

Abstract
Hexapeptide (des-N-methylleucyl) derivatives of LY264826 were prepared in order to examine further the role of N-substituted hydrophobic side chains in defining the mechanisms of action of semisynthetic glycopeptide antibiotics. The hexapeptide of LY264826 binds to the cell wall intermediate analog l-Lys-d-Ala-d-Ala with a 100-fold lower affinity than LY264826 and inhibits Micrococcus luteus almost 200-fold more poorly than LY264826 does. Alkylation of the 4-epi-vancosamine moiety of the disaccharide significantly enhanced the antibacterial activity of the hexapeptide. Alkylation did not affect the binding affinity for d-alanyl-d-alanine residues; however, it did enhance dimerization 7,000-fold and enhanced binding to bacterial membrane vesicles noticeably compared with the levels of dimerization and binding for the unsubstituted hexapeptide. The findings from this study complement those presented in an earlier report (N. E. Allen, D. L. LeTourneau, and J. N. Hobbs, Jr., J. Antibiot. 50:677-684, 1997) and are consistent with the conclusion that the enhanced antibacterial activities of semisynthetic glycopeptide antibiotics derive from the ability of the hydrophobic side chain to markedly affect both dimerization and binding to bacterial membranes.
The heptapeptide moiety of glycopeptide antibiotics such as vancomycin forms a carboxylate-binding pocket, which binds to d-alanyl-d-alanine (d-Ala-d-Ala) peptidyl residues of disaccharide pentapeptide cell wall intermediates (4, 26, 31, 33). Binding to d-Ala-d-Ala residues can lead to inhibition of transglycosylation and/or transpeptidation; these interactions in turn lead to bacterial growth inhibition (33). LY264826 (A82846B, chloroeremomycin, or chloroorienticin A) differs from vancomycin in having a 4-epi-vancosamine sugar substituted for vancosamine in the disaccharide attached at residue 4 and an additional 4-epi-vancosamine attached at residue 6 of the linear heptapeptide (Fig. 1) (29). The antibacterial activity of LY264826 is approximately four- to eightfold greater than that of vancomycin (24, 27, 35). Several derivatives of LY264826 with N-substituted alkyl hydrophobic side chains on the 4-epi-vancosamine of the disaccharide sugar and exquisite antibacterial activities against both vancomycin-susceptible and -resistant bacteria have been described (12, 25, 27, 28, 34).
FIG. 1.

Chemical structures of glycopeptide antibiotics included in this study. Me, methyl.
Despite differences in antibacterial activities, vancomycin, LY264826, and the hydrophobic side chain derivatives of LY264826 share nearly identical binding affinities for d-ala-d-Ala residues when they are measured in solution with a model peptide (2, 3). VanA and VanB-type strains of vancomycin-resistant enterococci (VRE) contain d-alanyl-d-lactate (d-Ala-d-Lac) depsipeptidyl residues to which vancomycin binds very poorly (8, 32). Many of the hydrophobic side chain derivatives of LY264826 have very good activities against VRE (27, 28), yet these same agents have nearly the same binding affinities as vancomycin and LY264826 for d-Ala-d-Lac residues in solution (2, 3). Clearly, the antibacterial activities of the side chain derivatives cannot be explained on the basis of simple binding affinities for d-Ala-d-Ala and d-Ala-d-Lac residues measured in solution.
LY264826 and its derivatives (as well as some other members of the glycopeptide class) have a strong tendency to self-associate and form homodimers (2, 3, 15, 18). The extent of dimerization of LY264826 is approximately 2 orders of magnitude greater than that of vancomycin (2, 15, 22). The N-substituted derivatives of LY264826 are even more strongly dimerized, with the extent of dimerization apparently determined by the nature of the side chain (3). The N-substituted derivatives also demonstrate a greater tendency to interact with bacterial membranes, a process that could serve to anchor these agents at the in vivo target site (2, 3, 5, 6, 11, 37). Our studies (3) revealed a relatively high degree of correlation between the extent of dimerization of glycopeptide antibiotics and antibacterial activity and a very high degree of correlation between antibacterial activity and the concentration of a d-Ala-d-Ala-containing tripeptide ligand needed in the growth medium to antagonize antibacterial activity. The antagonism experiments are particularly pertinent to the mechanisms of action of these glycopeptides because the results imply that growth inhibition is accompanied by stronger interactions with d-Ala-d-Ala-containing cell wall intermediates at the in vivo target site than with the d-Ala-d-Ala-containing tripeptide ligand in solution. The combined effect of dimerization and membrane anchoring can lead to cooperative interactions that facilitate strong intramolecular effects at the target site that can fully account for the enhanced antibacterial activities. These kinds of interactions are not predicted by estimating association constants in free solution. However, the contribution of intramolecular effects to the binding of glycopeptide antibiotics to d-Ala-d-Ala and d-Ala-d-Lac residues at the bacterial surface has been demonstrated by nuclear magnetic resonance and surface plasmon resonance techniques with a variety of model systems, namely, phosphatidylcholine vesicles (13, 37), lipid monolayers (10, 11), and a whole-cell antagonist binding assay (5).
