ins, present at about one-fourth the concentration of MSH in the parent strain. In stationary phase, the levels of CoA and Cys-GlcN-Ins approached that of fCys-GlcN-Ins. The level of succ-Cys-GlcN-Ins was comparable to that of MSH in the mutant, both rising in the transition to stationary phase. The level of Cys in the mutant during exponential growth was ∼5-fold lower than that of the parent strain but rose to the parental level during stationary phase.
Cellular thiols are normally maintained in a highly reduced state, but the thiol composition of the mshD::Tn5 mutant is markedly different from the parental strain and this might result in an altered thiol redox status. To calculate a thiol redox ratio (RSH/RSS) for the M. smegmatis strains, a measurement of the disulfide levels was necessary. We first reacted the thiols with N-ethylmaleimide, then reduced the soluble disulfides with dithiothreitol, and finally labeled the released thiols with mBBr for analysis by HPLC. This method cannot be used for CoA, because many acyl-CoA's in the cell will undergo transacylation with dithiothreitol to release CoA not derived from disulfides (8). In the parent strain (Fig. 3A), mycothiol was maintained in a highly reduced state, with a redox ratio ranging from 200 to 1,000. The redox ratio for Cys is generally lower by an order of magnitude. For the mshD::Tn5 mutant, the redox ratios of MSH, fCys-GlcN-Ins, and succ-Cys-GlcN-Ins were close to 100 and generally lower than those of MSH in the parental strain. The redox ratios for Cys were lower, as was the case with the parental strain (Fig. 3). However, Cys-GlcN-Ins, one of the three principal thiols in the mutant, was found to be substantially oxidized. During exponential growth, 5 to 10% of Cys-GlcN-Ins is present in disulfide forms and this increases to ∼40% in late stationary phase, the redox ratio approaching unity.
FIG. 3.

Cellular redox status during growth of M. smegmatis mc2155 (A) and M. smegmatis mshD::Tn5 (B). •, A600; ▾, MSH; ♦, Cys; ○, fCys-GlcN-Ins; □, succ-Cys-GlcN-Ins; ▪, Cys-GlcN-Ins. Results from two independent cultures are shown. The average deviation from the mean was ≤30%, except for the results shown in panel B, where the average deviation was 45% for MSH and 55% for succ-Cys-GlcN-Ins.
Production of acyl-Cys-GlcN-Ins via chemical transacylation of Cys-GlcN-Ins by acyl-CoA.
Transfer of an acyl group from one thiol to another is generally recognized as a facile process, even without enzyme catalysis. If the thiol is a derivative of cysteine with the amino group free, as is the case for Cys-GlcN-Ins, then the thermodynamically favored transfer of the acyl group to the amine can occur by an intramolecular rearrangement proceeding via a readily accessible five-member ring intermediate. We measured the nonenzymatic rates of acylation of Cys-GlcN-Ins by succinyl-CoA, acetyl-CoA, and propionyl-CoA to generate the corresponding acyl-Cys-GlcN-Ins derivatives at physiologic pH (Fig. 4). Acylation by succinyl-CoA was fastest, with an apparent second-order rate constant of 0.58 ± 0.07 M−1 s−1 (Fig. 4). Reaction of 250 μM Cys-GlcN-Ins with 1 mM succinyl-CoA was largely complete at 90 min, 17 μM Cys-GlcN-Ins (7%) remaining unreacted and 227 μM succ-Cys-GlcN-Ins (91%) being formed. This accounted for 98% of the starting Cys-GlcN-Ins concentration. Chemical synthesis of MSH from acetyl-CoA and propionyl (prop)-Cys-GlcN-Ins from propionyl-CoA was slower, the apparent second-order rates being 0.028 ± 0.001 and 0.031 ± 0.007 M−1 s−1, respectively (Fig. 4). These rates suggest that MSH and succ-Cys-GlcN-Ins may be produced in the mshD::Tn5 mutant by chemical rather than enzymatic processes (see Discussion).
FIG. 4.
