

Abstract
The glucocorticoid-induced tumor necrosis factor (TNF) receptor-related gene (GITR; TNFRSF18) modulates immune response activating coaccessory signals in T cells and is expressed at high levels in CD4+CD25+ cells. Its ligand (GITRL) is expressed in antigen-presenting cells, where it is capable of promoting signaling. We investigated the role of GITR/GITRL interaction during disseminated candidiasis in GITR knockout (GITR−/−) mice. GITR−/− mice survived longer and had a significantly decreased yeast load in kidneys and brain compared to GITR+/+ mice. Since protective immunity to the fungus is mediated by antigen-specific T helper (Th) 1 cells, we studied in vitro cytokine production following infection. CD4+ T cells of GITR−/− mice demonstrated a more efficient Th1 polarization as suggested by a two- to threefold decreased production of interleukin- (IL-)4 and IL-10 and a four- to fivefold increased production of gamma interferon compared to GITR+/+ mice. This effect was not due to differences in lymphocyte and dendritic cell (DC) subpopulations in infected mice as demonstrated by flow cytometric studies. To verify whether DC activity was differently modulated, DCs were cocultured with CD4+ T cells in the presence of heat-inactivated Candida albicans. DCs, cocultured with GITR+/+ CD4+CD25+ cells produced a lower amount of IL-12 than DCs cocultured with GITR−/− CD4+CD25+ T cells. These results suggest that GITR regulates susceptibility to systemic candidiasis by negatively modulating IL-12 production and promoting polarization of CD4+ T cells towards Th2 by analogy with OX40, another TNF receptor superfamily member.
The glucocorticoid-induced tumor necrosis factor (TNF) receptor-related gene (GITR; TNFRSF18) encodes a 35- to 40-kDa protein belonging to the TNF receptor superfamily (TNFRSF), cloned in a murine hybridoma T-cell line treated with dexamethasone (18). Given the striking homology of the GITR cytoplasmic region with other TNFRSF members (4-1BB, OX40, CD27, and CD40), GITR and these molecules form a new subfamily inside the TNFRSF (19, 29), characterized by coactivating activity and involved in the regulation of T lymphocyte function (25, 33, 34). GITR is expressed at different levels in normal T cells, including single-positive CD4+ and CD8+ thymocytes and spleen and lymph node T cells (18, 19). Upon T-cell activation, GITR is up-regulated in both CD4+ and CD8+ cells, suggesting it plays a role in the control of T-cell activation and immune response development (18, 25).
Moreover, GITR is expressed at high levels in CD4+CD25+ regulatory T (Treg) cells (13, 26), which exert suppressor activity on CD4+CD25− effector T cells and are involved in control of several autoimmune/inflammatory diseases. During T-cell activation, GITR costimulus potentiates CD4+CD25− effector activity and negatively modulates CD4+CD25+ Treg cell suppressor activity (13, 19, 25, 26, 33). Consequently, T-cell receptor/CD3-induced activation of T lymphocytes is deregulated in GITR-deficient mice (GITR−/−), although no significant differences were observed in the relative composition of lymphocyte subpopulations in the spleen and lymph nodes between GITR+/+ and GITR−/− mice (24).
GITR is activated by its ligand (GITRL), which is expressed in antigen-presenting cells, including macrophages, immature and mature dendritic cells (DCs), and B lymphocytes, but not in resting and activated T cells (8, 19, 33, 35). After DC activation, GITRL is transiently up-regulated (33). Recently, Choi's group demonstrated that, after the GITR-GITRL interaction, GITRL also delivers signals by means of its cytoplasmic domain (11, 27, 28).
Candida albicans is the leading cause of fungal opportunistic diseases in humans. Intravenous infection of mice with C. albicans blastospores causes a pathology that resembles the human one, characterized by active fungal replication in the kidney, brain, and heart, with death usually resulting from kidney failure (2). In murine systemic candidiasis, the development of the T helper (Th) type 1 response is correlated with induction of protective immunity to the fungal pathogen. In contrast, the occurrence of the Th2 response is associated with pathology progression (22).
Recent evidence suggests that Treg cells play a role in responses to infection. In fact, Treg cells may limit the magnitude of effector responses, which may result in failure to adequately control infection (3, 31, 32). In particular, in an experimental model of murine candidiasis, a reduction of Treg cell activity causes better control of the infection (16).
In this study, we investigated the role of GITR in the development of C. albicans infection using GITR−/− mice. The results indicate that GITR−/− mice are more resistant to infection than wild-type (GITR+/+) mice, due to an increase in Th1 response.
MATERIALS AND METHODS
Animals.
Sv129 GITR-deficient (GITR−/−) mice were generated by homologous recombination as previously described (24). Mice were maintained under specific-pathogen-free conditions in the Animal Care Facility of the University of Perugia. They were group housed under controlled temperature and photoperiod (12-h:12-h light-dark cycle) and allowed unrestricted access to standard mouse chow and tap water. Mice of both sexes were used at the age of 8 to 12 weeks. Protocols were approved by the Animal Study Committees of the University of Perugia according to governmental guidelines for animal care.
Infection.