In this report, we directly address the question of whether addition of an alkyl side chain can compensate for a major reduction in antibacterial activity due to a modification of the carboxylate-binding pocket. The des-N-methylleucyl derivative of LY264826 (LY312607) lacks the N-terminal amino acid of the heptapeptide core (24) which is crucial for tight binding of d-Ala-d-Ala residues into the carboxylate-binding pocket (38). The antibacterial activity and d-Ala-d-Ala residue binding affinity of LY312607 are 2 orders of magnitude lower than those of LY264826. We report here that N substitution of LY312607 results in significantly enhanced dimerization, membrane binding, and antibacterial activity, even though it has no effect on simple binding affinity for d-Ala-d-Ala-containing peptidyl residues in solution.
MATERIALS AND METHODS
Chemicals.
LY264826 was from Eli Lilly & Company. The semisynthetic derivative LY307599 (4-phenylbenzyl) was prepared as described previously (12). Analytical and preparative high-pressure liquid chromatography (HPLC) analyses were conducted as described previously (12). Hexapeptides were prepared by standard Edman degradation techniques (7, 24) as described below. N,N′-Diacetyl-l-Lys-d-Ala-d-Ala was purchased from Sigma Chemical Co.
Experimental procedures. (i) Preparation of LY312607.
The triacetate salt of LY264826 (A82846B) (1.0 g, 0.56 mmol) was dissolved in 20 ml of H2O-pyridine (1:1 [vol/vol]) and treated with phenyl isothiocyanate (0.10 ml, 0.84 mmol). The resulting mixture was stirred at room temperature for 1 h, at which time HPLC analysis indicated complete consumption of the starting material. The reaction mixture was concentrated in vacuo, and the crude product was purified by preparative HPLC to give 0.74 g (68% yield) of the desired thiourea intermediate. Fast atom bombardment mass spectrometry (FAB-MS): calculated for C80H93Cl2N11O26S, 1,725.5; obtained, 1,728.3. A sample of the purified thiourea intermediate (133.4 mg, 0.0772 mmol) was suspended in 25 ml of CH2Cl2, cooled to 0°C, and then treated with trifluoroacetic acid (0.25 ml). After 1 h the reaction mixture was warmed to room temperature and stirred for an additional 2 h. The solvent was removed in vacuo, and the crude product was purified by preparative HPLC to give 51.7 mg (46% yield) of LY312607 as a white powder. FAB-MS: calculated for C66H75Cl2N9O25, 1463.4; obtained, 1,466.7.
(ii) Preparation of LY314015.
LY307599 (41 mg, 0.023 mmol) was dissolved in 4 ml of H2O-pyridine (1:1 [vol/vol]) and treated with phenyl isothiocyanate (4.0 μl, 0.033 mmol). The resulting mixture was stirred at room temperature for 3 h, at which time HPLC analysis indicated complete consumption of the starting material. The reaction mixture was concentrated in vacuo to give the crude thiourea intermediate as a white solid. The thiourea derivative was suspended in 10 ml of CH2Cl2, cooled to 0°C, and then treated with trifluoroacetic acid (0.25 ml). After 30 min the reaction mixture was warmed to room temperature and stirred for an additional 1 h. The solvent was removed in vacuo, and the crude product was purified by preparative HPLC to give 14.0 mg (37% yield) of LY314015 as a white powder. FAB-MS: calculated for C79H85Cl2N9O25, 1,629.5; obtained, 1,632.5.
