

Abstract
Human immunodeficiency virus (HIV)-related opportunistic infections continue to occur in patients who are newly diagnosed with HIV infection, those in the early course of highly active antiretroviral therapy or nonadherent to HIV care, and other immunosuppressed individuals. One of the most common opportunistic infections in these patients is Pneumocystis pneumonia. CD8+ T cells are recruited to the lung after P. carinii infection and have been associated with both lung injury and host defense. This variability may be due to subpopulations of CD8+ T cells recruited to the lung. We have previously shown using adoptive transfer studies that in vivo-generated T-cytotoxic-1 (Tc1) CD8+ T cells, defined by the secretion of gamma interferon (IFN-γ), have effector activity against Pneumocystis spp. in vitro as well as in vivo. To better understand the mechanisms of these effects, we generated, expanded, and tested Tc1 and Tc2 CD8+ T cells specific for P. murina ex vivo. Tc1-polarized CD8+ T cells secreted higher levels of IFN-γ and granulocyte-macrophage colony-stimulating factor (GM-CSF) and lower levels of interleukin-4 (IL-4), IL-5, IL-10, and IL-13 than Tc2 CD8+ T cells when stimulated with P. murina antigen. Moreover, Tc1 CD8+ T cells demonstrated enhanced effector activity in a macrophage-mediated killing assay which was independent of cell contact. The augmentation in macrophage-mediated P. murina killing was significantly abrogated when GM-CSF was neutralized in the Tc1 CD8+ T cells. These data support the possibility that antigen-specific GM-CSF secretion is critical for effector activity of P. murina-specific Tc1 CD8+ T cells in vitro.
Pneumocystis jiroveci is an opportunistic pathogen of particular importance since the onset of the AIDS epidemic (19, 22). Chemoprophylaxis of human immunodeficiency virus (HIV)-infected patients at high clinical risk for Pneumocystis pneumonia and the introduction of highly active antiretroviral therapy have contributed to a reduced incidence of Pneumocystis pneumonia (19). Despite the success of these clinical interventions, Pneumocystis pneumonia remains one of the most common opportunistic pneumonias and the most common life-threatening infectious complication in HIV-infected patients (19). The first-line agent for effective therapy and chemoprophylaxis remains trimethoprim-sulfamethoxazole, and corticosteroids represent an important adjunctive agent in patients with hypoxemia (21). However, treatment failures, high rates of adverse drug reactions, drug intolerance to first-line antimicrobials, and drug resistance represent formidable challenges to the management and treatment of AIDS-related P. carinii pneumonia (19, 21).
The recruitment of CD4+ T cells into lung tissue is requisite for efficient host defense against Pneumocystis spp. (13, 24). The role of CD8+ T cells in Pneumocystis pneumonia is less clear (4, 7, 10, 14, 31). Our laboratory has examined host defense against P. murina in mice specifically depleted of CD4+ T lymphocytes, which mimics the immunologic defect of HIV infection (13, 24). We have demonstrated that gamma interferon (IFN-γ) overexpression using adenovirus-mediated gene transfer (AdIFN) results in the clearance of P. murina in the absence of CD4+ T cells (14, 18). The mechanism for the clearance of P. murina with AdIFN involves CD8+ T cells, as depletion of this T-cell subset abrogated the effect of AdIFN on P. murina clearance (14). AdIFN resulted in the pulmonary recruitment of P. murina-specific T-cytotoxic-1 (Tc1)-like CD8+ T cells, defined by high levels of endogenous IFN-γ production. These cells have in vitro effector activity against P. murina in a macrophage coculture assay and affect the clearance of the organism upon adoptive transfer into P. murina-infected scid mice (18). Moreover, CD8+ T cells without a Tc1 phenotype, from mice treated with a control adenovirus vector, lacked in vitro effector activity against P. murina and upon adoptive transfer into P. murina-infected scid mice resulted in significantly worse lung injury (18).
To gain a better understanding of the potential mechanisms by which Tc1 CD8+ T cells have effector activity against P. murina, we generated and expanded Tc1 and Tc2 CD8+ T cells specific for P. murina. Polarized CD8+ T-cell populations were tested directly against P. murina or in macrophage coculture assays. Tc1-polarized CD8+ T cells were found to elaborate significantly higher levels of IFN-γ and granulocyte-macrophage colony-stimulating factor (GM-CSF) in antigen recall assays and lower levels of interleukin-4 (IL-4) than Tc2 CD8+ T cells. Moreover, P. murina-specific Tc1 CD8+ T cells had significantly greater effector activity against P. murina, but only in the presence of macrophages. Conditioned medium from antigen-stimulated Tc1 CD8+ T cells also augmented macrophage-mediated killing of P. murina, suggesting that a soluble factor may be responsible for the increase in macrophage-mediated killing of P. murina by Tc1 CD8+ T cells. Among the cytokines differentially expressed between Tc1 CD8+ T cells with effector activity and Tc2 CD8+ T cells without effector activity were IFN-γ and GM-CSF. Antibody neutralization studies demonstrated that GM-CSF secretion by Tc1 cells is critical for effector activity in vitro. Moreover, recombinant GM-CSF was more potent at increasing macrophage-mediated killing of P. murina than IFN in peritoneal macrophages, resident alveolar macrophages, and monocyte chemoattractant protein 1 (MCP-1)-elicited alveolar macrophages. Taken together, these data suggest that the antigen-specific release of GM-CSF may be critical for host defense against P. murina.
