

Short abstract
A rationale for deciding about whole-genome sequencing projects is discussed, along with other important strategies for understanding the phenotypic divergence of species.
Abstract
Many decisions about genome sequencing projects are directed by perceived gaps in the tree of life, or towards model organisms. With the goal of a better understanding of biology through the lens of evolution, however, there are additional genomes that are worth sequencing. One such rationale for whole-genome sequencing is discussed here, along with other important strategies for understanding the phenotypic divergence of species.
Bioinformaticists and computational biologists working in the field of comparative genomics are largely dependent on datasets generated by others. Working with available data opens up desires for complementary datasets to fill knowledge gaps. In addition to writing grants for experimental laboratories and molecular biology supplies, one can also write an opinion piece to convince others to do some of the dirty work for you; this is what I am attempting to do here. Comparative genomics starts with sequencing. Many have suggested gaps in the tree of life, where additional genome projects will augment current knowledge, either to shorten long 'branches' on the tree of sequenced genomes or to complement existing genome projects. For example, there remain huge gaps in our knowledge of archaea. But with the faith that these gaps will ultimately be filled in, in this article I focus on alternative strategies for directing genomic resources so as to answer fundamental questions in evolution.
The tape of life
A whole class of genomic experiments can be hypothesized through what can be called the 'tape of life' question. Stephen J. Gould wrote in his book Wonderful Life [1], "Wind back the tape of life to the early days of the Burgess shale; let it play again from an identical starting point, and the chance becomes vanishingly small that anything like human intelligence would grace the replay". At the molecular level, the tape of life has been played in parallel. Different species have gone from a similar ancestral point to a similar derived phenotype. In these cases, are the same molecules and pathways driving the phenotypic evolution? Comparative genomics gives us unprecedented opportunities to answer such questions.
A few studies have tried to address the tape-of-life question through analysis of a single gene, such as the melanocortin-1 receptor (MC1R). This receptor plays a role in pigmentation and body/hair color, representing an obvious link between selectable genotype and phenotype. MC1R has been demonstrated to be under such selective pressure in various birds [2] and mammals [3]. In another set of studies, the transcription factor Pitx1, involved in hindlimb formation, has been implicated in parallel evolution of morphologically very distinct types of stickleback fish [4]. At a genomic level, there are whole classes of experiments that can be proposed where phenotypic evolution is the driving force.
As an example of the tape-of-life question played in parallel, terrestrial mammals have returned to the water in at least three independent lineages (see Figure 1). Seals diverged from dogs (which have an ongoing genome project); whales evolved from an ancestor shared with the hippopotamus (there are ongoing terrestrial Artiodactyl genome projects for the somewhat related pig and cow); and manatees evolved from an ancestor shared with hyrax (a small furry mammal also know as a dassie) and elephants (a genome project for elephants has now been funded) [5]. Systematic comparisons of parallel anatomic evolution have been made from several aquatic mammal lineages (see, for example, [6]). In the three branches suggested above, a close terrestrial relative has an ongoing or highly prioritized complete genome sequencing project, but sufficient sequences are not available from the aquatic mammals to allow thorough comparisons. Systematic study of gene sequences, relative expression levels of genes, alternative splicing changes, and other functional data in appropriate related species will allow the type of analysis that tests whether the same pathways, genes, and nucleotide positions were under similar selective pressures during re-adaptation of an ancestral terrestrial mammal to an aqueous environment.
Figure 1.
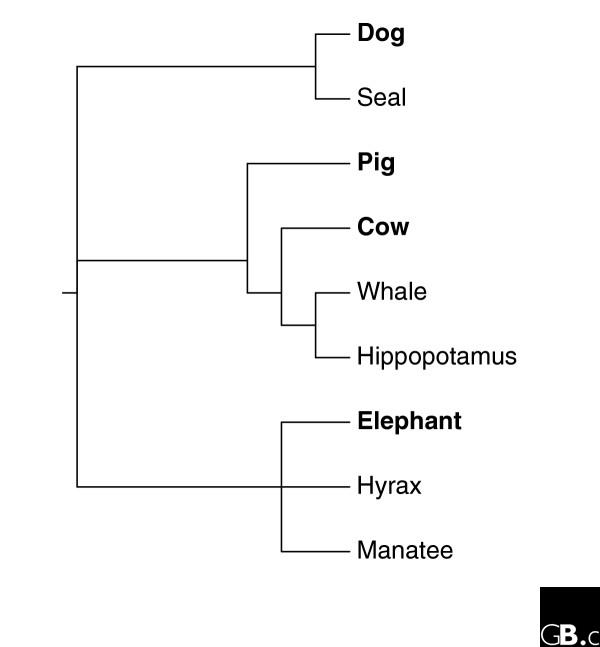
A standard rooted phylogenetic tree of eutherian mammals [5]. It indicates the branches where the aquatic species, seals, whales, and manatees, evolved together with their closest relatives that do (in bold) or do not (plain text) have complete genome sequencing projects. Some relationships are indicated in non-binary nodes where the branching order is not clear.
