

Abstract
Genetic variants of interleukin-3 (IL-3), a well-studied cytokine, may have a role in the pathophysiology of rheumatoid arthritis (RA); but reports on this association sometimes conflict. A case-control study was designed to investigate association between RA and a single-nucleotide polymorphism (SNP) in the IL-3 promoter region. Comparison of cases of RA versus control individuals yielded a χ2 value of 14.28 (P=.0002), with a genotype odds ratio of 2.24 (95% confidence interval [95%CI] 1.44–3.49). When female cases with earlier onset were compared with female control individuals, the SNP revealed an even more significant correlation, with χ2=21.75 (P=.000004) and a genotype odds ratio of 7.27 (95%CI 2.80–18.89). The stronger association that we observed in this clinically distinct subgroup (females with early onset), within a region where linkage disequilibrium was not significantly extended, suggested that the genuine RA locus should locate either within or close to the IL-3 gene. Combined genotype data on SNPs on eight other candidate genes were combined with our IL-3 results, to estimate relationships between pairs of loci and RA, by maximum-likelihood analysis. The utility of combining the genotype data in this way to identify possible contributions of various genes to this disease is discussed.
Introduction
Rheumatoid arthritis (RA [MIM 180300]) is one of the common clinical entities with complex genetic etiology; in fact, numerous genes are likely to be associated with this disease (Selden et al. 1999). Two independent sib-pair linkage analyses have reported multiple loci as candidates (Cornelis et al. 1998; Shiozawa et al. 1998), but association studies involving those genes have been inconclusive except in the case of human leukocyte antigen (HLA) class II (Nepom 1998).
The cytokine IL-3 represents a potential candidate for RA because it affects differentiation, proliferation, and function of three hematopoietic lineages in bone marrow and also has important physiological functions with regard to mature myeloid cells in the peripheral system (Harant and Lindley 1998). The human IL-3 gene lies on chromosome 5q23-31, together with genes encoding granulocyte monocyte-colony–stimulating factor (GM-CSF), IL-4, IL-5, IL-9, macrophage colony–stimulating factor, and several other genes related to the immune system. The GM-CSF gene is only 9 kb distant from IL-3 (Yang et al. 1988). Therefore, it seemed interesting to investigate the possible association between RA and polymorphisms in the region containing the IL-3 gene, not only to detect potential association between the gene itself and the disease but also to evaluate other genes in this region to determine their possible contributions to RA, if considerable linkage disequilibrium (LD) exists among them.
Although previous reports on association between IL-3 and RA were not conclusive (Firestein et al. 1988; Waalen et al. 1992; Heller et al. 1997; Levesque et al. 1998), we designed a case-control association study, using an a single-nucleotide polymorphism (SNP) in the promoter region of the IL-3 gene, and we evaluated the range of LD by genotyping two other SNPs close to the promoter SNP. In addition, the functional consequence that the promoter SNP had on gene expression was investigated.
Identification of genes associated with complex genetic traits is often very difficult because their individual contributions are likely to be small; optimization of data seems indispensable. Therefore, to estimate the combinatorial effect of pairs of loci by the maximum-likelihood method, we also genotyped our subjects for SNPs present in eight other candidate genes and investigated relationships between each of them and the IL-3 polymorphism, with regard to RA susceptibility.
Subjects and Methods
Subjects
Cases and controls were recruited through several medical institutes in Japan. All subjects were Japanese and consented to participate in the study according to the process approved by the Ethical Committee at the SNP Research Center, The Institute of Physical and Chemical Research (RIKEN), Tokyo. All cases met the American Rheumatism Association's revised criteria for the classification of RA (Arnett et al. 1988). A total of 254 cases and 881 controls were enrolled.
SNPs
Three SNPs in the IL-3 vicinity were genotyped. These sites are at nt −16 (T→C) in the 5′ flanking (promoter) region of the IL-3 gene, at nt 131 (T→C) of exon 1 of the IL-3 gene, and at nt 23 (C→T) of exon 4 of the GM-CSF gene; these sites match bases 4247, 4393, and 19375 on the GenBank sequence AC004511.1 (gi 3002474). Hereafter, we will refer to them as “rIL-3-16,” “cIL-3-131,” and “cGM-CSF-23,” respectively (fig. 1). cIL-3-131 substitutes the 8th serine for proline in the IL-3 product (Jeong et al. 1998), and cGM-CSF-23 substitutes the 117th threonine for isoleucine in the GM-CSF protein (Yamada et al. 2000). SNPs on eight other genes were also assessed for association with RA, to evaluate possible relationships to IL-3. The eight genes were those for metalloproteinase 2 (MMP2), tumor necrosis–factor receptor 1 (TNFR1), signal transducer and activator of transcription-6 (STAT6), interleukin-15 (IL-15), interleukin-1β (IL-1β), CD44, interleukin-4 receptor α (IL-4α), and tissue inhibitor of metalloproteinase 1 (TIMP1). The accession number of the GenBank reference sequence, the location on the sequence, the bases potentially substituted in the eight SNPs, as well as the SNPs in IL-3 and in GM-CSF were summarized in table 1. These SNPs were identified by the same procedure that we had used earlier for cGM-CSF-23 (Yamada et al. 2000).
Figure 1.

Map of IL-3 and GM-CSF genes, showing locations of the three SNPs genotyped.
Table 1.