MATERIALS AND METHODS
Reagents.
Murine IFN-γ, IL-2, IL-4, IL-12, anti-IFN-γ, anti-GM-CSF, and anti-IL-4 were from Biosource, Camarillo, California. Murine GM-CSF was from R&D Systems (Minneapolis, MN).
Mice.
Male BALB/c mice, 6 to 8 weeks of age, were obtained from Jackson Laboratories (Bar Harbor, ME). They were kept in the American Association for Laboratory Animal Science-certified animal facility of the Rangos Research Center and housed in microisolator caging in keeping with National Institutes of Health guidelines for animal care in a specific-pathogen-free environment. Animal protocols for these experiments were approved by the Children's Hospital of Pittsburgh Animal Research and Care Committee.
P. murina antigen vaccine and vaccination protocol.
The schema for the generation of antigen-specific CD8+ T cells is outlined in Fig. 1. P. murina organisms were isolated from lung tissue of BALB/c scid mice that were previously inoculated with P. murina and purified by differential centrifugation as previously described (18, 34), and protein antigen was produced by sonication for 5 min. For vaccine preparation, antigen was adsorbed on complete Freund adjuvant (for Tc1 cells; Sigma, St. Louis, MO) or aluminum hydroxide gel (for Tc2 cells; Sigma). For Tc1 booster inoculations, incomplete Freund adjuvant (IFA) was used instead of colonization factor antigen (CFA). The final concentration of antigen was 0.25 mg/ml. Male BALB/c mice, 6 to 8 weeks old, were vaccinated with 25 μg of P. murina antigen adsorbed on CFA or alum via intraperitoneal injection. At day 14, mice were boosted with the same dose of antigen adsorbed on IFA or alum.
FIG. 1.

Protocol for Tc1 and Tc2 clone generation. Male BALB/c mice were immunized with P. murina antigen in CFA (Tc1) or alum (Tc2), followed by boosting with P. murina antigen in IFA (Tc1) or alum (Tc2). At day 21, after the first vaccine inoculation, the mice were sacrificed and splenocytes were harvested. After isolation, CD8+ T cells were grown in Tc1 or Tc2 medium for 7 days, followed by 48 h of antigen resting. After this period, CD8+ T cells were restimulated in vitro with ConA or with DCs pulsed with antigen (DC/PC pulsed) (antigen recall assays). Cells and supernatants were collected 48 h later and assayed for in vitro effector activity against P. murina prior to phenotyping. IP, intraperitoneal.
Generation of P. murina-specific Tc1 and Tc2 CD8+ T cells.
Mice were sacrificed at day 7 after the booster vaccination. Spleens were collected and passed through a 70-μm filter and a 40-μm filter (BD Falcon, Franklin Lakes, NJ). After centrifugation at 1,800 rpm for 10 min, the cell pellet was resuspended in ice-cold red blood cell lysis buffer (8.02 g of NH4Cl, 0.84 g of NaHCO3, 0.37 g of EDTA, 100 ml of H2O at pH 7.4 and 4°C) for 3 min. After being centrifuged and double washed, cells were resuspended in medium and enumerated on a hemacytometer. Trypan blue staining was used for viability determination. CD8+ T cells were then isolated using CD8a (Ly-2) microbead columns (Miltenyi Biotec, Auburn, CA), following the manufacturer's protocol. In brief, cells were exposed to microbeads and incubated for 15 min at 6 to 12°C. After being washed, cells were applied to columns, and negative cells were allowed to pass through. Columns were removed from the separator, an appropriate amount of buffer was added, and the positive fraction was recovered. To determine the purity of the separation, cells were stained with phycoerythrin-conjugated anti-CD8a (BD PharMingen, San Diego, CA). The purity of double-selected cells was >96% by flow cytometry and contained <1% CD4+ T cells.
Bone marrow-derived dendritic cells (DCs) were used as antigen-presenting cells and were obtained from hematopoietic progenitors from the femurs of 6- to 8-week-old male BALB/c mice (Charles River Laboratories, Worcester, Massachusetts). The cells were grown in complete RPMI 1640 medium (10% fetal bovine serum, 2 mM l-glutamine, 100 mg/ml streptomycin, and 100 units/ml penicillin) supplemented with 100 units/ml recombinant mouse GM-CSF and 20 ng/ml recombinant mouse IL-4 (both from R&D Systems Inc., Minneapolis, Minnesota). Loosely adherent cells were harvested on day 6 by gentle pipetting. DC preparations were over 90% positive for major histocompatibility complex class II (I-A) and CD11c, with less than 1% of the cells staining for CD4, CD8, CD19, or DX-5 (BD PharMingen, San Diego, California). DCs were matured with 1 μg/ml of soluble CD40 ligand (R&D Systems, Minneapolis, MN). DCs were pulsed with P. murina antigen 24 h later.