Cichlid fish (together with Darwin's finches) may be the textbook example(s) of parallel evolution (reviewed in [7,8]). As seen in Figure 2a, haplochromine cichlids from Lake Tanganyika gave rise to a whole diversity of cichlids in Lake Malawi and in Lake Victoria. The entire 600 species of Lake Victoria cichlids diverged from a single lineage of Lake Tanganyika cichlids in about 100,000 years [7]. A similar origin of Lake Malawi cichlids has resulted in species closely resembling more distantly related Lake Tanganyika cichlids, as seen in Figure 2b. Cichlids are another ideal system in which to study the link between selectable genotype and phenotype; many other adaptive regimes, for example cold adaptation, can also be examined in this context, and a draft cichlid genome is now planned by the US Joint Genome Institute [9].
Figure 2.
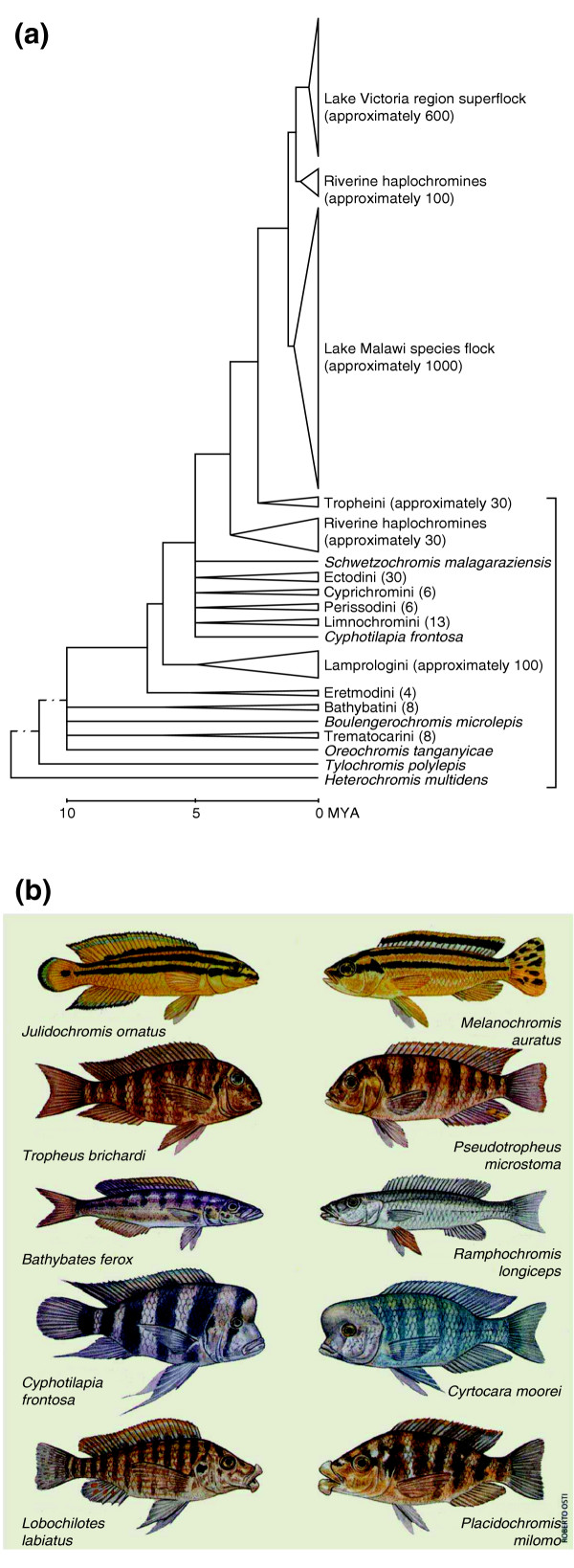
The evolution of cichlid fish. (a) A phylogenetic tree adapted with permission from [7] indicates the origin of Lake Malawi and Lake Victoria cichlids from a single lineage of Lake Tanganyika cichlids; the bracket indicates the Lake Tanganyika and riverine cichlids. MYA, millions of years ago; numbers of species are indicated in parentheses; all species shown are cichlids. (b) A single lineage from the diverse cichlid species of Lake Tanganyika (left) recapitulates a diverse group of cichlid species in Lake Malawi (right) from a single lineage. Many of the Lake Malawi cichlids evolved to fill similar niches to more distantly related species in Lake Tanganyika, and species in similar niches have a surprisingly similar appearance (reproduced from [8] with permission from Roberto Osti).