The origin and characteristics of the highly virulent C. albicans (CA-6) strain used in this study have been previously described (23). For infection, C. albicans cells were washed twice in saline and diluted to the desired density to be injected intravenously via the lateral tail vein in a volume of 0.5 ml/mouse. Littermate GITR+/+ and GITR−/− mice were used in the experiments. Infected animals were monitored for survival every day and at days 2 and 6 after infection for organ clearance. Quantification of fungal growth in the organs of infected mice (four to six mice per group) was performed by plating serial dilutions of homogenized organs in Sabouraud dextrose agar, and results (mean ± standard error of the mean) were expressed as CFU per organ.
Culture medium.
The cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum, streptomycin (100 μg/ml), 10 mM HEPES, 0.1% nonessential amino acids, 1 mM sodium pyruvate, and 50 μM 2-mercaptoethanol (complete medium).
Cell cultures.
Dendritic cells (at a concentration of 105 cells/ml) and CD4+ or CD4+CD25− T cells (at a concentration of 106 cells/ml) were cocultured in 1 ml of complete medium (24-well plates). Cocultures were stimulated with heat-inactivated C. albicans (DC-C. albicans ratio, 1:10) for 18 h. C. albicans yeast cells were inactivated by heating to 65°C for 1 h. The fungal inactivation was confirmed by plating the suspension onto Sabouraud agar.
DCs (at a concentration of 105 cells/ml) and CD4+CD25+ T cells were cocultured (DC-CD4+CD25+ ratio, 1:1 or 1:2) in 1 ml of complete medium (24-well plates) and were stimulated with heat-inactivated C. albicans (DC-C. albicans ratio, 1:10) for 18 h.
Cytokine production.
Spleen cells (2 × 106 cells/ml in 1 ml of complete medium in wells of flat-bottomed 24-well plates) were stimulated with heat-inactivated C. albicans (spleen cell-C. albicans ratio, 1:10). CD4+ T cells (1.25 × 106 cells/ml) were plated in wells (24-well plates, 1 ml/well) on a layer of irradiated splenocytes (5 × 106 cells/well) and cultured in the presence of heat-inactivated C. albicans (CD4+ cell-C. albicans ratio, 1:10) for 48 h. Interleukin- (IL-)12p70, IL-10, IL-4, and gamma interferon (IFN-γ) were evaluated in cell culture supernatant fluids according to a previously described method (25). The antibodies (capture and detection) used for the evaluation of the above cytokines were purchased from Pharmingen (San Diego, CA) and the enzyme-linked immunosorbent assays were performed according to the manufacturer's instructions.
Cell purification.
CD4+ T cells were purified by positive selection from spleen of infected and noninfected mice using CD4 (L3T4) microbeads (Miltenyi Biotech, Bologna, Italy), according to the manufacturer's instructions. Microbeads allow a purification level higher than 95% and do not activate cells.
CD4+CD25− cells were purified from CD25+-depleted spleen cells. For depletion, spleen cells were incubated with phycoerythrin-conjugated anti-mouse CD25 (IL-2 receptor α chain, p55) (clone 7D4), and the stained cells were incubated with anti-phycoerythrin microbeads (Miltenyi Biotech). The depletion of CD25+ was detected by flow cytometry (less than 0.1% of the depleted cells expressed CD25). After CD25+ cell depletion, CD4 were isolated using CD4 (L3T4) microbeads.
CD4+CD25+ cells were purified from lymph nodes by using a CD4+CD25+ T-cell isolation kit (Miltenyi Biotech) following the manufacturer's instructions. Briefly, after isolating CD4+ T cells with the (L3T4) microbeads, the CD25+ phycoerythrin-labeled cells were magnetically labeled with anti-phycoerythrin microbeads according to the manufacturer's instructions (25). The cell suspension was loaded on a column, which was placed in the magnetic field of a MACS separator. The retained cells eluted from the column were CD4+CD25+ cells. To achieve the highest purities, two consecutive column runs were performed. The purity of T-cell subpopulations was >98% as evaluated by flow cytometry analysis.
DCs were separated from spleens of wild-type or GITR−/− mice using N-418 monoclonal antibody-conjugated microbeads (Miltenyi Biotech), and magnetic separation was done according to manufacturer's instructions.
Flow cytometry analysis.
We pelleted 5 × 105 cells in a round-bottomed centrifuge tube, resuspended in 10 μl of labeling buffer containing either fluorescein isothiocyanate- or phycoerythrin-conjugated antibodies, and incubated at 4°C for 20 min in the dark. The cells were then washed twice and resuspended in 1X phosphate-buffered saline. The samples were then analyzed using a FACScan flow cytometer (Becton Dickinson, Sunnyvale, CA).
The following monoclonal antibodies were used: fluorescein isothiocyanate-conjugated anti-mouse CD3ɛ (clone 145-2C11), fluorescein isothiocyanate-conjugated anti-mouse CD8α (clone 53-6.7), phycoerythrin-conjugated anti-mouse CD4 (L3T4) (clone H129.19), and phycoerythrin-conjugated anti-mouse B220. All monoclonal antibodies were purchased from Pharmingen.