CD8+ T cells (2 × 105) were added to unpulsed or P. murina-pulsed DC cultures, and conditioned medium was added at the initiation of the culture to achieve optimal growth and complete polarization. IL-2 (50 U/ml), IL-12 (100 U/ml), and anti-IL-4 (10 μg/ml) were present in medium for Tc1 polarization. IL-2 (50 U/ml), IL-4 (200 U/ml), and anti-IFN-g (10 U/ml) were present in medium for Tc2 polarization. After 7 days, Tc1 and Tc2 cells were harvested and cultured for 48 h in the presence of IL-2 (50 U/ml) only as an antigen resting period. Polarized T-cell populations were restimulated with fresh DCs pulsed with P. murina antigen or with concanavalin A (ConA).
Cytokine analysis.
Supernatants were collected from the cultures after 48 h of T-cell restimulation, and levels of IL-4, IL-5, IL-10, IL-13, IFN-γ, and GM-CSF were assayed by using BioPlex (Bio-Rad Laboratories, Inc.) following the manufacturer's instructions. The data were analyzed using BioPlex Manager software.
Peritoneal and alveolar macrophage isolation.
To isolate peritoneal macrophages, male BALB/c mice were administered 3% sterile thioglycolate intraperitoneally. Five days after injection, the animals were euthanized, and a peritoneal lavage was performed using 10 ml RPMI 1640 medium. The lavage fluid was centrifuged at 300 × g for 10 min, and the subsequent cell pellets were enumerated using a hemacytometer. For residential alveolar macrophage isolation, male BALB/c mice were euthanized by administration of intraperitoneal pentobarbital and exsanguination. Thereafter, lungs were lavaged via an intratracheal catheter with prewarmed (37°C) calcium and magnesium-free phosphate-buffered saline supplemented with 0.6 mM EDTA. A total of 10 ml was used in each mouse in 0.5-ml increments with a 30-s dwell time. The lavage fluids were pooled and centrifuged at 300 × g for 10 min, and the cells were collected for the coculture assay. For MCP-1-elicited alveolar macrophages, BALB/c mice were injected intratracheally with recombinant MCP-1 protein (5 μg/mouse). Forty-eight hours after MCP-1 administration, the animals were euthanized and lavaged as described for residential alveolar macrophage isolation. To ensure that each cell preparation was >97% macrophages, 25,000 cells were cytospun onto slides and stained with Dif-Quik (Baxter, McGaw Park, IL).
P. murina killing assay.
To evaluate the in vitro effector activity of CD8+ T cells, we performed an in vitro killing assay of P. murina as previously described (18, 26, 34). Macrophages were resuspended at a concentration of 106/ml. Cells (100 μl) were cocultured with intact P. murina organisms (103 cysts) with or without CD8+ T cells for 24 h at 37°C and 5% CO2. Controls included P. murina incubated with medium alone. Total RNA was extracted from each well using Trizol LS reagent (Invitrogen, Carlsbad, CA). The viability of P. murina was analyzed through real-time PCR measurement of rRNA copy number and quantified by plotting a standard curve of known copy numbers of P. murina rRNA as previously described (18, 26, 34). This methodology detects viable P. murina organisms as confirmed by the absence of detectable P. murina rRNA in samples subjected to heat inactivation or exposure to trimethoprim-sulfamethoxazole. Percent killing was defined by the following equation:
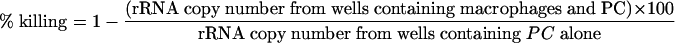 |
where PC is Pneumocystis.
In selected experiments, murine GM-CSF or IFN-γ was dissolved in RPMI 1640 medium with 10% fetal bovine serum and 2 mM l-glutamine, or supernatants from Tc1 or Tc2 cells were added directly to the macrophage killing assays at various concentrations. For GM-CSF and IFN-γ neutralizing studies, rat monoclonal anti-murine GM-CSF antibody, rat monoclonal anti-mouse IFN-γ antibody, or mouse immunoglobulin G1 isotype control was added to the cultures at 2 μg/ml.
Statistical analysis.
Data were analyzed using StatView statistical software (Brainpower, Inc., Calabasas, Calif.). Comparisons between groups where data were normally distributed were made with Student's t test, and comparisons among multiple groups or nonparametric data were made with analyses of variance. Scheffe's test was the post hoc test used.
RESULTS
Generation of in vitro-polarized P. murina-specific CD8+ T-cell populations.
The schema for the generation of antigen-specific CD8+ T cells is outlined in Fig. 1. CD8+ T-cell supernatants were assayed for ConA- or antigen-specific cytokine responses using BioPlex reagents. Tc1-polarized CD8+ T cells that were restimulated with ConA or DCs pulsed with antigen showed significantly higher levels of IFN-γ and GM-CSF than Tc2 cells (Fig. 2). In the case of Tc1 cells, the antigen-stimulated levels of IFN-γ approached those observed with ConA, while GM-CSF antigen-stimulated levels were significantly higher than those observed with ConA. In contrast, Tc2 cells showed higher levels of IL-4, IL-5, IL-10, and IL-13 after antigen recall or ConA stimulation (Fig. 2). Again, for IL-10 and IL-13, antigen-induced responses approached those of ConA, whereas IL-4 secretion in response to antigen was significantly lower than those of ConA (Fig. 2). Tc1 or Tc2 cells cultured with unpulsed DCs secreted less than 100 pg/ml of IFN-γ, GM-CSF, IL-4, IL-5, IL-10, and IL-13 (data not shown).