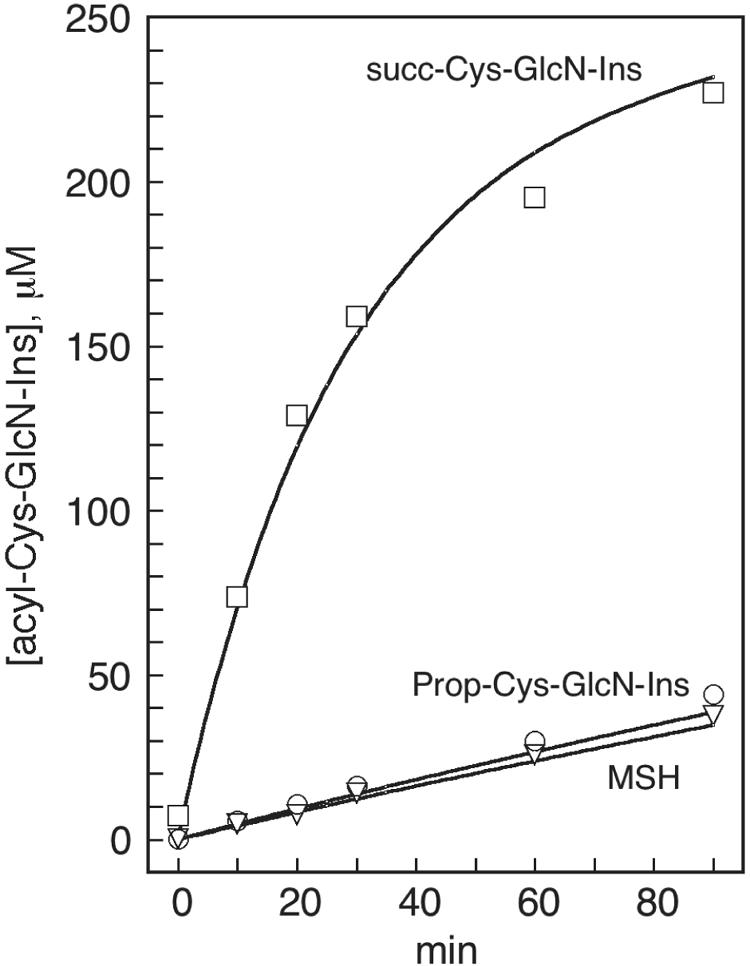
Chemical synthesis of acyl-Cys-GlcN-Ins from a reaction of 250 μM Cys-GlcN-Ins with 1 mM of propionyl-CoA to produce prop-Cys-GlcN-Ins (○), of acetyl-CoA to produce MSH (▿) and of succinyl-CoA to produce succ-Cys-GlcN-Ins (□) in 50 mM HEPES, pH 7.4, containing 1 mM DTT at 30°C. Lines correspond to the second-order rates calculated from the rate constants given in the text. The data shown are representative of three independent experiments which produced similar results.
Autoxidation of Cys-GlcN-Ins.
The relatively oxidized state of Cys-GlcN-Ins found in the mshD::Tn5 mutant strain (Fig. 3) may result from Cys-GlcN-Ins undergoing autoxidation in the cell at an unusually rapid rate or from lack of an adequate system to reduce its oxidized products. To explore the susceptibility of Cys-GlcN-Ins to autoxidation, we compared the copper catalyzed autoxidation of Cys-GlcN-Ins with that of MSH (Fig. 5). The initial rate (0 to 8 min) of oxidation of Cys-GlcN-Ins was ∼11-fold faster than that of MSH when tested independently at 200 μM initial thiol concentration (Fig. 5A). When a mixture of 100 μM (each) Cys-GlcN-Ins and MSH was examined (Fig. 5B), the initial rate of total thiol loss (5.5 ± 0.2 μM min−1; three experiments) was the same as measured for Cys-GlcN-Ins alone (4.7 ± 0.2 μM min−1; three experiments). The initial rate of MSH loss in the presence of Cys-GlcN-Ins was ∼4-fold greater than that for MSH alone, while the rate of Cys-GlcN-Ins loss in the presence of MSH was ∼1.3-fold slower than for Cys-GlcN-Ins alone. Thus, Cys-GlcN-Ins facilitates the oxidation of other thiols.
FIG. 5.