For flow cytometric analysis of dendritic subpopulation, purified DCs (106/ml) from the spleens of GITR+/+ and GITR−/− mice before and after infection with C. albicans, were washed twice in phosphate-buffered saline, fixed in 10% formalin, and labeled for 40 min on ice with phycoerythrin-conjugated rat monoclonal antibody to mouse CD8α (Chemicon Int., Temecula, CA; clone KT15, isotype immunoglobulin G2a). After incubation, cells were washed twice in fluorescence buffer (phosphate-buffered saline containing 0.5% bovine serum albumin and 0.4% sodium azide), resuspended, and analyzed by FACScan. Data are expressed as percentage of positive cells.
Intracellular staining.
For analysis of intracellular IL-12, purified DCs (105/ml) were cocultured overnight in complete medium at 37°C with 5% CO2 with autologous or heterologous CD4+ T cells (106/ml) from naïve or C. albicans CA-6-infected mice in the presence of heat-inactivated CA-6. After incubation the cells were washed twice in phosphate-buffered saline, fixed in 10% formalin, permeabilized for 10 min at room temperature with 0.1% saponin (Sigma Chemical Co., St Louis, MO) in phosphate-buffered saline, and labeled for 40 min on ice with biotin goat anti-mouse interleukin-12 (Cederlane, Hornby, Ontario, Canada). After incubation, cells were reacted for 40 min on ice with avidin-fluorescein isothiocyanate, washed with phosphate-buffered saline containing 0.1% saponin, resuspended in fluorescence buffer and analyzed by FACScan. Data are expressed as percentage of positive cells.
Statistical analysis.
Student's t test was used to determine the statistical significance of differences in organ clearance and in in vitro assays. Data reported were pooled from three experiments with similar results. Survival data were analyzed by the Mantel-Cox logrank test. Significance was defined as P < 0.05.
RESULTS
GITR−/− mice are more resistant to systemic C. albicans infection.
To investigate the role of GITR in C. albicans infection, we intravenously infected GITR−/− and GITR+/+ mice with virulent C. albicans cells. As shown in Fig. 1A, GITR−/− mice survived significantly longer than GITR+/+ mice (21 days median survival time of GITR−/− versus 13 days of GITR+/+ mice, P < 0.01). At the end of the monitoring period, all mice of both groups were dead.
FIG. 1.

Decreased susceptibility to systemic C. albicans infection in GITR−/− mice. GITR+/+ and GITR−/− mice were intravenously infected with 5 × 105 C. albicans cells. A: Survival was assessed daily for 40 days. % survival of infected mice was evaluated according to Mantel-Cox Logrank test and the difference between GITR−/− and GITR+/+ mice resulted significant (P < 0.01). B: For the quantification of fungal burden, mice were sacrificed 2 and 6 days after infection, serial dilutions of organ homogenates were plated onto Sabouraud agar, and CFU were counted after 48 h incubation. Fungal burden is expressed as CFU/organ. Data represent means ± standard error of the mean of data from three independent experiments. **, P < 0.01, GITR−/− versus GITR+/+, according to Student's t test.
Fungal growth in kidneys and brain, the target organs of murine candidiasis, was also determined. For this purpose, infected mice were sacrificed at day +2 and day +6 postinfection, and kidney and brain homogenates were plated on Sabouraud agar. The fungal burden was expressed as CFU/organ. GITR−/− mice showed significantly less (P < 0.01) fungal burden in kidneys with respect to control mice at both time points (Fig. 1B, left). No differences were found in fungal growth between GITR−/− and GITR+/+ mice at day +2 in the brain. However, at day +6 GITR−/− mice showed a decrease (P < 0.01) in fungal burden compared to control mice (Fig. 1B, right).
These results indicate that GITR−/− mice are less susceptible to systemic C. albicans infection than GITR+/+ mice because of more efficient clearance of C. albicans.
C. albicans infection increases Th1 responses in GITR−/− mice.
Since low susceptibility to C. albicans infection is linked to Th1 response, while high susceptibility is linked to Th2 response, we evaluated cytokine production of T cells. In particular, IL-4, IL-10, IL-12 p70, and IFN-γ levels were detected on supernatants of spleen cells from C. albicans infected mice (day +6) after in vitro restimulation with heat-inactivated C. albicans. We found that the levels of IL-4 and IL-10 were significantly lower (P < 0.05) in GITR−/− mice than in GITR+/+ mice, while IFN-γ production was significantly higher (P < 0.05) in GITR−/− mice compared to GITR+/+ mice (Fig. 2A). IL-12 p70 was slightly, but not significantly, increased in GITR−/− mice. Similar results were obtained using purified spleen CD4+ T cells (Fig. 2B), suggesting that a higher percentage of Th1 polarized T cells are present in the spleen of GITR−/− mice.
FIG. 2.

In vitro production of cytokines in GITR+/+ and GITR−/− mice during systemic C. albicans infection. GITR+/+ and GITR−/− mice were infected with 5 × 105 C. albicans cells and 6 days after infection spleen cells (A) or CD4+ T cells (B) were harvested. The cells were restimulated with heat-inactivated C. albicans (cell-C. albicans ratio, 1:10). Following 48 h incubation supernatants were harvested and Th1 (IL-12 p70 and IFN-γ) and Th2 (IL-4 and IL-10) cytokines were measured by enzyme-linked immunosorbent assay. Data represent means ± standard error of the mean of data from three independent experiments. *, P < 0.05 GITR−/− versus GITR+/+, according to Student's t test.