FIG. 2.

CD8+ T-cell polarization. Cytokines were measured in supernatants collected 48 h after Tc1 and Tc2 CD8+ T-cell restimulation with either ConA (Con A) or DCs pulsed with antigen (DC/PC). A third group of cells was exposed to medium, and the cytokine concentrations obtained were subtracted from those of the stimulated samples. Cytokines levels were measured using the BioPlex system from Bio-Rad (Hercules, CA) as described in Materials and Methods. Concentrations were expressed in pg/ml. (*, P of <0.05 compared to respective control).
In vitro effector activity of Tc1 CD8+ T cells against P. murina.
To evaluate whether these ex vivo-polarized CD8+ T-cell populations had in vitro effector activity similar to in vivo-generated Tc1 CD8+ T cells (18), we tested them in in vitro killing assays in the presence or absence of macrophages. Similar to what we have observed for in vivo-generated CD8+ T cells purified from lung tissue (18), we observed no direct killing activity of CD8+ T cells harvested on day 30 or 32 against P. murina (data not shown). However, in the presence of thioglycolate-elicited peritoneal macrophages, Tc1 CD8+ T cells significantly augmented the killing of P. murina (Fig. 3A). We titrated the macrophage-to-P. murina ratio such that macrophage-mediated killing was approximately 20 to 30% so we could assay for augmentation as well as suppression by the T-cell populations (Fig. 3A). The addition of Tc1 cells at a ratio of 1:1 significantly augmented the killing of P. murina from 20 ± 3.5% to 40 ± 2.5%, whereas the identical number of Tc2 cells suppressed macrophage-mediated killing to 9.6 ± 6.7% (Fig. 3A). Moreover, the killing was dose dependent, as reducing the CD8+ T cell/macrophage ratio from 1:1 to 1:10 or 1:100 reduced the effector activity of Tc1 cells (Fig. 3A).
FIG. 3.

In vitro effector activity of Tc1 CD8+ T cells. Tc1 and Tc2 in vitro-polarized CD8+ T cells (A) purified from the spleens of P. murina-vaccinated-boosted BALB/c mice, as well as the supernatants obtained from Tc1 and Tc2 cultures (B), were incubated with P. carinii and thioglycolate-induced peritoneal macrophages for 24 h. Controls included P. murina cultured without macrophages. Thereafter, RNA was isolated from the contents of each well, and quantitative real-time PCR for P. carinii rRNA copy number was performed as described in Materials and Methods. Percent killing at 24 h was assayed by quantifying the integrity of the P. murina rRNA subunit by TaqMan in comparison to P. murina organisms cultured without macrophages (*, P < 0.05, analysis of variance [ANOVA]).
Since we observed no direct effect of CD8+ T cells against P. murina in vitro, similar to what has been reported for human CD8+ T cells against Candida spp. through granulysin (16), we investigated whether a soluble factor was required for macrophage-mediated killing of P. murina. Cell-free supernatants from Tc1 or Tc2 cells were obtained 48 h after antigen restimulation and added to the macrophage-mediated killing assay. Cell-free conditioned medium from antigen-restimulated Tc1 cells significantly augmented killing from 21% to 53.2% (Fig. 3B). Furthermore, the addition of cell-free supernatants from Tc2 CD8+ T cells suppressed macrophage-mediated killing of P. murina (Fig. 3B). Again, the killing was dose dependent, as reducing the ratio of conditioned Tc1 medium to unconditioned medium from 1:2 to 1:10 or 1:100 reduced the effector activity of Tc1 cells (Fig. 3B). These data suggested that the augmentation of macrophage-mediated killing of P. murina induced by Tc1 CD8+ T cells was mediated by a soluble factor.
GM-CSF and IFN-γ up-regulate the killing of P. murina by inflammatory peritoneal macrophages in a dose-dependent fashion.
Of the cytokines that we observed to be differentially expressed in antigen-stimulated Tc1 CD8+ T cells, both IFN-γ and GM-CSF were putative candidates for mediating enhanced macrophage-mediated killing of P. murina. Although both aerosolized IFN-γ (3) and overexpression of IFN-γ using adenovirus-mediated gene transfer (14) augment the clearance of P. murina, endogenous IFN-γ is not essential (11). GM-CSF appears to play a more critical role in endogenous clearance (20) and has been reported to be therapeutic in CD4+-deficient mice (17). Utilizing our in vitro P. murina killing assay, we observed significantly enhanced macrophage-mediated killing by both recombinant IFN-γ and GM-CSF (Fig. 4A and B). However, the dose of IFN-γ required to observe significant killing above that mediated by macrophages alone was quite high at 5 ng/ml (Fig. 4A). However, GM-CSF had activity in this assay at levels as low as 1 ng/ml (Fig. 4B). In dose-response studies, we also showed that the increased killing mediated by the cytokines is dose dependent and that maximum responses were reached with 500 ng/ml of IFN-γ and 10 ng/ml of GM-CSF (Fig. 4A and B).