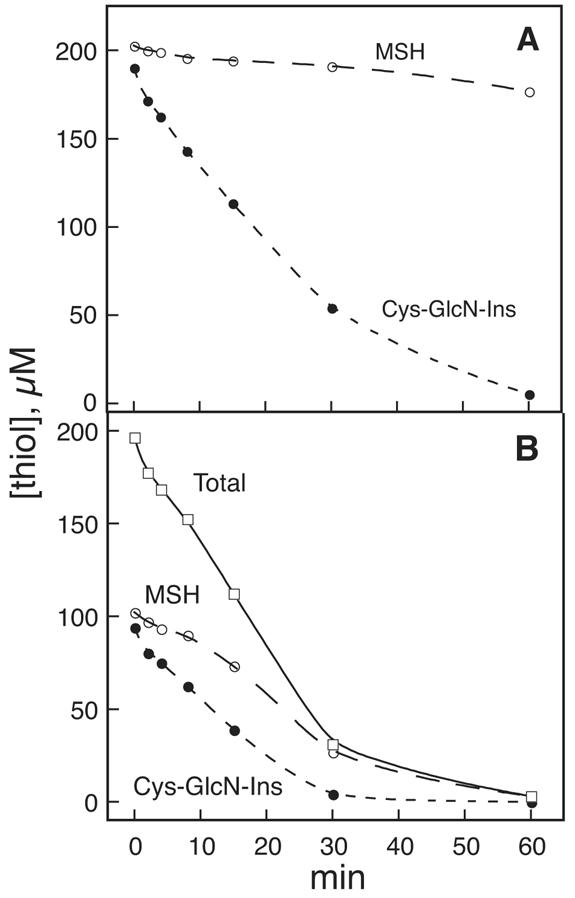
Autoxidation of thiols in the presence of 50 mM HEPES, pH 7.3, containing 0.3 μM CuSO4. (A) ○, 200 μM MSH; •, 200 μM Cys-GlcN-Ins. (B) 100 μM MSH (○) plus 100 μM Cys-GlcN-Ins (•); total thiol (□). Three independent experiments all produced results essentially identical to the one shown.
Substrate specificity of Mtr.
Actinomycete genomes contain a gene (mtr) encoding a mycothione reductase (17, 18) homologous to glutathione reductase. To elaborate the role played by Mtr in the redox status of the mshD::Tn5 mutant, we prepared the symmetrical disulfides of the major thiols and tested them as substrates for M. tuberculosis Mtr. The mtr gene from M. tuberculosis was expressed and purified as the C-terminal His6-tagged protein (18). Sufficient MSSM was available to determine a Km value of 54 ± 12 μM by least-squares fit to the Michaelis-Menten equation of the initial rate determined at a saturating concentration of NADPH (100 μM) for 12 disulfide concentrations ranging from 10 to 500 μM. This Km value is similar to that (70 ± 30 μM) determined for the non-His-tagged M. tuberculosis enzyme (17). The other thiols were available in limited quantities, and this allowed only an estimate of the rates relative to MSSM. For the disulfides of fCys-GlcN-Ins and succ-Cys-GlcN-Ins, the rates doubled from 50 to 100 μM disulfide, producing rates relative to MSSM (100%) of 13 ± 1 and 0.8 ± 0.1, respectively. For the disulfide of Cys-GlcN-Ins, the rate did not increase with disulfide concentration and only an upper limit of ≤4 could be put as its relative rate.
To examine the reactivity of mixed disulfides of the most- and least-reactive thiols as substrates for Mtr, we tested the effect of 30 μM fCys-GlcN-Ins or MSH added to 100 μM succ-Cys-GlcN-Ins disulfide 30 min prior to addition of Mtr (Fig. 6). This allows thiol-disulfide exchange with the added thiol prior to initiation of the enzymatic reaction. No increased rate of reaction was observed with fCys-GlcN-Ins, but MSH produced a marked increase in the rate of reaction during the first 2 min followed by a slower, but still enhanced, rate of reduction from 2 to 10 min (Fig. 6). Although insufficient succ-Cys-GlcN-Ins disulfide was available for replicate experiments, the qualitative difference in effect between MSH and fCys-GlcN-Ins was clear in the results shown Fig. 6. This demonstrates that MSH can facilitate reduction of succ-Cys-GlcN-Ins disulfide through thiol-disulfide exchange to generate the mixed disulfide of MSH with succ-Cys-GlcN-Ins and MSSM, thereby facilitating reduction by Mtr. However, the analogous reactions of the thiol fCys-GlcN-Ins were ineffective in facilitating reduction of succ-Cys-GlcN-Ins disulfide.
FIG. 6.