These results show that the better outcome of C. albicans infection in GITR−/− mice is correlated to an increase in Th1 responses.
Phenotypic analysis of GITR−/− and GITR+/+ mice.
To evaluate whether the different cytokine patterns were due to a different lymphocyte phenotype developed in response to candidiasis, we performed flow cytometric analysis of spleens from GITR+/+ and GITR−/− mice 6 days after systemic infection with C. albicans. No differences were observed in terms of percentage of B, T, CD4+ and CD8+ cells in GITR−/− mice compared to GITR+/+ mice (data not shown).
Because CD8α− DCs stimulate Th2 responses, whereas CD8α+ DCs produce IL-12 and induce Th1 responses, we investigated the phenotype of DCs in noninfected and infected mice. However, no differences in the percentage of CD8α+ cells from the spleens of GITR+/+ and GITR−/− mice before infection and on day 6 of infection were observed (data not shown).
These results indicate that, during C. albicans infection, lymphocyte and DC subpopulations are present at comparable levels in GITR−/− and GITR+/+ mice.
GITR−/− DCs produce more IL-12 than GITR+/+ DCs.
To gain insight into the mechanisms involved in the increase of Th1 response in GITR−/− mice during candidiasis, we performed in vitro experiments by coculturing DCs with CD4+ T cells from naive and infected mice in the presence of heat-inactivated C. albicans. Intracellular staining of IL-12 shows that GITR−/− DCs produce a significantly higher level of this cytokine than GITR+/+ DCs (P < 0.05), when cocultured with CD4+ T cells from infected mice (Fig. 3, column 5 versus 4). Interestingly, no differences were observed in cocultures of DCs with CD4+ T cells from noninfected mice (Fig. 3, column 2 versus 1).
FIG. 3.

Expression of IL-12 in DCs is modulated by GITR+/+ CD4+ T cells from infected mice. DCs from GITR+/+ and GITR−/− mice were cocultured with autologous or heterologous CD4+ T cells from untreated or infected mice in the presence of heat-inactivated C. albicans. IL-12 production was evaluated by DC intracellular staining. Data represent means ± standard error of the mean from three independent experiments. *, P < 0.05 column 5 versus 4 and 5 versus 6, according to Student's t test.
To investigate whether the increased IL-12 production was due to a GITR−/− DC phenotype different from that of GITR+/+ DCs, we cocultured GITR−/− DCs with CD4+ T cells from GITR+/+ mice. Figure 3 shows that IL-12 production of GITR−/− DCs in heterologous cocultures (GITR−/− DCs + GITR+/+ CD4+ T cells, column 6) was significantly less (P < 0.05) than in autologous cocultures (GITR−/− DCs + GITR−/− CD4+ T cells, column 5), reaching the levels of GITR+/+ autologous cocultures (GITR+/+ DCs + GITR+/+ CD4+ T cells, column 4). Therefore, during candidiasis, GITR+/+ CD4+ T cells inhibit IL-12 production in DCs.
Activated CD4+ T cells and Treg cells inhibit IL-12 production.
GITR is expressed at basal levels in CD4+CD25− T cells and at high levels in CD4+CD25+ T cells either derived from CD4+CD25− cells following activation (effector T cells) or constitutively expressing CD25 (Treg cells) (13, 18, 19, 26). Therefore, we performed experiments to evaluate whether the modulation of IL-12 production in DCs was mainly due to CD4+CD25− T cells or to CD4+CD25+ cells. For that purpose, DCs were cocultured with purified CD4+ T-cell population (including both CD25− and CD25+ cells) or purified CD4+CD25− T cells from spleen of infected mice in the presence of heat-inactivated C. albicans. Figure 4 shows that in GITR+/+ DC depletion of CD4+CD25+ cells significantly increased (P < 0.05) the IL-12 production (column 3 versus 1), that reaches levels similar to those observed in GITR−/− DCs (columns 2 and 4). In fact, no significant differences of IL-12 production were observed in GITR+/+ and GITR−/− DCs when cocultured with autologous CD4+CD25− (column 4 versus 3). In summary, only cells expressing high levels of GITR (GITRhigh) were able to negatively modulate IL-12 production in DCs (Fig. 4, column 1), while both GITRlow and GITR−/− cells failed to negatively modulate IL-12 production (Fig. 4, columns 2, 3, and 4).
FIG. 4.