FIG. 4.

IFN-γ and GM-CSF up-regulate macrophage-mediated P. murina killing. Dose-dependent increases in macrophage-mediated P. murina killing by recombinant IFN-γ (A) and GM-CSF (B) are shown. Results are expressed as the means ± standard errors of the mean (SEMs) of results for triplicate samples from one representative experiment. *, P < 0.05 by ANOVA. Dotted line denotes baseline killing.
GM-CSF neutralization decreases the activity of Tc1-conditioned medium on P. murina killing mediated by macrophages.
Based on the recombinant cytokine data, we performed neutralization of both IFN-γ and GM-CSF in conditioned medium from P. murina antigen-stimulated Tc1 CD8+ T cells in the in vitro killing assay (Fig. 1). Again, peritoneal macrophages were seeded with P. murina to achieve approximately 20% killing (Fig. 5). The addition of conditioned medium from P. murina-stimulated Tc1 CD8+ T cells augmented killing from a baseline of 21 ± 2.6% to 63.4 ± 8.3%. The neutralization of GM-CSF resulted in an 85% decrement in the enhanced macrophage-mediated killing of the Tc1 CD8+ T-cell conditioned medium (Fig. 5). The neutralization of IFN-γ also decreased macrophage-mediated killing induced by Tc1 CD8+ T cells, although not to the same degree as the neutralization of GM-CSF. Finally, the neutralization of both cytokines GM-CSF and IFN-γ did not improve the effect on killing produced by Tc1 cells more than GM-CSF neutralization alone (Fig. 5).
FIG. 5.

Effects of GM-CSF and IFN-γ blocking in the presence of Tc1 supernatants. Thyoglycolate-induced peritoneal macrophages were added to P. murina in the presence of Tc1 supernatant (Tc1 sup.). Neutralization of GM-CSF and IFN-γ was performed using 2 μg/ml of murine anti-GM-CSF and/or the same concentration of murine anti-IFN-γ antibodies (Biosource, Camarillo, California). Controls included P. murina cultured with macrophages in medium alone. Results are expressed as the means ± SEMs of results for triplicate samples from one representative experiment (*, P < 0.05, ANOVA).
GM-CSF up-regulates the killing of P. murina by alveolar macrophages.
Since P. murina is essentially a lung pathogen, we confirmed our findings with two populations of alveolar macrophages, resident alveolar macrophages (RAM) and macrophages elicited by intratracheal MCP-1 administration (MCP-1 AM). Both RAM and MCP-1 AM showed the same baseline killing of P. murina (Fig. 6). However, when recombinant GM-CSF was added to the coculture at 10 ng/ml, MCP-1 AM showed a significantly higher P. murina killing percentage than RAM (Fig. 6). IFN-γ at a dose of 50 ng/ml also significantly increased the killing of P. murina by MCP-1 AM but was less effective at increasing killing by RAM. Moreover, the effect with IFN-γ was again less substantial than that observed with 10 ng/ml GM-CSF (Fig. 6).
FIG. 6.

GM-CSF up-regulates alveolar macrophage-mediated P. murina killing. RAM or MCP-1 AM were treated with vehicle or recombinant GM-CSF (10 mg/ml) or IFN-γ (50 ng/ml). Results are expressed as the means ± SEMs of results for triplicate samples from one representative experiment (*, P < 0.05, ANOVA; **, P < 0.01, ANOVA).
DISCUSSION
The recruitment of immune effector cells into the lungs is critical for host defense against pulmonary infections. The influx of inflammatory cells into a site of infection may result in either clearance of the infection or, in some cases, tissue damage. Studies of the onset and progression of Pneumocystis pneumonia in both human patients and animal models have suggested that the host's ability to mount an inflammatory response correlates directly with the severity of the clinical manifestation of Pneumocystis pneumonia (2, 5, 30, 31, 33).
It has been previously reported that CD8+ T cells are recruited in significant numbers into the lungs of experimental animals and patients with Pneumocystis pneumonia (2, 4, 7, 10, 14, 31). However, their role in host defense and lung injury remains unclear. Beck and colleagues have previously shown that the depletion of CD8+ T cells in the CD4+-depleted mouse model of P. murina infection exacerbated P. murina infection, suggesting a role for CD8+ T cells in host defense against P. murina (4). Moreover, γδ T-cell-receptor knockout mice, which clear P. murina more rapidly, demonstrated augmented recruitment of IFN-producing CD8+ T cells, and the depletion of CD8+ T cells in this model reverses the augmented clearance seen in this mouse strain (27). Furthermore, we confirmed that overexpression of IFN resulted in the recruitment of Tc1 phenotype CD8+ T cells into the lung with in vitro augmentation of macrophage-mediated clearance of P. murina and in vivo effector activity in scid mice (18). In contrast, non-Tc1 CD8+ T cells exacerbated P. murina-mediated lung injury upon scid mouse reconstitution (18). Consistent with these data, Harmsen and colleagues have shown that CD8+ T-cell depletion can also attenuate lung injury in the CD4+-depleted P. murina model (1). Therefore, the balance between resolution of P. murina infection and the development of lung injury may be due to the phenotype of the CD8+ T cells that are recruited to the lung.