Ligand binding.
Affinity capillary electrophoresis was used to measure the level of binding of N,N′-diacetyl-l-Lys-d-Ala-d-Ala to glycopeptides by published procedures (9, 17). Electrophoretic mobilities were measured with a P/ACE 5000 (Beckman Instruments) equipped with a fused-silica capillary (57 cm [50 cm to the detector] by 75 μm [inner diameter]). Mesityl oxide was used as a neutral marker. Analytes were detected at 214 nm. The run buffer was sodium phosphate; the ionic strength of the buffer was varied from 20 to 75 mM and the pH was varied from 6.7 to 7.1 to optimize the peak shape. The voltage was consequently varied from 30 kV for buffer with a low ionic strength to 7.5 kV for buffer with a high ionic strength; polarity was positive to negative.
The difference between the electrophoretic mobility of a glycopeptide with a given ligand concentration in the run buffer and the electrophoretic mobility of the glycopeptide in the absence of ligand in the run buffer (ΔμG,L) was calculated by the equation
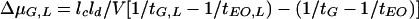 |
(1) |
where lc is the total length of the capillary; ld is the length of the capillary to the detector; V is voltage, tG and tEO are the migration times of the glycopeptide and the neutral marker, respectively, in run buffer with no ligand added; and tG,L tEO,L are the migration times of the glycopeptide and the neutral marker, respectively, with ligand added (17). Binding constants (Kb) were estimated from the relationship
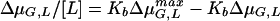 |
(2) |
where [L] is the ligand concentration, and ΔμG,Lmax is the mobility of the glycopeptide when it is saturated with ligand. Kb was determined as the negative slope from a plot of ΔμG,L/[L] against ΔμG,L (17).
Dimerization.
The extent of dimerization of the glycopeptide antibiotics was measured by capillary zone electrophoresis. The methodology used to measure the mobility of glycopeptide antibiotics as a function of their concentration and the mathematics used to estimate dimerization constants (Kdim) have been described previously (21).The electrophoretic mobility (μobs) of a glycopeptide was calculated by the equation
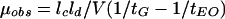 |
(3) |
The concentrations of the glycopeptides as they passed the detector were determined by using the peak height, as explained previously (1). Kdim were calculated by the relationship
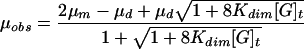 |
(4) |
where μd is the electrophoretic mobility of a hypothetical sample containing all dimer, μm is the electrophoretic mobility of a hypothetical sample containing all monomer, and [G]t is the concentration of glycopeptide (monomer plus dimer) (21). Equation 4 was fitted to μobs and [G]t by nonlinear regression analysis with a curve-fitting program (JMP; SAS Institute).
Membrane binding.
Membrane vesicles were prepared by treatment of Bacillus megaterium with N-acetylmuramidase, DNase, and RNase by previously published methods (20). Binding of glycopeptides to membrane vesicles was measured as described previously (2).
Antibacterial activity and antagonism assays.
MICs were determined for Micrococcus luteus ATCC 9341 grown in TY broth (10 g of tryptone per liter, 5 g of yeast extract per liter, 5 g of NaCl per liter) in a microplate format. Each well contained 200 μl of TY broth, and the inoculum was a 1:40,000 dilution of an overnight culture. The plates were incubated overnight at 35°C and read at 650 nm in a ThermoMax plate reader (Molecular Devices). Antagonism of antibacterial activity by N,N′-diacetyl-l-Lys-d-Ala-d-Ala was determined by using the same microplate format and procedure, except that N,N′-diacetyl-l-Lys-d-Ala-d-Ala was added to the wells prior to inoculation. Each glycopeptide concentration was tested for inhibition in the presence of a range of N,N′-diacetyl-l-Lys-d-Ala-d-Ala concentrations. The molar excess (ME) is the ratio of [N,N′-diacetyl-l-Lys-d-Ala-d-Ala]:[glycopeptide] in the well containing the lowest N,N′-diacetyl-l-Lys-d-Ala-d-Ala concentration that completely suppressed growth inhibition. The values given in Table 1 represent the mean ME calculated for several inhibitory concentrations of glycopeptide. ME values varied by 1 or 2 dilutions when they were tested with different glycopeptide concentrations. Therefore, ME values were determined over a fivefold range around the MIC and were averaged to maximize the accuracy of the ME estimate.