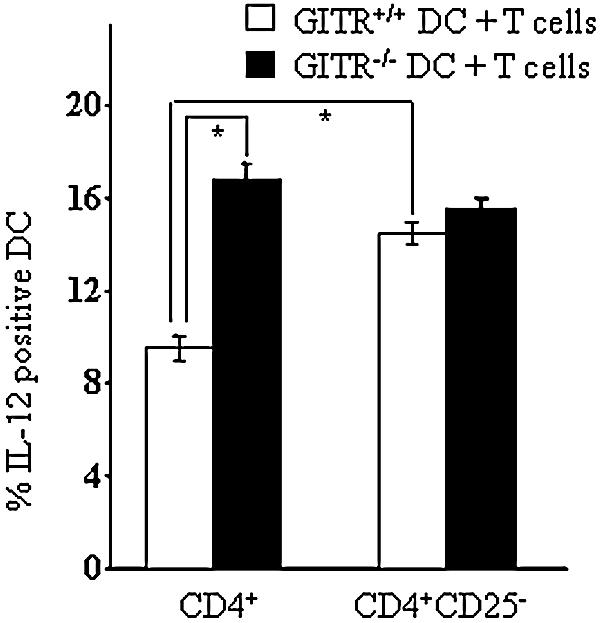
Increase of IL-12 production after depletion of GITR+/+ CD4+CD25+ T cells. DCs from GITR+/+ and GITR−/− mice were cocultured with autologous CD4+ or CD4+CD25− T cells from infected mice in the presence of heat-inactivated C. albicans. IL-12 production was evaluated by DC intracellular staining. Data represent means ± standard error of the mean from three independent experiments. *, P < 0.05 column 2 versus 1 and 3 versus 1, according to Student's t test.
Since it is likely that the phenotype of CD4+CD25+ cells is modified in several ways during infection, we wondered whether the difference in the modulation of IL-12 production in DCs was due to the high level of expression of GITR in T cells from infected mice or to the expression of different surface markers in response to infection in GITR+/+ and GITR−/− T cells. Therefore, to verify whether GITR expressed at high levels in nonprimed cells was able to modulate IL-12 production in DCs, we used CD4+CD25+ cells from noninfected mice (Treg cells), which express constitutively high levels of GITR. These cells, from GITR+/+ and GITR−/− mice, were cocultured with autologous DCs in the presence of heat-inactivated C. albicans. As shown in Fig. 5, GITR−/− DCs cocultured with GITR−/− Treg produce significantly more (P < 0.05) IL-12 than GITR+/+ DCs cocultured with GITR+/+ Treg both at 1:1 and 1:2 DC/Treg cell ratio. These results further suggest that high levels of GITR are responsible for a decreased IL-12 production in DCs.
FIG. 5.

Unprimed Treg cells from GITR+/+ mice modulate production of IL-12 in DCs. DCs from C. albicans-infected GITR+/+ and GITR−/− mice were cocultured at the indicated ratio (DC:Treg) with autologous CD4+CD25+ Treg cells from noninfected mice in the presence of heat-inactivated C. albicans. IL-12 production was evaluated by intracellular staining on DCs. Data represent means ± standard error of the mean from three independent experiments. *, P < 0.05 GITR−/− versus GITR+/+, according to Student's t test.
DISCUSSION
Here we show that GITR−/− mice are more resistant to C. albicans infection compared to GITR+/+ mice, and this is due to a more efficient clearance of the fungus. The increased resistance is correlated to a skew to a Th1 phenotype in GITR−/− mice, related, at least in part, to increased IL-12 production by DCs.
GITR is a molecule participating in the immune response by costimulating CD4+ and CD8+ effector T cells (19, 25, 33) and abolishing the suppressor activity of Treg cells (13, 19, 26). Accordingly, GITR is involved in autoimmune diseases and potentiates antiviral and antitumor responses (4, 19). However, the data presented here cannot be explained by looking at GITR as a molecule stimulating the immune system in all its components. In fact, GITR−/− mice demonstrate a longer survival time than control mice because of a higher efficiency of C. albicans clearance. Thus, GITR expression hampers the reaction against C. albicans. This is not surprising since several TNFRSF members with costimulatory function do not operate as generic boosters of cell activation, but as specialized players of immune system response, favoring or depotentiating immune response to specific pathogens. As an example, the CD40/CD40L system potentiates host defense against C. albicans and Cryptococcus neoformans (15, 21), while the blockade of OX40/OX40L ameliorates progressive leishmaniasis (1).
A recent study demonstrates that GITR triggering potentiates the graft-versus-host disease mediated by CD8+CD25− cells while inhibiting the graft-versus-host disease mediated by CD4+CD25− cells (14), suggesting that GITR is a sophisticated player in the immune response. The data here presented assign another specialized function to GITR. It is known that Th1/Th2 polarization derives from the balance among cytokines: high levels of IFN-γ and IL-12 correlate with a Th1 response, while high levels of IL-4 and IL-10 correlate with a Th2 response. The prevalent Th1 polarization in infected GITR−/− mice demonstrated by a lower production of IL-4 and IL-10 and a higher production of IFN-γ suggests that GITR/GITRL system favors Th2 polarization in C. albicans infection. This is in line with the function of other TNFRSF members, such as OX40 and CD30, that bias cytokine production to Th2 polarization in several infection models (1, 7, 10, 34). However, caution has to be applied in drawing conclusions from in vitro data for several reasons, including, in this case, the possibility that cell stimulation by heat-inactivated C. albicans may not cause the same effects obtained with virulent C. albicans. Further in vivo studies will be performed to confirm our hypothesis.
We also focused on how GITR absence was able to skew cytokine production to Th1 in C. albicans infection. It is well known that CD4+ polarization is favored by messages delivered by antigen-presenting cells, including DCs, during CD4+ T-cell priming, and that T cells deliver signals to DCs favoring their differentiation. Several members of TNFRSF (including GITR and OX40) are expressed on T cells, and their ligands (including GITRL and OX40L) are expressed on antigen-presenting cells (10, 19, 35). OX40L ligation on CD40-activated monocyte-derived antigen-presenting cells enhances cytokine production including that of IL-12 (20). GITRL is also able to promote signaling as demonstrated in macrophages and osteoclasts (11, 27, 28).