The present data support the concept that antigen-specific Tc1 CD8+ T-cell lymphocytes are capable of effector activity against P. murina. Tc1-polarized CD8+ T cells produced significantly higher levels of IFN-γ and GM-CSF and demonstrated enhanced killing of P. murina in a macrophage coculture assay compared to cells cultured with P. murina alone. Prior studies with Tc1 CD8+ T cells generated in vivo failed to show augmented killing when these cells were cultured with P. murina alone (18). These data suggest that in situ antigen presentation and/or soluble factors may be required for increased macrophage-mediated killing of P. murina. In support of a role for soluble factors was the fact that in vitro-generated Tc1-conditioned medium also augmented the killing of P. murina by macrophages. Murine recombinant IFN-γ and GM-CSF improved macrophage-mediated P. murina killing in a dose-dependent fashion, although GM-CSF was effective at much lower concentrations. Antibody neutralization confirmed the critical role of GM-CSF in augmenting macrophage-mediated killing of P. murina by Tc1 CD8+ T cells. Furthermore, the addition of GM-CSF was more potent than the addition of IFN-γ at increasing macrophage-mediated killing of P. murina by both RAM and MCP-1 AM. The latter population of macrophages is likely a critical effector against P. murina, as P. murina elicits MCP-1 in the lung (6, 32), and recent evidence supports a role of P. murina in inducing apoptosis of RAM (15). Taken together, these data suggest that recruited macrophages are likely the cells that eliminate P. murina from the lower respiratory tract. It is of interest that the concentration of antigen-elicited GM-CSF is lower than that required to achieve the same level of killing with recombinant GM-CSF. These data would suggest that other soluble factors are made by Tc1 cells that may synergize with GM-CSF or have independent activity on their own.
In vivo administration of murine GM-CSF recombinant protein to CD4+-depleted mice has been shown to decrease the intensity of murine Pneumocystis pneumonia possibly by enhancing alveolar macrophage tumor necrosis factor alpha (TNF-α) production (2, 17). Moreover, mice with homozygous deletion of the GM-CSF gene (GM-CSF−/−) that were depleted of CD4+ cells show greater intensities of P. murina infection and inflammation than do mice with the GM-CSF gene intact (20). In contrast, transgenic expression of GM-CSF directed solely to the lungs of GM-CSF−/− mice dramatically decreased the intensity of infection and inflammation (20). Furthermore, alveolar macrophages from GM-CSF−/− mice demonstrated impaired phagocytosis of P. murina organisms as well as the absence of TNF-α production in response to P. murina in vitro (2, 20). TNF-α production and phagocytic capacity of alveolar macrophages were restored by transgenic overexpression of GM-CSF in the lung. These defects may be due to the ability of GM-CSF to increase the expression of PU.1, a critical transcription factor in macrophage development, and may result in increased expression of many cell surface receptors, including the macrophage receptor with collagenase structure (12), mannose receptors (8, 28), and the Dectin-1 beta-glucan receptor (26, 29). Interestingly, IFN does not increase Dectin-1 or glucan binding, and this may explain, in part, the differential activity of GM-CSF in this assay. Thus, one possibility is that GM-CSF increases the uptake of P. murina. Additionally, GM-CSF also increases the expression of NADPH oxidases (9, 25) as well as superoxide anion production, which may also be responsible for the enhanced microbicidal activity of Tc1 CD8+ T cells with macrophages. In fact, GM-CSF has recently been shown to improve the clearance of P. murina in neonatal mice, in part by increasing antigen presentation (23). It is of interest that the pathology of Pneumocystis pneumonia is similar to that of pulmonary alveolar proteinosis, suggesting that disruptions in local GM-CSF activity may play a role in the pathogenesis of Pneumocystis pneumonia. The ultimate cellular sources of GM-CSF for host defenses against P. murina remain to be defined; however, our data suggest a role of antigen-specific T cells in addition to other known sources such as epithelial cells. Taken together, our data support the concept that GM-CSF secreted by subpopulations of CD8+ T cells may play a critical role in the inflammatory response to P. murina.
Acknowledgments
This work was supported by Public Health Service grants P01HL076100, HL61721, and HL62052 from the National Institutes of Health, National Heart, Lung, and Blood Institute.