TABLE 1.
Binding of cell wall ligand, dimerization, antibacterial activity, and antagonism by heptapeptide and hexapeptide derivatives of glycopeptide antibiotics
| Glycopeptide | Side chaina | Kb (M−1)b | Kdim (M−1)c | MIC (μM)d | MEe |
|---|---|---|---|---|---|
| LY264826 (heptapeptide) | None (H) | 1.5 × 105 | 8.6 × 103 | 0.057 | 302 |
| LY312607 (hexapeptide) | None (H) | 1.3 × 103 | 7.5 × 101 | 11 | 12 |
| LY307599 (heptapeptide) | N-Phenylbenzyl | 5.9 × 105 | 3.2 × 105 | 0.0084 | 16,500 |
| LY314015 (hexapeptide) | N-Phenylbenzyl | 1.0 × 103 | 5.6 × 105 | 0.48 | 1,032 |
See Fig. 1 for chemical structures.
Kb, binding constant to N,N′-diacetyl-l-Lys-d-Ala-d-Ala.
Kdim, dimerization constant.
Lowest concentration required to inhibit growth of M. luteus ATCC 9341.
ME (molar excess) of N,N′-diacetyl-l-Lys-d-Ala-d-Ala ligand required to completely suppress growth inhibition of M. luteus ATCC 9341.
RESULTS AND DISCUSSION
N-substituted derivatives of LY264826 have greater antibacterial activities than the unsubstituted parent compound derived, at least in part, from the augmentation of dimerization and membrane anchoring (3, 5, 37). Strongly dimerized glycopeptides have the advantage that their interactions with d-Ala-d-Ala residues (and the d-Ala-d-Lac residues of vancomycin-resistant bacteria) can be influenced and driven by intramolecular effects (5, 6, 10, 11, 23, 30, 37). These kinds of interactions are much stronger and more complex than the bimolecular interactions that typify the actions of more weakly dimerized glycopeptides such as vancomycin. Findings from this laboratory (2, 3) and others (5, 11, 37) support the notion that the enhanced antibacterial activities of LY264826 and its N-substituted derivatives against both vancomycin-susceptible and -resistant bacteria derive from the cooperative effects of dimerization and membrane anchoring.
Hexapeptide derivatives of glycopeptide antibiotics lack the N-terminal amino acid of the carboxylate-binding pocket and therefore lack one of the hydrogen bonds necessary for binding to cell wall precursors. Loss of this amino acid is accompanied by a 100-fold reduction in binding affinity and a comparable loss in antibacterial activity (38). We have examined the influence of the addition of a hydrophobic side chain to the hexapeptide of LY264826 on its antibacterial properties. Table 1 presents the binding constants for N,N′-diacetyl-l-Lys-d-Ala-d-Ala, dimerization constants, MICs for M. luteus, and a measure of the ME of N,N′-diacetyl-l-Lys-d-Ala-d-Ala required to completely antagonize growth inhibition for LY264826, LY312607 (the des-N-methylleucyl hexapeptide analog of LY264826), LY307599 (the N-phenylbenzyl side chain derivative of LY264826), and LY314015 (LY312607 substituted with an N-phenylbenzyl side chain) (see Fig. 1 for the structures of these compounds).
The hexapeptide LY312607 is nearly 200 times less active than LY264826 against M. luteus. The MICs of LY312607 for other gram-positive bacteria, including vancomycin-susceptible and -resistant bacteria, were higher but followed the same pattern (data not shown). The binding affinity for N,N′-diacetyl-l-Lys-d-Ala-d-Ala was reduced 100-fold. Interestingly, the extent of dimerization of LY312607 was approximately 100-fold less than that of LY264826, and less free N,N′-diacetyl-l-Lys-d-Ala-d-Ala ligand was required for suppression of bacterial growth inhibition by LY312607 than for suppression of bacterial growth inhibition by LY264826. The intermolecular hydrogen bond formed between the amino group of the 4-epi-vancosamine on residue 6 and the carbonyl on residue 2 of the complementary half of the dimer is important for dimerization (18, 22); loss of the N-methylleucine may interfere with this interaction and compromise dimerization. The increased MIC of LY312607 could be the result of a reduction in both the binding affinity and the extent of dimerization of this compound.