Here we demonstrate that autologous DCs cocultured with GITR+/+ CD4+CD25+ T cells from both infected and noninfected mice produce a lower amount of IL-12 compared to DCs cocultured with GITR−/− T cells (Fig. 4 and 5). This effect is not due to differences between GITR+/+ and GITR−/− DCs since GITR−/− and GITR+/+ DCs cocultured with GITR+/+ CD4+ T cells from infected mice (thus including activated CD4+CD25+ T cells) produced a similar amount of IL-12 (Fig. 3). Thus, we may hypothesize that GITR triggers GITRL present on DCs and promotes a decreased IL-12 production. Interestingly, CD4+ T cells from noninfected GITR+/+ mice did not negatively modulate IL-12 production in DCs, probably as a consequence of the lower percentage of GITRhigh T cells compared to that present in CD4+ cells from infected mice. In fact, GITR expression is impressively up-regulated during T-cell activation (18, 24, 25).
A different explanation of the in vitro results may be that GITR+/+ CD4+CD25+ cells are different from GITR−/− CD4+CD25+ cells concerning their activity and/or the expression of surface markers other than GITR. Alternatively, GITR triggering, modulating TNFR-associated factors (TRAFs) and NF-κB activity (5, 6, 9, 19), may induce the expression of surface markers other than GITR in CD4+CD25+ cells, which, in turn, modulate DC function. A clear picture will be drawn only when GITRL−/− mice and antibodies against GITRL and/or GITR fusion proteins clearly functioning as agonists are available.
We here demonstrate that GITR/GITRL system activation negatively modulate the synthesis of IL-12 in DCs, contributing to Th2 polarization. However, GITR triggering in responder CD4+ T cells may also directly cause a Th2 polarization, as suggested for another TNFRSF member, OX40 (1, 12). A summary of the possible mechanisms underlying the role of the GITR/GITRL system in Th2 polarization is presented in Fig. 6.
FIG. 6.

Hypothetical mechanisms by which GITR lack promotes a more efficient clearance of C. albicans. In panel A, the low level of IL-12 observed following T-cell receptor stimulation by C. albicans antigens (Ags) in the presence of GITR/GITRL signaling may be explained by three (1, 2, and 3) concurrent intracellular pathways activated following binding of GITRL (expressed on antigen presenting cells) with GITR (expressed at high levels in Treg and activated T cells). IL-12 and other ligands (Ls), including other cytokines (Cks), activate the respective receptors (IL-12R and other R's) either in antigen-presenting cells or in activated CD4+ T cells, skewing to Th2 cytokine production. Also, activation of GITR in activated CD4+ cells (4) might play a role. In panel B, the lack of GITR causes a lack of both GITR and GITRL signaling, high IL-12 levels, and cytokine production promoting a prevalent Th1 polarization. The high efficiency of C. albicans clearance in GITR−/− mice might be obtained in GITR+/+ mice using antibodies or fusion proteins with antagonist function.
Treg cells are crucial in the response to infection. In fact, Treg cells may limit the magnitude of effector responses, which result in failure to adequately control infection (3, 32). As an example, mice lacking toll-like receptor (TLR) 2 are more resistant to C. albicans infection, despite the pivotal role of TLR2 in the recognition of C. albicans, due to a 50% decrease in the Treg cells in TLR−/− mice (16). Ronchetti et al. have found a lower percentage (about 25%) of Treg cells in GITR−/− compared to GITR+/+ Sv129 mice that, however, resulted in nonsignificance upon statistical evaluation (19, 25). Stevens et al. also found in GITR−/− mice of mixed background a difference (about 33%) in Treg presence (30). Thus, although our experiments have been performed in Sv129 mice, GITR−/− mice may have a lower percentage of Treg cells and this may contribute to the more efficient clearance of C. albicans.
Finally, this work supports the hypothesis that GITR/GITRL system hampers C. albicans clearance by skewing to Th2 phenotype. The same effect has been described for OX40/OX40L system in other models (1, 7, 10). This is not surprising if we consider the striking homology in the cytoplasmic domains of the two receptors (17). Furthermore, we have analyzed the short cytoplasmic domains of GITRL and OX40L and here we show that they share a 70% similarity and a 55% identity, with an acid region at the beginning of the protein (E-x [0, 1]-E-x[4, 5]-[ED]) and a strong basic region (R-x[1, 2]-[RK]-x[1]-K-K) next to the transmembrane domain (Fig. 7). The homology of both receptors and ligands suggests that the GITR/GITRL and OX40/OX40L systems may play redundant and/or integrated functions. Further studies will be devoted to addressing the possible interacting functions of GITR/GITRL and OX40/OX40L systems in infection resistance.
FIG. 7.

High similarity between the cytoplasmic domains of murine GITRL and OX40L. Identical amino acids and those with similar function (R and Ki; S and T; and M and V) are in boxes. Identical amino acids and conserved charges are reported in the consensus sequence.