Rapid phenotypic evolution
Just as genome sequencing projects can be directed at interesting examples of parallel evolution, large-scale sequencing efforts can also be directed at points where phenotypic change appears to have been particularly rapid. This will improve the signal-to-noise ratio in attempting to detect those substitutions that drove phenotypic change. Studies of parallelism in the cichlid fish, especially in Lake Victoria, fall into this category (as well as the parallel evolution category) [7]. In another example, polar bears diverged from brown bears only a little more than 100,000 years ago. The oldest polar bear fossil is less than 100,000 years old [10]. From phylogenetics, polar bears fall within the brown bear clade (see Figure 3), indicating that some brown bears are more closely related to polar bears than they are to other brown bears [11]. During the past 100,000 years, polar bears have undergone changes in body size and morphology, hair color, dietary preference, and habitat, as well as multiple behavioral changes. Morphologists can probably point to other similar examples of rapid phenotypic evolution. Sequencing from species such as these will enable better detection of the links between genotype and phenotype using a comparative approach.
Figure 3.

The relationship of polar bears to other bears. (a) A rooted phylogenetic tree adapted from [11]. Polar bears are thought to have diverged from brown bears from the ABC Islands in Canada after these brown bears had diverged from other brown bears. Black bears and other bear species are more distantly related to both polar bears and brown bears. The pictures show (b) a brown bear (not from the ABC islands) (Ursus arctos) and (c) a polar bear (Ursus maritimus). Original bear images courtesy of Peter Haase, Carl Lund and Michael Petersen (Copenhagen Zoo).
Examination of the tape-of-life question or rapid phenotypic evolution does not need to involve entire genome sequencing. Large-scale full-length cDNA [12,13] and upstream promoter sequence can be generated more cheaply but contains much of the relevant functional information. The molecular basis for changes in coding sequence function, gene expression, and possibly alternative splicing is likely to be contained within such data. Ultimately, population-level data in the form of single nucleotide polymorphisms (SNPs) linked to biogeography will also be desirable, to shed light on the process of speciation.
Regulatory evolution
In addition to coding-sequence evolution, changes in alternative splicing patterns and gene-expression levels and patterns can also contribute to lineage-specific diversification. Large-scale inter-specific datasets that characterize relative splice-site usage or splice-variant frequencies would be valuable. An initial study comparing alternative splicing patterns in mouse, rat, and human led to the conclusion that alternative splice variants, like gene duplicates, have been used as a testbed for evolutionary novelty [14].
Changes in gene expression have become the leading candidates as drivers of evolutionary novelty, dating back to Allan Wilson's attempt to explain the phenotypic divergence between human and chimpanzee [15]. Pioneering work on the evolution of regulatory networks in echinoderms has pointed to a major role for changes in the expression of key regulatory proteins during development in driving morphological change [16]. A systematic examination of gene-expression changes in higher primates has also been presented [17]. The molecular variation in the human population that affects gene expression that is subject to the diversifying selection and fixation seen in inter-specific studies is also being characterized [18] and can be related to chimpanzee sequences in a bid to understand lineage-specific evolution. Extending this in a well controlled study across larger portions of the tree of life (initially at the inter-specific level) is warranted.
Both relative gene-expression levels and relative alternative splicing levels are continuous variables, unlike sequences that are discretely A, C, G or T. There are methods for reconstructing such values over a phylogenetic tree and parsing changes onto branches, coupled to a reconstruction of the regulatory sequences that govern such processes (see, for example, [19]). The power of harnessing phylogenetic information not only provides an understanding of the molecular basis for organismal phenotypic divergence but can also be used to reduce the background 'noise' in attempts to understand basic principles of transcriptional regulation, mRNA splicing, and protein folding and function [19,20].
Even within the completed genomes that we already have, there are many unknown genes. Phylogenetic focusing (systematically attempting to sequence such genes from closely related species) will help us understand how they evolved, their function, and the evolution of novel genes in general. This can also be applied to rare protein structures, in order to understand the process of neostructuralization by searching for phylogenetic intermediates that provide a 'missing link' sequence. Phylogenetic focusing will be greatly aided by the establishment of local DNA banks containing genomic DNA from regionally specific species. This will also aid nations and their regions in understanding local biodiversity.
Ohno [21], and subsequently Lynch and Conery [22], proposed a major role for gene duplication in the generation of evolutionary novelty. Wilson and Davidson and colleagues have done the same for gene expression [15,16]; the Lee lab has done the same for alternative splicing [14]. All are probably right to some degree, as evolution is opportunistic and different regulatory mechanisms have potential different selectable outcomes. Generating datasets that enable us to integrate such knowledge and output better models (also drawing on work in population genetics, structural genomics, and systems biology) will allow a better understanding of biology, with evolution at its core. This article aims to continue a dialog between experimental and computational researchers towards the aim of a better understanding of genomes, and to encourage experimentalists to provide the community with even more varieties of genomic data.