The hexapeptide LY314015 contains an N-phenylbenzyl side chain substitution. The presence of the hydrophobic side chain on this molecule did not affect the binding affinity for N,N′-diacetyl-l-Lys-d-Ala-d-Ala but did enhance the extent of dimerization by 4 orders of magnitude over that for the hexapeptide without a side chain. LY314015 dimerized as well as LY307599 (a heptapeptide) (Fig. 1 and Table 1), consistent with the side chain playing a significant role in dimerization (3). The antibacterial activity of LY314015 was also an order of magnitude greater than that of LY312607 but was still more than an order of magnitude less than that of LY307599. The fact that a 1,032-fold ME of N,N′-diacetyl-l-Lys-d-Ala-d-Ala was sufficient for suppression of growth inhibition by LY314015 whereas a 16,500-fold ME was required for suppression of growth inhibition by LY307599 may be related to the reduced binding affinity that LY314015 has for d-Ala-d-Ala-containing peptidyl residues.
Hydrophobic side chains on glycopeptide antibiotics facilitate interactions with membranous structures (2, 3, 5, 6, 11, 22). We and others have proposed that hydrophobic side chains act as anchors at the membrane outer interface and facilitate intramolecular effects, leading to very tight interactions with d-Ala-d-Ala residues of cell wall intermediates. The N-phenylbenzyl hexapeptide derivative (LY314015) showed greater binding than the hexapeptide without a side chain to B. megaterium membrane vesicles (Fig. 2). Increased membrane anchoring and enhanced dimerization by LY314015 can reasonably account for the increased ME of N,N′-diacetyl-l-Lys-d-Ala-d-Ala required to suppress in vitro antibacterial activity. The MIC of LY314015 is higher than that of LY307599, and the ME of N,N′-diacetyl-l-Lys-d-Ala-d-Ala needed to antagonize inhibition by LY314015 is lower than the amount needed to antagonize inhibition by LY307599. Nevertheless, the differences between the values measured for the unsubstituted hexapeptide and the hexapeptide with a side chain are dramatic and signify that addition of the side chain to the hexapeptide was able to compensate for the reduced binding affinity of the modified carboxylate-binding pocket to d-Ala-d-Ala residues.
FIG. 2.

Binding of hexapeptide derivatives to membrane vesicles.
N-substituted derivatives of vancomycin (with alkyl and aryl substitutions on the vancosamine sugar) with enhanced activity against vancomycin-susceptible and -resistant bacteria have been reported (14, 16). N-Chlorophenylbenzyl-vancomycin was found to be 30 times more active than vancomycin against vancomycin-susceptible enterococci and 100 times more active than vancomycin against VRE. The same group (14, 19) reported that the hexapeptide (des-N-methylleucyl) derivative of N-chlorophenylbenzyl-vancomycin was nearly as active as the heptapeptide parent compound against VRE. The investigators proposed that the unexpected antibacterial activity of the hexapeptide of N-chlorophenylbenzyl-vancomycin against both vancomycin-susceptible and -resistant bacteria was due to direct inhibition of the transglycosylase enzyme and argued that the weak binding of the hexapeptide derivative to dipeptidyl residues could not explain the observed inhibitory effects. The fact that both N-chlorophenylbenzyl-vancomycin and N-chlorophenylbenzyl-des-N-methylleucyl-vancomycin inhibited peptidoglycan formation and induced lipid II accumulation in an in vitro particulate membrane-based assay with either a pentapeptide or a tetrapeptide substrate was cited as further evidence that these agents were acting directly on transglycosylase without directly binding to d-Ala-d-Ala or d-Ala-d-Lac residues (14, 16).
We have shown that N substitution of LY264826 with a hydrophobic side chain dramatically enhances dimerization, membrane interactions, and antibacterial activity without affecting simple binding affinity for d-Ala-d-Ala or d-Ala-d-Lac residues (1-3). Oritavancin (LY333328) is the N-chlorophenylbenzyl derivative of LY264826 undergoing clinical trials for the treatment of serious infections caused by gram-positive bacteria, including those involving VRE (28, 36). We (2, 3) and others (5, 6, 11, 37) have proposed that classic chelate phenomena and the attendant intramolecular effects can explain the enhanced antibacterial activities of these derivatives in vivo. We have not directly measured the effects that dimerization and membrane anchoring have on interactions with cell wall intermediates at the in vivo target site. Nevertheless, it is difficult to reconcile the results from antagonism experiments (note the ME values in Table 1) with a mechanism that does not accommodate a role for a direct interaction with dipeptidyl and didepsipeptidyl residues. It should be noted that the antagonism studies were conducted with d-Ala-d-Ala-containing bacteria. A role for dimerization and anchoring in the mechanism of action against d-Ala-d-Lac-containing bacteria derives from previous work of others (10, 11, 13, 37) with various model systems and by inference from our studies reported here.