Acknowledgments
We thank Gabriella F. Mansi for editorial assistance.
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC), Milan, and by a Marie Curie Network Grant, Galar Fungail II Project, to A.V.
Editor: A. Casadevall
REFERENCES
- 1.Akiba, H., Y. Miyahira, M. Atsuta, K. Takeda, C. Nohara, T. Futagawa, H. Matsuda, T. Aoki, H. Yagita, and K. Okumura. 2000. Critical contribution of OX40 ligand to T helper cell type 2 differentiation in experimental leishmaniasis. J. Exp. Med. 191:375-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashman, R. B. 1998. Candida albicans: pathogenesis, immunity and host defence. Res. Immunol. 149:281-288. [DOI] [PubMed] [Google Scholar]
- 3.Belkaid, Y., and B. T. Rouse. 2005. Natural regulatory T cells in infectious disease. Nat. Immunol. 6:353-360. [DOI] [PubMed] [Google Scholar]
- 4.Cuzzocrea, S., E. Ayroldi, R. Di Paola, M. Agostini, E. Mazzon, S. Bruscoli, T. Genovese, S. Ronchetti, A. P. Caputi, and C. Riccardi. 2005. Role of glucocorticoid-induced TNF receptor family gene (GITR) in collagen-induced arthritis. FASEB J. 19:1253-1265. [DOI] [PubMed] [Google Scholar]
- 5.Esparza, E. M., and R. H. Arch. 2004. TRAF4 functions as an intermediate of GITR-induced NF-kappaB activation. Cell Mol. Life Sci. 61:3087-3092. [DOI] [PubMed] [Google Scholar]
- 6.Hauer, J., S. Puschner, P. Ramakrishnan, U. Simon, M. Bongers, C. Federle, and H. Engelmann. 2005. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc. Natl. Acad. Sci. USA 102:2874-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ito, T., R. Amakawa, M. Inaba, T. Hori, M. Ota, K. Nakamura, M. Takebayashi, M. Miyaji, T. Yoshimura, K. Inaba, and S. Fukuhara. 2004. Plasmacytoid dendritic cells regulate Th cell responses through OX40 ligand and type I IFNs. J. Immunol. 172:4253-4259. [DOI] [PubMed] [Google Scholar]
- 8.Kim, J. D., B. K. Choi, J. S. Bae, U. H. Lee, I. S. Han, H. W. Lee, B. S. Youn, D. S. Vinay, and B. S. Kwon. 2003. Cloning and characterization of GITR ligand. Genes Immun. 4:564-569. [DOI] [PubMed] [Google Scholar]
- 9.Kwon, B., K. Y. Yu, J. Ni, G. L. Yu, I. K. Jang, Y. J. Kim, L. Xing, D. Liu, S. X. Wang, and B. S. Kwon. 1999. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J. Biol. Chem. 274:6056-6061. [DOI] [PubMed] [Google Scholar]
- 10.Lane, P. 2000. Role of OX40 signals in coordinating CD4 T-cell selection, migration, and cytokine differentiation in T helper (Th)1 and Th2 cells. J. Exp. Med. 191:201-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee, H. S., H. H. Shin, B. S. Kwon, and H. S. Choi. 2003. Soluble glucocorticoid-induced tumor necrosis factor receptor (sGITR) increased MMP-9 activity in murine macrophage. J. Cell Biochem. 88:1048-1056. [DOI] [PubMed] [Google Scholar]
- 12.Linton, P. J., B. Bautista, E. Biederman, E. S. Bradley, J. Harbertson, R. M. Kondrack, R. C. Padrick, and L. M. Bradley. 2003. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion in vivo. J. Exp. Med. 197:875-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McHugh, R. S., M. J. Whitters, C. A. Piccirillo, D. A. Young, E. M. Shevach, M. Collins, and M. C. Byrne. 2002. CD4+CD25+ immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity 16:311-323. [DOI] [PubMed] [Google Scholar]
- 14.Muriglan, S. J., T. Ramirez-Montagut, O. Alpdogan, T. W. Van Huystee, J. M. Eng, V. M. Hubbard, A. A. Kochman, K. H. Tjoe, C. Riccardi, P. P. Pandolfi, S. Sakaguchi, A. N. Houghton, and M. R. Van Den Brink. 2004. GITR activation induces an opposite effect on alloreactive CD4+ and CD8+ T cells in graft-versus-host disease. J Exp. Med. 200:149-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Netea, M. G., J. W. Meer, I. Verschueren, and B. J. Kullberg. 2002. CD40/CD40 ligand interactions in the host defense against disseminated Candida albicans infection: the role of macrophage-derived nitric oxide. Eur. J. Immunol. 32:1455-1463. [DOI] [PubMed] [Google Scholar]
- 16.Netea, M. G., R. Sutmuller, C. Hermann, C. A. Van der Graaf, J. W. Van der Meer, J. H. van Krieken, T. Hartung, G. Adema, and B. J. Kullberg. 2004. Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J. Immunol. 172:3712-3718. [DOI] [PubMed] [Google Scholar]