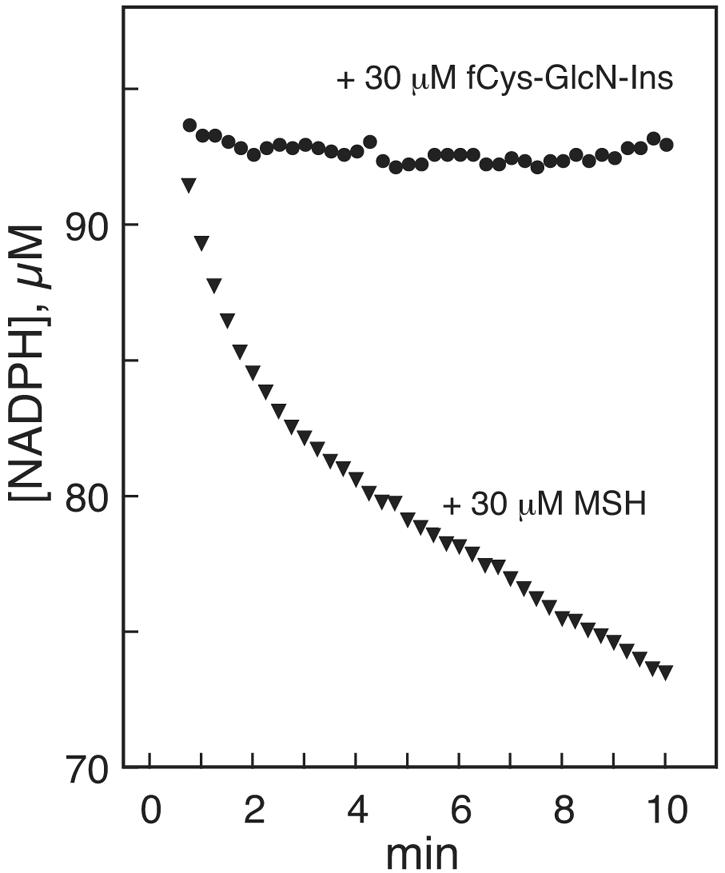
Reduction of 100 μM succ-Cys-GlcN-Ins disulfide preincubated for 30 min with 30 μM of the thiol fCys-GlcN-Ins (•) or MSH (▾) by Mtr as monitored by NADPH absorption at 340 nm.
Resistance of the mshD::Tn5 mutant to peroxide stress.
The low but measurable MSH content of the mshD::Tn5 mutant and production of novel thiols related to MSH suggested that the mshD::Tn5 mutant might be more resistant to oxidative stress than MSH mutants blocked at earlier steps in the MSH biosynthetic pathway. To test this, we determined the sensitivity of the mshD::Tn5 mutant and its parental strain to several peroxides by the broth dilution assay. No difference between the strains was found in the MIC of hydrogen peroxide (3 to 4 mM), tert-butyl hydroperoxide (0.3 mM), and cumene hydroperoxide (0.1 mM).
DISCUSSION
The dominant thiol in M. smegmatis mc2155 is MSH, followed by CoA and Cys at ∼10% and ∼1% the level of MSH, respectively (Fig. 2A). In contrast, the mutant mshD::Tn5 produces a surprising array of thiols, with three being dominant. The most abundant and unexpected thiol is fCys-GlcN-Ins, which is produced at a level 20 to 30% of that of MSH in the parent strain (Fig. 2B). CoA, the second most abundant thiol, and Cys are found at levels similar to those found in the parent strain. Cys-GlcN-Ins, as expected in the absence of MshD, is present at a level that is orders of magnitude higher than in the parent strain (<0.01 μmol/g [dry weight]). In the mshD::Tn5 mutant, MSH and succ-Cys-GlcN-Ins are produced at levels ∼1% of that of MSH in the parent strain. In mc2155, Cys-GlcN-Ins, fCys-GlcN-Ins, and succ-Cys-GlcN-Ins are undetectable.
We considered first how MSH and succ-Cys-GlcN-Ins might be produced in the mshD::Tn5 mutant. It seems likely that MSH and succ-Cys-GlcN-Ins are generated from Cys-GlcN-Ins by reaction with succinyl- and acetyl-CoA, respectively, either by chemical or enzymatic reactions. The chemical transacylation reactions are quite facile under physiologic conditions (Fig. 4). The much faster reaction of succinyl-CoA than that of acetyl- or propionyl-CoA likely reflects the ability of the free carboxyl group in the succinyl residue to facilitate reaction by intramolecular catalysis of the slow step, leading to generation of succ-Cys-GlcN-Ins.