This study further emphasizes the significant role played by hydrophobic side chains in the mechanisms of action of semisynthetic glycopeptide antibiotics. We have no evidence for a direct effect of any of the N-substituted derivatives of LY264826 on transglycosylation or transpeptidation. However, despite the high correlations among the extent of dimerization, the MIC, and the degree of antagonism (3), our findings do not rule out alternative mechanisms including those that do not rely on interaction with d-Ala-d-Ala or d-Ala-d-Lac residues (14, 16, 19). We believe that the most straightforward explanation for the extraordinary activities of these compounds is through an association with cell wall residues made possible by dimerization and membrane anchoring. We previously reported (3) that an analog of vancomycin with an N-phenylbenzyl substitution (the vancomycin analog of LY307599) dimerized readily (2 orders of magnitude more readily than vancomycin, as measured by capillary electrophoresis) and showed significant binding to membrane vesicles. Although we did not examine des-N-methylleucyl derivatives of vancomycin in detail, we note that N-substituted derivatives of both vancomycin and LY264826 have enhanced activities against vancomycin-susceptible and -resistant bacteria, show enhanced extents of dimerization and membrane anchoring, and higher ME values. The findings argue in favor of related mechanisms of action for both the vancomycin- and LY264826-type glycopeptides at the in vivo target site.
REFERENCES
- 1.Allen, N. E., J. N. Hobbs, Jr., and T. I. Nicas. 1996. Inhibition of peptidoglycan biosynthesis in vancomycin-susceptible and -resistant bacteria by a semisynthetic glycopeptide antibiotic. Antimicrob. Agents Chemother. 40:2356-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen, N. E., D. L. LeTourneau, and J. N. Hobbs, Jr. 1997. Molecular interactions of a semisynthetic glycopeptide antibiotic with d-alanyl-d-alanine and d-alanyl-d-lactate residues. Antimicrob. Agents Chemother. 41:66-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, N. E., D. L. LeTourneau, and J. N. Hobbs, Jr. 1997. The role of hydrophobic side chains as determinants of antibacterial activity of semisynthetic glycopeptide antibiotics. J. Antibiot. 50:677-684. [DOI] [PubMed] [Google Scholar]
- 4.Barna, J. C. J., and D. H. Williams. 1984. The structure and mode of action of glycopeptide antibiotics of the vancomycin group. Annu. Rev. Microbiol. 38:339-357. [DOI] [PubMed] [Google Scholar]
- 5.Beauregard, D. A., A. J. Maguire, D. H. Williams, and P. E. Reynolds. 1997. Semiquantitation of cooperativity in binding of vancomycin-group antibiotics to vancomycin-susceptible and -resistant organisms. Antimicrob. Agents Chemother. 41:2418-2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beauregard, D. A., D. H. Williams, M. N. Gwynn, and D. J. Knowles. 1995. Dimerization and membrane anchors in extracellular targeting of vancomycin group antibiotics. Antimicrob. Agents Chemother. 39:781-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Booth, P. M., D. J. M. Stone, and D. W. Williams. 1987. The Edman degradation of vancomycin: preparation of vancomycin hexapeptide. J. Chem. Soc. Chem. Commun. 22:1694-1695. [Google Scholar]
- 8.Bugg, T. D. H., G. D. Wright, S. Dutka-Malen, M. Arthur, P. Courvalin, and C. T. Walsh. 1991. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 30:10408-10415. [DOI] [PubMed] [Google Scholar]
- 9.Chu, Y.-H., and G. M. Whitesides. 1992. Affinity capillary electrophoresis can simultaneously measure binding constants of multiple peptides to vancomycin. J. Org. Chem. 57:3524-3525. [Google Scholar]