Editor: T. R. Kozel
REFERENCES
- 1.An, C. L., X. P. Su, and A. G. Harmsen. 2000. The role of CD8+ T cells in the pathogenesis of Pneumocystis carinii pneumonia in mice depleted of CD4+ T cells. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 18:207-212. [PubMed] [Google Scholar]
- 2.Beck, J. M., and A. G. Harmsen. 1998. Lymphocytes in host defense against Pneumocystis carinii. Semin. Respir. Infect. 13:330-338. [PubMed] [Google Scholar]
- 3.Beck, J. M., H. D. Liggitt, E. N. Brunette, H. J. Fuchs, J. E. Shellito, and R. J. Debs. 1991. Reduction in intensity of Pneumocystis carinii pneumonia in mice by aerosol administration of gamma interferon. Infect. Immun. 59:3859-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck, J. M., R. L. Newbury, B. E. Palmer, M. L. Warnock, P. K. Byrd, and H. B. Kaltreider. 1996. Role of CD8+ lymphocytes in host defense against Pneumocystis carinii in mice. J. Lab. Clin. Med. 128:477-487. [DOI] [PubMed] [Google Scholar]
- 5.Beck, J. M., M. L. Warnock, J. L. Curtis, M. J. Sniezek, S. M. Arraj-Peffer, H. B. Kaltreider, and J. E. Shellito. 1991. Inflammatory responses to Pneumocystis carinii in mice selectively depleted of helper T lymphocytes. Am. J. Respir. Cell Mol. Biol. 5:186-197. [DOI] [PubMed] [Google Scholar]
- 6.Benfield, T. L., B. Lundgren, J. H. Shelhamer, and J. D. Lundgren. 1999. Pneumocystis carinii major surface glycoprotein induces interleukin-8 and monocyte chemoattractant protein-1 release from a human alveolar epithelial cell line. Eur. J. Clin. Investig. 29:717-722. [DOI] [PubMed] [Google Scholar]
- 7.Board, K. F., S. Patil, I. Lebedeva, S. Capuano III, A. M. Trichel, M. Murphey-Corb, P. A. Rajakumar, J. L. Flynn, C. G. Haidaris, and K. A. Norris. 2003. Experimental Pneumocystis carinii pneumonia in simian immunodeficiency virus-infected rhesus macaques. J. Infect. Dis. 187:576-588. [DOI] [PubMed] [Google Scholar]
- 8.Bonfield, T. L., B. Raychaudhuri, A. Malur, S. Abraham, B. C. Trapnell, M. S. Kavuru, and M. J. Thomassen. 2003. PU.1 regulation of human alveolar macrophage differentiation requires granulocyte-macrophage colony-stimulating factor. Am. J. Physiol. Lung Cell. Mol. Physiol. 285:L1132-L1136. [DOI] [PubMed] [Google Scholar]
- 9.Dang, P. M., C. Dewas, M. Gaudry, M. Fay, E. Pedruzzi, M. A. Gougerot-Pocidalo, and J. El Benna. 1999. Priming of human neutrophil respiratory burst by granulocyte/macrophage colony-stimulating factor (GM-CSF) involves partial phosphorylation of p47(phox). J. Biol. Chem. 274:20704-20708. [DOI] [PubMed] [Google Scholar]
- 10.Fleury, J., E. Escudier, M. J. Pocholle, C. Carre, and J. F. Bernaudin. 1985. Cell population obtained by bronchoalveolar lavage in Pneumocystis carinii pneumonitis. Acta Cytol. 29:721-726. [PubMed] [Google Scholar]
- 11.Garvy, B. A., R. A. B. Ezekowitz, and A. G. Harmsen. 1997. Role of gamma interferon in the host immune and inflammatory responses to Pneumocystis carinii infection. Infect. Immun. 65:373-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Granucci, F., F. Petralia, M. Urbano, S. Citterio, F. Di Tota, L. Santambrogio, and P. Ricciardi-Castagnoli. 2003. The scavenger receptor MARCO mediates cytoskeleton rearrangements in dendritic cells and microglia. Blood 102:2940-2947. [DOI] [PubMed] [Google Scholar]
- 13.Harmsen, A. G., and M. Stankiewicz. 1990. Requirement for CD4+ cells in resistance to Pneumocystis carinii pneumonia in mice. J. Exp. Med. 172: 937-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolls, J. K., S. Habetz, M. K. Shean, C. Vazquez, J. A. Brown, D. Lei, P. Schwarzenberger, P. Ye, S. Nelson, W. R. Summer, and J. E. Shellito. 1999. IFN-gamma and CD8+ T cells restore host defenses against Pneumocystis carinii in mice depleted of CD4+ T cells. J. Immunol. 162:2890-2894. [PubMed] [Google Scholar]
- 15.Lasbury, M. E., P. J. Durant, and C. H. Lee. 2003. Decrease in alveolar macrophage number during Pneumocystis carinii infection. J. Eukaryot. Microbiol. 50(Suppl.):630-631. [DOI] [PubMed] [Google Scholar]
- 16.Ma, L. L., J. C. Spurrell, J. F. Wang, G. G. Neely, S. Epelman, A. M. Krensky, and C. H. Mody. 2002. CD8 T cell-mediated killing of Cryptococcus neoformans requires granulysin and is dependent on CD4 T cells and IL-15. J. Immunol. 169:5787-5795. [DOI] [PubMed] [Google Scholar]
- 17.Mandujano, J. F., N. B. D'Souza, S. Nelson, W. R. Summer, R. C. Beckerman, and J. E. Shellito. 1995. Granulocyte-macrophage colony stimulating factor and Pneumocystis carinii pneumonia in mice. Am. J. Respir. Crit. Care Med. 151:1233-1238. [DOI] [PubMed] [Google Scholar]