- 17.Nocentini, G., A. Bartoli, S. Ronchetti, L. Giunchi, A. Cupelli, D. Delfino, G. Migliorati, and C. Riccardi. 2000. Gene structure and chromosomal assignment of mouse GITR, a member of the tumor necrosis factor/nerve growth factor receptor family. DNA Cell Biol. 19:205-217. [DOI] [PubMed] [Google Scholar]
- 18.Nocentini, G., L. Giunchi, S. Ronchetti, L. T. Krausz, A. Bartoli, R. Moraca, G. Migliorati, and C. Riccardi. 1997. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T-cell receptor-induced apoptosis. Proc. Natl. Acad. Sci. USA 94:6216-6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nocentini, G., and C. Riccardi. 2005. GITR: a multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur. J. Immunol. 35:1016-1022. [DOI] [PubMed] [Google Scholar]
- 20.Ohshima, Y., Y. Tanaka, H. Tozawa, Y. Takahashi, C. Maliszewski, and G. Delespesse. 1997. Expression and function of OX40 ligand on human dendritic cells. J. Immunol. 159:3838-3848. [PubMed] [Google Scholar]
- 21.Pietrella, D., P. Lupo, S. Perito, P. Mosci, F. Bistoni, and A. Vecchiarelli. 2004. Disruption of CD40/CD40L interaction influences the course of Cryptococcus neoformans infection. FEMS Immunol. Med. Microbiol. 40:63-70. [DOI] [PubMed] [Google Scholar]
- 22.Romani, L. 1999. Immunity to Candida albicans: Th1, Th2 cells and beyond. Curr. Opin. Microbiol. 2:363-367. [DOI] [PubMed] [Google Scholar]
- 23.Romani, L., A. Mencacci, E. Cenci, R. Spaccapelo, P. Mosci, P. Puccetti, and F. Bistoni. 1993. CD4+ subset expression in murine candidiasis. Th responses correlate directly with genetically determined susceptibility or vaccine-induced resistance. J. Immunol. 150:925-931. [PubMed] [Google Scholar]
- 24.Ronchetti, S., G. Nocentini, C. Riccardi, and P. P. Pandolfi. 2002. Role of GITR in activation response of T lymphocytes. Blood 100:350-352. [DOI] [PubMed] [Google Scholar]
- 25.Ronchetti, S., O. Zollo, S. Bruscoli, M. Agostini, R. Bianchini, G. Nocentini, E. Ayroldi, and C. Riccardi. 2004. GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur. J. Immunol. 34:613-622. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu, J., S. Yamazaki, T. Takahashi, Y. Ishida, and S. Sakaguchi. 2002. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3:135-142. [DOI] [PubMed] [Google Scholar]
- 27.Shin, H. H., S. J. Kim, D. S. Lee, and H. S. Choi. 2005. Soluble glucocorticoid-induced tumor necrosis factor receptor (sGITR) stimulates osteoclast differentiation in response to receptor activator of NF-kappaB ligand (RANKL) in osteoclast cells. Bone 36:832-839. [DOI] [PubMed] [Google Scholar]
- 28.Shin, H. H., S. J. Kim, H. S. Lee, and H. S. Choi. 2004. The soluble glucocorticoid-induced tumor necrosis factor receptor causes cell cycle arrest and apoptosis in murine macrophages. Biochem. Biophys. Res. Commun. 316:24-32. [DOI] [PubMed] [Google Scholar]
- 29.Spinicelli, S., G. Nocentini, S. Ronchetti, L. T. Krausz, R. Bianchini, and C. Riccardi. 2002. GITR interacts with the pro-apoptotic protein Siva and induces apoptosis. Cell Death Differ. 9:1382-1384. [DOI] [PubMed] [Google Scholar]
- 30.Stephens, G. L., R. S. McHugh, M. J. Whitters, D. A. Young, D. Luxenberg, B. M. Carreno, M. Collins, and E. M. Shevach. 2004. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J. Immunol. 173:5008-5020. [DOI] [PubMed] [Google Scholar]
- 31.Suffia, I., S. K. Reckling, G. Salay, and Y. Belkaid. 2005. A role for CD103 in the retention of CD4+CD25+ Treg and control of Leishmania major infection. J. Immunol. 174:5444-5455. [DOI] [PubMed] [Google Scholar]
- 32.Taylor, M. D., L. LeGoff, A. Harris, E. Malone, J. E. Allen, and R. M. Maizels. 2005. Removal of regulatory T-cell activity reverses hyporesponsiveness and leads to filarial parasite clearance in vivo. J. Immunol. 174:4924-4933. [DOI] [PubMed] [Google Scholar]
- 33.Tone, M., Y. Tone, E. Adams, S. F. Yates, M. R. Frewin, S. P. Cobbold, and H. Waldmann. 2003. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc. Natl. Acad. Sci. USA 100:15059-15064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watts, T. H. 2005. TNF/TNFR family members in costimulation of T-cell responses. Annu. Rev. Immunol. 23:23-68. [DOI] [PubMed] [Google Scholar]
- 35.Yu, K. Y., H. S. Kim, S. Y. Song, S. S. Min, J. J. Jeong, and B. S. Youn. 2003. Identification of a ligand for glucocorticoid-induced tumor necrosis factor receptor constitutively expressed in dendritic cells. Biochem. Biophys. Res. Commun. 310:433-438. [DOI] [PubMed] [Google Scholar]