The apparent second-order rate constant for production of succ-Cys-GlcN-Ins from succinyl-CoA (0.58 M−1 s−1) can be used to estimate the concentrations of succinyl-CoA required to produce the observed succ-Cys-GlcN-Ins content. We used the concentration of Cys-GlcN-Ins during exponential growth, estimated at 200 μM from the data shown in Fig. 2B, a water content for M. smegmatis of 4 μl per mg residual (dry weight) (G. L. Newton, unpublished data), and observed concentration of ∼15 μM for succ-Cys-GlcN-Ins calculated from the data shown in Fig. 2B. From these values, we calculated that 9 μM succinyl-CoA is required to produce the observed concentration of succ-Cys-GlcN-Ins during the 4-h cellular doubling time. An analogous calculation from the transacylation rate with acetyl-CoA (0.04 M−1 s−1) and the MSH content (also ∼15 μM) gives an estimate of 130 μM for the acetyl-CoA content. Although production of methylmalonic acid and succinic acid from propionyl-CoA and CO2 by extracts of M. smegmatis was described in 1962, implying the production of succinyl-CoA (28), no data for succinyl-CoA content or even the acetyl-CoA content appear to have been measured for mycobacteria. Recently reported values for Escherichia coli (26), after corrections were made for the water content of the cells (20), provide estimates of ∼80 μM succinyl-CoA and ∼600 μM acetyl-CoA. These are 5- to 10-fold higher than the values required for generation of succ-Cys-GlcN-Ins and MSH by chemical transacylation reactions. Thus, it seems most probable that succ-Cys-GlcN-Ins and MSH are generated chemically as a consequence of the high cellular levels of Cys-GlcN-Ins in the mutant.
Enzyme catalyzed processes may also contribute to the transacylations producing MSH and succ-Cys-GlcN-Ins. The GNAT family of acetyltransferases to which MshD belongs is large (31). A member of this group may utilize Cys-GlcN-Ins as substrate when it accumulates to a high level in the mshD::Tn5 mutant. Enzymes with succinyltransferase activity are less common. Succinyl-CoA:tetrahydrodipicolinate N-succinyltransferase produces l-2-(succinylamino)-6-oxopimelic acid (2). This compound is an intermediate in the production of diaminopimelic acid, a key component of the bacterial cell wall (25). However, this succinyltransferase has been shown to be quite specific for the 2-aminopimelic acid moiety (2), which has two free carboxyl groups. Thus, it seems highly unlikely that this succinyltransferase would exhibit significant activity with Cys-GlcN-Ins, which lacks free carboxyl groups.
Production of the high level of fCys-GlcN-Ins found for the mshD::Tn5 mutant is probably the result of an enzyme-catalyzed reaction. Possible candidates include N10-formyl tetrahydrofolate-dependent methionyl-tRNA transformylase (29) and the glycinamide ribonucleotide transformylases PurT (11) and PurN (34), which utilize formate/ATP and N10-formyl tetrahydrofolate, respectively. These enzymes catalyze formylation of an amino group on an amino acid linked to a sugar via its carboxyl group. It is plausible that these enzymes might recognize Cys-GlcN-Ins as an acceptor. Homologs of the genes for these enzymes are present in the tuberculosis genome (http://genolist.pasteur.fr/TubercuList), but none of these proteins has been characterized from mycobacteria.
We consider next the redox status of the thiols in the mshD::Tn5 mutant and its parent strain. As expected, MSH is maintained in a highly reduced state in both strains by the inherent resistance of MSH to autoxidation (Fig. 5) (13) and the action of the mycothiol specific reductase Mtr (17, 18). Cys exists in a more oxidized state in both strains, presumably reflecting the fact that it autoxidizes substantially faster than MSH (13). Cys-GlcN-Ins is appreciably oxidized in the mutant. This probably reflects its high rate of autoxidation (Fig. 5) and the low activity of its disulfide as a substrate for Mtr (≤3% of the activity of MSSM). The mechanism of copper catalyzed thiol autoxidation is complex when studied for a single thiol (19). It is even more complex in cells where copper proteins are likely involved (33) and especially for the M. smegmatis mshD::Tn5 mutant with its complex mixture of thiols. But from the present results (Fig. 5B), it is clear that Cys-GlcN-Ins promotes thiol oxidation. This makes Cys-GlcN-Ins a liability to the cell and provides a rationale for why MshD evolved so as to maintain the concentration Cys-GlcN-Ins at a very low level in the parent strain (<0.01 μmol per g or ∼3 μM). The presence of a high level of Cys-GlcN-Ins in the mutant strain must require a significant diversion of resources to counteract its facile autoxidation, but this stress is not sufficient to significantly slow the growth of the mshD::Tn5 mutant relative to the parent strain.