- 10.Cooper, M. A., M. T. Fiorini, C. Abell, and D. H. Williams. 2000. Binding of vancomycin group antibiotics to d-alanine and d-lactate presenting self-assembled monolayers. Bioorg. Med. Chem. 8:2609-2616. [DOI] [PubMed] [Google Scholar]
- 11.Cooper, M. A., and D. H. Williams. 1999. Binding of glycopeptide antibiotics to a model of a vancomycin-resistant bacterium. Chem. Biol. 6:891-899. [DOI] [PubMed] [Google Scholar]
- 12.Cooper, R. D. G., N. J. Snyder, M. J. Zweifel, M. A. Staszak, S. C. Wilkie, T. I. Nicas, D. L. Mullen, T. F. Butler, M. J. Rodriguez, B. E. Huff, and R. C. Thompson. 1996. Reductive alkylation of glycopeptide antibiotics: synthesis and antibacterial activity. J. Antibiot. 49:575-581. [DOI] [PubMed] [Google Scholar]
- 13.Entress, R. M. H., R. J. Dancer, D. P. O'Brien, A. C. Try, M. A. Cooper, and D. H. Williams. 1998. 19F NMR in the measurement of binding affinities of chloroeremomycin to model bacterial cell-wall surfaces that mimic VanA and VanB resistance. Chem. Biol. 5:329-337. [DOI] [PubMed] [Google Scholar]
- 14.Ge, M., Z. Chen, H. R. Onishi, J. Kohler, L. L. Silver, R. Kerns, S. Fukuzawa, C. Thompson, and D. Kahne. 1999. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding d-Ala-d-Ala. Science (Washington, D.C.) 284:507-511. [DOI] [PubMed] [Google Scholar]
- 15.Gerhard, U., J. P. Mackay, R. A. Maplestone, and D. H. Williams. 1993. The role of the sugar and chlorine substituents in the dimerization of vancomycin antibiotics. J. Am. Chem. Soc. 115:232-237. [Google Scholar]
- 16.Goldman, R. C., E. R. Baizman, C. B. Longley, and A. A. Branstrom. 2000. Chlorobiphenyl-desleucyl-vancomycin inhibits the transglycosylation process required for peptidoglycan synthesis in bacteria in the absence of dipeptide binding. FEMS Microbiol. Lett. 183:209-214. [DOI] [PubMed] [Google Scholar]
- 17.Gomez, F. A., L. Z. Avila, Y.-H. Chu, and G. M. Whitesides. 1994. Determination of binding constants of ligands to proteins by affinity capillary electrophoresis: compensation for electroosmotic flow. Anal. Chem. 66:1785-1791. [DOI] [PubMed] [Google Scholar]
- 18.Groves, P., M. S. Searle, J. P. Mackay, and D. H. Williams. 1994. The structure of an asymmetric dimer relevant to the mode of action of the glycopeptide antibiotics. Structure (London) 2:747-754. [DOI] [PubMed] [Google Scholar]
- 19.Kerns, R., S. D. Dong, S. Fukuzawa, J. Carbeck, J. Kohler, L. Silver, and D. Kahne. 2000. The role of hydrophobic substituents in the biological activity of glycopeptide antibiotics. J. Am. Chem. Soc. 122:12608-12609. [Google Scholar]
- 20.Konings, W. N., A. Bisschop, M. Veenhuis, and C. A. Vermeulen. 1973. New procedure for the isolation of membrane vesicles of Bacillus subtilis and an electron microscopy study of their ultrastructure. J. Bacteriol. 116:1456-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.LeTourneau, D. L., and N. E. Allen. 1997. Use of capillary electrophoresis to measure dimerization of glycopeptide antibiotics. Anal. Biochem. 246:62-66. [DOI] [PubMed] [Google Scholar]
- 22.Mackay, J. P., U. Gerhard, D. A. Beauregard, R. A. Maplestone, and D. H. Williams. 1994. Dissection of the contributions towards dimerization of glycopeptide antibiotics. J. Am. Chem. Soc. 116:4573-4580. [Google Scholar]
- 23.Mackay, J. P., U. Gerhard, D. A. Beauregard, M. S. Westwell, M. S. Searle, and D. H. Williams. 1994. Glycopeptide antibiotic activity and the possible role of dimerization: a model for biological signalling. J. Am. Chem. Soc. 116:4581-4590. [Google Scholar]
- 24.Nagarajan, R. 1994. Structure-activity relationships of vancomycin antibiotics, p. 195-218. In R. Nagarajan (ed.), Glycopeptide antibiotics. Marcel Dekker, Inc., New York, N.Y.