- 18.McAllister, F., C. Steele, M. Zheng, E. Young, J. E. Shellito, L. Marrero, and J. K. Kolls. 2004. T cytotoxic-1 CD8(+) T cells are effector cells against Pneumocystis in mice. J. Immunol. 172:1132-1138. [DOI] [PubMed] [Google Scholar]
- 19.Morris, A., J. D. Lundgren, H. Masur, P. D. Walzer, D. L. Hanson, T. Frederick, L. Huang, C. B. Beard, and J. E. Kaplan. 2004. Current epidemiology of Pneumocystis pneumonia. Emerg. Infect. Dis. 10:1713-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paine, R., III, A. M. Preston, S. Wilcoxen, H. Jin, B. B. Siu, S. B. Morris, J. A. Reed, G. Ross, J. A. Whitsett, and J. M. Beck. 2000. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J. Immunol. 164:2602-2609. [DOI] [PubMed] [Google Scholar]
- 21.Patel, N., and H. Koziel. 2004. Pneumocystis jiroveci pneumonia in adult patients with AIDS: treatment strategies and emerging challenges to antimicrobial therapy. Treat. Respir. Med. 3:381-397. [DOI] [PubMed] [Google Scholar]
- 22.Phair, J., A. Munoz, R. Detels, R. Kaslow, C. Rinaldo, and A. Saahet. 1990. The risk of Pneumocystis carinii pneumonia among men infected with human immunodeficiency virus type I. N. Engl. J. Med. 322:155-161. [DOI] [PubMed] [Google Scholar]
- 23.Qureshi, M. H., K. M. Empey, and B. A. Garvy. 2005. Modulation of proinflammatory responses to Pneumocystis carinii f. sp. muris in neonatal mice by granulocyte-macrophage colony-stimulating factor and IL-4: role of APCs. J. Immunol. 174:441-448. [DOI] [PubMed] [Google Scholar]
- 24.Shellito, J., V. V. Suzara, W. Blumenfeld, J. M. Beck, H. J. Steger, and T. H. Ermak. 1990. A new model of Pneumocystis carinii infection in mice selectively depleted of helper T lymphocytes. J. Clin. Investig. 85:1686-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimizu, T., R. Kodama, S. Tsunawaki, and K. Takeda. 2002. GM-CSF induces expression of gp91phox and stimulates retinoic acid-induced p47phox expression in human myeloblastic leukemia cells. Eur. J. Haematol. 68:382-388. [DOI] [PubMed] [Google Scholar]
- 26.Steele, C., L. Marrero, S. Swain, A. G. Harmsen, M. Zheng, G. D. Brown, S. Gordon, J. E. Shellito, and J. K. Kolls. 2003. Alveolar Macrophage-mediated killing of Pneumocystis carinii f. sp. muris involves molecular recognition by the Dectin-1 beta-glucan receptor. J. Exp. Med. 198:1677-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steele, C., M. Zheng, E. Young, L. Marrero, J. E. Shellito, and J. K. Kolls. 2002. Increased host resistance against Pneumocystis carinii pneumonia in γδ T-cell-deficient mice: protective role of gamma interferon and CD8+ T cells. Infect. Immun. 70:5208-5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tazawa, R., E. Hamano, T. Arai, H. Ohta, O. Ishimoto, K. Uchida, M. Watanabe, J. Saito, M. Takeshita, Y. Hirabayashi, I. Ishige, Y. Eishi, K. Hagiwara, M. Ebina, Y. Inoue, K. Nakata, and T. Nukiwa. 2005. Granulocyte-macrophage colony stimulating factor and lung immunity in pulmonary alveolar proteinosis. Am. J. Respir. Crit. Care Med. 171:1142-1149. [DOI] [PubMed] [Google Scholar]
- 29.Willment, J. A., H. H. Lin, D. M. Reid, P. R. Taylor, D. L. Williams, S. Y. Wong, S. Gordon, and G. D. Brown. 2003. Dectin-1 expression and function are enhanced on alternatively activated and GM-CSF-treated macrophages and are negatively regulated by IL-10, dexamethasone, and lipopolysaccharide. J. Immunol. 171:4569-4573. [DOI] [PubMed] [Google Scholar]
- 30.Wislez, M., E. Bergot, M. Antoine, A. Parrot, M. F. Carette, C. Mayaud, and J. Cadranel. 2001. Acute respiratory failure following HAART introduction in patients treated for Pneumocystis carinii pneumonia. Am. J. Respir. Crit. Care Med. 164:847-851. [DOI] [PubMed] [Google Scholar]
- 31.Wright, T. W., F. Gigliotti, J. N. Finkelstein, J. T. McBride, C. L. An, and A. G. Harmsen. 1999. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J. Clin. Investig. 104:1307-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright, T. W., C. J. Johnston, A. G. Harmsen, and J. N. Finkelstein. 1999. Chemokine gene expression during Pneumocystis carinii-driven pulmonary inflammation. Infect. Immun. 67:3452-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright, T. W., R. H. Notter, Z. Wang, A. G. Harmsen, and F. Gigliotti. 2001. Pulmonary inflammation disrupts surfactant function during Pneumocystis carinii pneumonia. Infect. Immun. 69:758-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng, M., J. E. Shellito, L. Marrero, Q. Zhong, S. Julian, P. Ye, V. Wallace, P. Schwarzenberger, and J. K. Kolls. 2001. CD4(+) T cell-independent vaccination against Pneumocystis carinii in mice. J. Clin. Investig. 108:1469-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]