The facile autoxidation of Cys has been attributed to the presence of a free amino group located beta to the thiol group that facilitates binding of heavy metals to catalyze thiol autoxidation. Cys-GlcN-Ins also possesses this functionality. In fCys-GlcN-Ins, succ-Cys-GlcN-Ins, and MSH, the amino group is acylated so that autoxidation is expected to be much slower. Our data show that all of these thiols are maintained in a substantially reduced state (Fig. 3). But even slow autoxidation requires a reductase to maintain the reduced state. The activity of Mtr is substantial with the disulfides of fCys-GlcN-Ins and MSH but is low with the disulfides of Cys-GlcN-Ins and succ-Cys-GlcN-Ins and the mixed disulfide of succ-Cys-GlcN-Ins with fCys-GlcN-Ins (Fig. 6). However, thiol-disulfide exchange of succ-Cys-GlcN-Ins disulfide with MSH facilitates the reduction of succ-Cys-GlcN-Ins disulfide (Fig. 6); this may contribute to maintenance of cellular succ-Cys-GlcN-Ins in the reduced state (Fig. 3). It is also possible that the reduced state of succ-Cys-GlcN-Ins results from reduction of its disulfides by thioredoxins (1) in combination with a slow autoxidation rate for its thiol form.
The mshD::Tn5 mutant exhibited no enhanced sensitivity to hydrogen peroxide, tert-butyl hydroperoxide, or cumene hydroperoxide relative to the parent strain mc2155. Other M. smegmatis mutants completely blocked in MSH production exhibit at least 12-fold increased sensitivity to peroxides (16, 21). However, even 1% of the normal MSH content suffices to provide ∼4-fold protection against hydrogen peroxide (21). The cellular thiol levels applicable to the current broth dilution peroxide sensitivity assays are those found in very early-log-phase mshD::Tn5 cells. In those cells, we find MSH and succ-Cys-GlcN-Ins at ∼0.3%, Cys-GlcN-Ins at ∼5%, and fCys-GlcN-Ins at ∼35% of the wild-type mycothiol level. With MSH at such a low level in early-log-phase mshD::Tn5 cells, we would expect an easily measured sensitivity to hydrogen peroxide. Since we did not detect an increased sensitivity to hydrogen peroxide, we must consider whether one of the predominant thiols present in the mutant, Cys-GlcN-Ins or fCys-GlcN-Ins, substitutes for MSH to provide normal peroxide resistance.
To evaluate the ability of Cys-GlcN-Ins or fCys-GlcN-Ins to substitute for MSH, we examined the activity of these compounds with mycothiol-dependent enzymes. One such activity is catalyzed by M. tuberculosis mycothiol S-conjugate amidase, and the activity of this enzyme with the bimane derivatives of Cys-GlcN-Ins and fCys-GlcN-Ins has been measured (27). Activities with CySmB-GlcN-Ins and fCySmB-GlcN-Ins were 0.1% and 5%, respectively, relative to the bimane derivative of mycothiol, MSmB. For this enzyme, fCySmB-GlcN-Ins is a 50-fold-better substrate than CySmB-GlcN-Ins. In the present work, we compared the activity of the disulfides of Cys-GlcN-Ins and fCys-GlcN-Ins with Mtr and showed that they have ≤4% and 13%, respectively, of the activity of MSSM. Thus, fCys-GlcN-Ins is a better substitute for MSH than is Cys-GlcN-Ins. Since it is present at ∼7-fold-higher concentrations than Cys-GlcN-Ins in early-log-phase growth, it is the most likely candidate to substitute for MSH in protecting against hydrogen peroxide toxicity. MSH and fCys-GlcN-Ins are closely related structures, differing by the substitution of the acetyl residue in the former by a formyl group in the latter. By contrast, the other related thiols present in the mutant have charges associated to the Cys residue, a positively charged ammonium residue in the case of Cys-GlcN-Ins and a negatively charged succinyl residue in the case of succ-Cys-GlcN-Ins. This likely makes them poor substrates for mycothiol-dependent enzymes.
The elevated Cys-GlcN-Ins content produced by inactivation of MshD in the mycothiol synthase mutant has a number of unexpected consequences. MSH production is not fully blocked because chemical transacylation of Cys-GlcN-Ins by acetyl-CoA is sufficiently rapid to support slow MSH synthesis. The high level of Cys-GlcN-Ins also facilitates the production of two novel thiols, fCys-GlcN-Ins and succ-Cys-GlcN-Ins. Although these are maintained in a substantially reduced state, Cys-GlcN-Ins itself is substantially oxidized in the cell, apparently because of its facile autoxidation coupled with the absence of an efficient reductase for its disulfide forms. It also appears that the levels of reduced acyl-Cys-GlcN-Ins compounds in the mshD::Tn5 M. smegmatis mutant are sufficient for normal growth and protection from peroxide toxicity.
Mycothiol is not required for growth of M. smegmatis, whereas it is required for growth of M. tuberculosis (23). High-density mutagenesis studies have identified the mshD gene (Rv0819) as nonessential for the growth of M. tuberculosis in medium (24). It was recently reported that a mshD mutant was severely attenuated for survival of M. tuberculosis in activated and nonactivated mouse macrophages (22). Thus, it would appear that in M. tuberculosis the expected thiol changes produced by inactivation of mshD are not capable of protecting M. tuberculosis from the oxidative challenge of the macrophage. A detailed characterization of the thiols produced by a M. tuberculosis mshD mutant will address this question.