Acknowledgments
Acknowledgements
I thank Axel Meyer for interesting discussions and for providing Figure 2, Peter Haase (Copenhagen Zoo) for providing Figure 3b and 3c and Marie Skovgaard, Matthew Betts, Janos Kodra, and Stephen Liberles for comments and suggestions.
References
- Gould SJ. Wonderful Life: The Burgess Shale and the Nature of History. New York: W.W. Norton & Company; 1989. [Google Scholar]
- Mundy NI, Badcock NS, Hart T, Scribner K, Janssen K, Nadeau NJ. Conserved genetic basis of a quantitative plumage trait involved in mate choice. Science. 2004;303:1870–1873. doi: 10.1126/science.1093834. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Hoekstra HE, D'Agostino SL. The genetic basis of adaptive melanism in pocket mice. Proc Natl Acad Sci USA. 2003;100:5268–5273. doi: 10.1073/pnas.0431157100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jonsson B, Schluter D, Kingsley DM. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Natur. 2004;428:717–723. doi: 10.1038/nature02415. [DOI] [PubMed] [Google Scholar]
- Liu FG, Miyamoto MM, Freire NP, Ong PQ, Tennant MR, Young TS, Gugel KF. Molecular and morphological supertrees for eutherian (placental) mammals. Science. 2001;291:1786–1789. doi: 10.1126/science.1056346. [DOI] [PubMed] [Google Scholar]
- Hatfield JR, Samuelson DA, Lewis PA, Chisholm M. Structure and presumptive function of the iridocorneal angle of the West Indian manatee (Trichechus manatus), short-finned pilot whale (Globicephala macrorhynchus), hippopotamus (Hippopotamus amphibius), and African elephant (Loxodonta africana). Vet Opthalmol . 2003;6:35–43. doi: 10.1046/j.1463-5224.2003.00262.x. [DOI] [PubMed] [Google Scholar]
- Salzburger W, Meyer A. The species flocks of East African cichlid fishes: recent advances in molecular phylogenetics and population genetics. Naturwissenschaften. 2004;91:277–290. doi: 10.1007/s00114-004-0528-6. [DOI] [PubMed] [Google Scholar]
- Stiassny MLJ, Meyer A. Cichlids of the rift lakes. Sci Am. 1999:64–69. [Google Scholar]
- DOE Joint Genome Institute - Why Sequence Cichlid Fish? http://www.jgi.doe.gov/sequencing/why/CSP2006/cichlids.html
- Kurten B. The evolution of the polar bear, Ursus maritimus. Acta Zoologica Fennica. 1964;108:1–26. [Google Scholar]
- Talbot SL, Shields GF. A phylogeny of the bears (Ursidae) inferred from complete sequences of three mitochondrial genes. Mol Phylogenet Evol. 1996;5:567–575. doi: 10.1006/mpev.1996.0051. [DOI] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNA. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Crawford DL. Functional genomics does not have to be limited to a few select organisms. Genome Biol. 2001;2:interactions1001.1–1001.2. doi: 10.1186/gb-2001-2-1-interactions1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nature Genet. 2003;34:177–180. doi: 10.1038/ng1159. [DOI] [PubMed] [Google Scholar]
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- Hinman VF, Nguyen AT, Cameron RA, Davidson EH. Developmental gene regulatory network architecture across 500 million years of echinoderm evolution. Proc Natl Acad Sci USA. 2003;100:13356–13361. doi: 10.1073/pnas.2235868100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard W, Khaitovich P, Klose J, Zollner S, Heissig F, Giavalisco P, Nieselt-Struwe K, Muchmore E, Varki A, Ravid R, et al. Intra- and interspecific variation in primate gene expression patterns. Science. 2002;296:340–343. doi: 10.1126/science.1068996. [DOI] [PubMed] [Google Scholar]
- Rockman MV, Wray GA. Abundant raw material for cis-regulatory evolution in humans. Mol Biol Evol. 2002;19:1991–2004. doi: 10.1093/oxfordjournals.molbev.a004023. [DOI] [PubMed] [Google Scholar]
- Rossnes R, Eidhammer I, Liberles DA. Phylogenetic reconstruction of ancestral character states for gene expression and mRNA splicing data. BMC Bioinformatics. 2005;6:127. doi: 10.1186/1471-2105-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukami-Kobayashi K, Schreiber DR, Benner SA. Detecting compensatory covariation signals in protein evolution using reconstructed ancestral sequences. J Mol Biol. 2002;319:729–743. doi: 10.1016/S0022-2836(02)00239-5. [DOI] [PubMed] [Google Scholar]
- Ohno S. Evolution by Gene Duplication. Berlin: Springer; 1970. [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]