- 25.Nagarajan, R. 1993. Structure-activity relationships of vancomycin-type glycopeptide antibiotics. J. Antibiot. 46:1181-1195. [DOI] [PubMed] [Google Scholar]
- 26.Nicas, T. I., and N. E. Allen. 1994. Resistance and mode of action, p. 219-241. In R. Nagarajan (ed.), Glycopeptide antibiotics. Marcel Dekker, Inc., New York, N.Y.
- 27.Nicas, T. I., D. L. Mullen, J. E. Flokowitsch, D. A. Preston, N. J. Snyder, R. E. Stratford, and R. D. D. Cooper. 1995. Activities of the semisynthetic glycopeptide LY191145 against vancomycin-resistant enterococci and other gram-positive bacteria. Antimicrob. Agents Chemother. 39:285-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicas, T. I., D. L. Mullen, J. E. Flokowitsch, D. A. Preston, N. J. Snyder, M. J. Zweifel, S. C. Wilkie, M. J. Rodriguez, R. C. Thompson, and R. D. G. Cooper. 1996. Semisynthetic glycopeptide antibiotics derived from LY264826 active against vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 40:2194-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicolaou, K. C., C. N. C. Boddy, S. Brase, and N. Winssinger. 1999. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. 38:2097-2152. [DOI] [PubMed] [Google Scholar]
- 30.O'Brien, D. P., R. M. H. Entress, M. A. Cooper, S. W. O'Brien, A. Hopkinson, and D. H. Williams. 1999. High affinity surface binding of a strongly dimerizing vancomycin-group antibiotic to a model of resistant bacteria. J. Am. Chem. Soc. 121:5259-5265. [Google Scholar]
- 31.Perkins, H. R., and M. Nieto. 1974. The chemical basis for the action of the vancomycin group of antibiotics. Ann. N. Y. Acad. Sci. 235:348-363. [DOI] [PubMed] [Google Scholar]
- 32.Popienick, P. H., and R. F. Pratt. 1987. A fluorescent ligand for binding studies with glycopeptide antibiotics of the vancomycin class. Anal. Biochem. 165:108-113. [DOI] [PubMed] [Google Scholar]
- 33.Reynolds, P. E. 1989. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 8:943-950. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez, M. J., N. J. Snyder, M. J. Zweifel, S. C. Wilkie, D. R. Stack, R. D. G. Cooper, T. I. Nicas, D. L. Mullen, T. F. Butler, and R. C. Thompson. 1998. Novel glycopeptide antibiotics: N-alkylated derivatives active against vancomycin-resistant enterococci. J. Antibiot. 51:560-569. [DOI] [PubMed] [Google Scholar]
- 35.Rolston, K. V. I., H. Nguyen, and M. Messer. 1990. In vitro activity of LY264826, a new glycopeptide antibiotic, against gram-positive bacteria isolated from patients with cancer. Antimicrob. Agents Chemother. 34:2137-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rupp, M. E., P. D. Fey, and G. M. Longo. 2001. Effect of LY333328 against vancomycin-resistant Enterococcus faecium in a rat central venous catheter-associated infection model. J. Antimicrob. Chemother. 47:705-707. [DOI] [PubMed] [Google Scholar]
- 37.Sharman, G. J., A. C. Try, R. J. Dancer, Y. R. Cho, T. Staroske, B. Bardsley, A. J. Maguire, M. A. Cooper, D. P. O'Brien, and D. H. Williams.. 1997. The roles of dimerization and membrane anchoring in activity of glycopeptide antibiotics against vancomycin-resistant bacteria. J. Am. Chem. Soc. 119:12041-12047. [Google Scholar]
- 38.Walsh, C. T., S. L. Fisher, I. S. Park, M. Prahalad, and Z. Wu. 1996. Bacterial resistance to vancomycin: five genes and one missing hydrogen bond tell the story. Chem. Biol. 3:21-28. [DOI] [PubMed] [Google Scholar]