Acknowledgments
This research was supported by Public Health Service grant AI49174 from the National Institute of Allergy and Infectious Diseases.
John S. Blanchard of the Albert Einstein School of Medicine kindly provided plasmid pMV261-MycRHis6 used to generate M. tuberculosis mycothione reductase. We thank Teresa Koledin for technical assistance, Nancy Buchmeier for constructive comments on the manuscript, and Daniel V. Santi of Kosan Biosciences for helpful comments on acyl-CoA analyses.
REFERENCES
- 1.Beckwith, J., and F. Aslund. 2001. Roles of thiol-redox pathways in bacteria. Annu. Rev. Biochem. 55:21-48. [DOI] [PubMed] [Google Scholar]
- 2.Berges, D. A., W. E. DeWolf, Jr., G. L. Dunn, D. J. Newman, S. J. Schmidt, J. J. Taggart, and C. Gilvarg. 1986. Studies on the active site of succinyl-CoA:tetrahydrodipicolinate N-succinyltransferase. Characterization using analogs of tetrahydrodipicolinate. J. Biol. Chem. 261:6160-6167. [PubMed] [Google Scholar]
- 3.Bornemann, C., M. A. Jardine, H. S. C. Spies, and D. J. Steenkamp. 1997. Biosynthesis of mycothiol: elucidation of the sequence of steps in Mycobacterium smegmatis. Biochem. J. 325:623-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchmeier, N. A., G. L. Newton, T. Koledin, and R. C. Fahey. 2003. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol. Microbiol. 47:1723-1732. [DOI] [PubMed] [Google Scholar]
- 5.Deutsch, J., and H. J. Niclas. 1993. A convenient procedure for the formylation of amines and alcohols using cyanomethylformate. Synthetic Commun. 23:1561-1568. [Google Scholar]
- 6.Ellman, G. L. 1959. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82:70-77. [DOI] [PubMed] [Google Scholar]
- 7.Fahey, R. C., and G. L. Newton. 1987. Determination of low-molecular-weight thiols using monobromobimane fluorescent labeling and high-performance liquid chromatography. Methods Enzymol. 143:85-96. [DOI] [PubMed] [Google Scholar]
- 8.Fenton, S. S., and R. C. Fahey. 1986. Analysis of biological thiols: determination of thiol components of disulfides and thioesters. Anal. Biochem. 154:34-42. [DOI] [PubMed] [Google Scholar]
- 9.Frost, A. A., and R. G. Pearson. 1961. Kinetics and mechanism, 2nd ed., p. 17. John Wiley & Sons, Inc., New York, N.Y.
- 10.Koledin, T., G. L. Newton, and R. C. Fahey. 2002. Identification of the mycothiol synthase gene (mshD) encoding the acetyltransferase producing mycothiol in actinomycetes. Arch. Microbiol. 178:331-337. [DOI] [PubMed] [Google Scholar]
- 11.Marolewski, A., J. M. Smith, and S. J. Benkovic. 1994. Cloning and characterization of a new purine biosynthetic enzyme: a non-folate glycinamide ribonucleotide transformylase from E. coli. Biochemistry 33:2531-2537. [DOI] [PubMed] [Google Scholar]
- 12.Newton, G. L., K. Arnold, M. S. Price, C. Sherrill, S. B. delCardayré, Y. Aharonowitz, G. Cohen, J. Davies, R. C. Fahey, and C. Davis. 1996. Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 178:1990-1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newton, G. L., C. A. Bewley, T. J. Dwyer, R. Horn, Y. Aharonowitz, G. Cohen, J. Davies, D. J. Faulkner, and R. C. Fahey. 1995. The structure of U17 isolated from Streptomyces clavuligerus and its properties as an antioxidant thiol. Eur. J. Biochem. 230:821-825. [DOI] [PubMed] [Google Scholar]
- 14.Newton, G. L., and R. C. Fahey. 2002. Mycothiol biochemistry. Arch. Microbiol. 178:388-394. [DOI] [PubMed] [Google Scholar]
- 15.Newton, G. L., and R. C. Fahey. 1987. Purification of thiols from biological samples. Methods Enzymol. 143:96-101. [DOI] [PubMed] [Google Scholar]
- 16.Newton, G. L., M. D. Unson, S. J. Anderberg, J. A. Aguilera, N. N. Oh, S. B. delCardayré, J. Davies, Y. Av-Gay, and R. C. Fahey. 1999. Characterization of a Mycobacterium smegmatis mutant defective in 1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside and mycothiol biosynthesis. Biochem. Biophys. Res. Commun. 255:239-244. [DOI] [PubMed] [Google Scholar]
- 17.Patel, M. P., and J. S. Blanchard. 1999. Expression, purification, and characterization of Mycobacterium tuberculosis mycothione reductase. Biochemistry 38:11827-11833. [DOI] [PubMed] [Google Scholar]
- 18.Patel, M. P., and J. S. Blanchard. 2001. Mycobacterium tuberculosis mycothione reductase: pH dependence of the kinetic parameters and kinetic isotope effects. Biochemistry 40:3119-3126. [DOI] [PubMed] [Google Scholar]
- 19.Pawelec, M., G. Stochel, and R. van Eldik. 2004. Mechanistic information on the copper-catalysed autoxidation of mercaptosuccinic acid in aqueous solution. Dalton Trans. 2004:292-298. [DOI] [PubMed] [Google Scholar]
- 20.Prise, K. M., N. E. Gillies, A. Whelan, G. L. Newton, R. C. Fahey, and B. D. Michael. 1995. Role of charge in the radioprotection of E. coli by thiols. Int. J. Radiat. Biol. 67:393-401. [DOI] [PubMed] [Google Scholar]
- 21.Rawat, M., G. L. Newton, M. Ko, G. J. Martinez, R. C. Fahey, and Y. Av-Gay. 2002. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents Chemother. 46:3348-3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rengarajan, J., B. R. Bloom, and E. J. Rubin. 2005. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. USA 102:8327-8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sareen, D., G. L. Newton, R. C. Fahey, and N. A. Buchmeier. 2003. Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J. Bacteriol. 185:6736-6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sassetti, C. M., D. H. Boyd, and E. J. Rubin. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77-84. [DOI] [PubMed] [Google Scholar]
- 25.Schleifer, K. H., and O. Kandler. 1972. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36:407-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimazu, M., L. Vetcher, J. L. Galazzo, P. Licari, and D. V. Santi. 2004. A sensitive and robust method for quantification of intracellular short-chain coenzyme A esters. Anal. Biochem. 328:51-59. [DOI] [PubMed] [Google Scholar]
- 27.Steffek, M., G. L. Newton, Y. Av-Gay, and R. C. Fahey. 2003. Characterization of Mycobacterium tuberculosis mycothiol S-conjugate amidase. Biochemistry 42:12067-12076. [DOI] [PubMed] [Google Scholar]
- 28.Stjernholm, R. L., R. E. Noble, and D. Koch-Weser. 1962. Formation of methylmalonyl-CoA and succinyl-CoA by extracts of Mycobacterium smegmatis. Biochim. Biophys. Acta 64:174-177. [DOI] [PubMed] [Google Scholar]
- 29.Takeuchi, N., T. Ueda, and K. Watanabe. 1998. Expression and characterization of bovine mitochondrial methionyl-tRNA transformylase. J. Biochem. (Tokyo) 124:1069-1071. [DOI] [PubMed] [Google Scholar]
- 30.Unson, M. D., G. L. Newton, C. Davis, and R. C. Fahey. 1998. An immunoassay for the detection and quantitative determination of mycothiol. J. Immunol. Methods 214:29-39. [DOI] [PubMed] [Google Scholar]
- 31.Vetting, M. W., L. P. S. de Carvalho, M. Yu, S. S. Hegde, S. Magnet, S. L. Roderick, and J. S. Blanchard. 2005. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433:212-226. [DOI] [PubMed] [Google Scholar]
- 32.Vetting, M. W., S. L. Roderick, M. Yu, and J. S. Blanchard. 2003. Crystal structure of mycothiol synthase (Rv0819) from Mycobacterium tuberculosis shows structural homology to the GNAT family of N-acetyltransferases. Protein Sci. 12:1954-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winterbourn, C. C., A. V. Peskin, and H. N. Parsons-Mair. 2002. Thiol oxidase activity of copper, zinc superoxide dismutase. J. Biol. Chem. 277:1906-1911. [DOI] [PubMed] [Google Scholar]
- 34.Zhang, Y., J. Desharnais, S. E. Greasley, G. P. Beardsley, D. L. Boger, and I. A. Wilson. 2002. Crystal structures of human GAR Tfase at low and high pH and with substrate beta-GAR. Biochemistry 41:14206-14215. [DOI] [PubMed] [Google Scholar]