Genotyped SNPs
|
GenBank Reference Sequence |
Base |
|||
| Gene Containing SNP(s) | Accession Number | Location of SNP | Major Allele | Minor Allele |
| IL-3 (rIL-3-16) | AC004511.1 (gi 3002474) | 4247 | T | C |
| IL-3 (cIL-3-131) | AC004511.1 (gi 3002474) | 4393 | T | C |
| GM-CSF (cGM-CSF-23) | AC004511.1 (gi 3002474) | 19375 | C | T |
| MMP2 | U96098.1 (gi 2459743) | 932 | C | T |
| TNFR1 | AC006057.1 (gi 4731048) | 34201 | A | G |
| STAT6 | AF067575.1 (gi 3789866) | 4671 | G | A |
| IL-15 | AF038163.1 (gi 2708695) | 560 | T | A |
| IL-1β | X04500.1 (gi 33788) | 1423 | C | T |
| CD44 | L05423.1 (gi 337955) | 674 | C | T |
| IL-4Rα | AC004525.1 (gi 3219333) | 94272 | G | A |
| TIMP1 | D11139.1 (gi 220124) | 600 | G | C |
Genotyping of SNPs
DNA samples were extracted from whole peripheral blood by standard methods and were genotyped for SNPs by PCR–allele-specific oligonucleotide hybridization (PCR-ASO). For that procedure, 10 segments, one containing two SNPs on the IL-3 gene and one from each of the other nine genes, were amplified by PCR using the primers listed in table 2. ASO hybridizations were performed as follows: Each PCR product containing an SNP locus was transferred to a BIODYNE (B) membrane (PALL), was chemically denatured, and was fixed by UV light. Each allele-specific genotyping primer was labeled with [32P]-ATP by polynucleotide kinase. Membranes were prehybridized in hybridization solution for 2 h at 35°C; then they were hybridized with genotyping primers for 4 h at 35°C; and then they were washed in 6 × SSC under the conditions described in table 2.
Table 2.
PCR-ASO Primers and Conditions
|
PCR Primer |
ASO-Genotyping Primer Specific to |
Washing Temperature(°C) |
||||
| SNP/GeneContaining SNP | 1 | 2 | Major Allele | Minor Allele | MajorAllele | MinorAllele |
| rIL-3-16 | GCCAGGGTAGTCCAGGTGAT | CATTGAGGTTGTTGAAGTCC | TTGGCAACAACCTCC | GGAGGCTGTTGCCAA | 50 | 50 |
| cIL-3-131 | GCCAGGGTAGTCCAGGTGAT | CATTGAGGTTGTTGAAGTCC | TTCAAGGACGTTGTC | GACAACGCCCTTGAA | 50 | 50 |
| cGM-CSF-23 | GAGTTCTAAGAGGCAGTAGAG | TTCTTCTGCCATGCCTGTAT | CCAGACTATCACC | GTGATAATCTGGGT | 35 | 49 |
| MMP2 | CCTGTGACCGAGAATGCGGAC | CTCCTGGGAGTGCAGCCCAG | GCGGACCCTCC | AGGAGAGTCCGC | 42 | 35 |
| TNFR1 | CTGTGTGGTTGTTTTCTGT | GATGGGTGGGATGGATGGAC | AGGAGAAGTGACC | GGTCACCTCTCC | 50 | 35 |
| STAT6 | CTAGTACAGGTTTTGCCCTG | CTATGACCCCTGCCTTGGG | CAGCTATACATTTAAC | TTAAATATATAGCTGG | 40 | 40 |
| IL-15 | GCAGAGAGGCTCATTGCTC | CGGGAGCATAGGCGAAGAC | GTTGGACTTCAAAG | GTTGGACATCAAAG | 35 | 49 |
| IL-1β | TGGTCTTGCAGGGTTGTGT | CGTTGTGCAGTTGATGTCC | GCTCCCGAGGCA | TGCCTCAGGAGCT | 38 | 38 |
| CD44 | AGATGGTTTCTCATAAGGTAAAA | TATTGTGTGCAATCAGCCCTTT | TGCTGTTACAATAATT | AATTATTATAACAGCA | 42 | 42 |
| IL-4Rα | TCCTCCTGCTGTTGCTATGA | AAGAGTCTGATGCGGTTCCT | TCAGGGACACACGTG | CACGTGTATCCCTGA | 53 | 53 |
| TIMP1 | GGAGTGGGAGGATTATGTCAGT | CATTCCTCACAGCCAACAGT | GCCTTTGGCAGC | GCCTTTGCCAGC | 42 | 35 |
Typing of HLA-DRB1
All cases were typed for HLA-DRB1, with the HLA-DRB BigDye Terminator Sequencing-Based Typing Kit (PE Biosystems).
Assay of IL-3 Promoter
We performed luciferase assays to evaluate whether the SNP at the rIL-3-16 locus influences promoter function, using two promoter segments of different lengths. The longer segment ranged from base −357 in the 5′ flanking region to base 55 in exon 1, and the shorter one ranged from base −59 in the 5′ flanking region to base 55 in exon 1. A target segment corresponding to each allele was amplified by PCR using genotype-known genomic DNAs as templates; primers for this procedure were, for the longer segment, 5′-AAGATCTAGGGTAGTCCAGGTGATGGCA-3′ and 5′-AAAGCTTCATGTTTGGATCGGCAGGAGG-3′ and, for the shorter segment, 5′-TTAAGATCTCGGGGTTGTGGGCACCTTGCTG-3′ and 5′-AAAGCTTCATGTTTGGATCGGCAGGAGG-3′. PCR products were digested with BglII and HindIII and were cloned into Photinus luciferase reporter plasmid pGVB2 (Nippon Gene). Cloned DNAs were validated by dye-terminator DNA sequencing. The plasmid DNAs were transfected into Jurkat cells, by Superfectant transfection reagent (QIAGEN). Transfected cells were stimulated with 40 ng of phorbol myristate acetate/ml and 4 μg of ionomycin/ml, and luciferase activities were measured 48 h later by a chemiluminescence assay (PicaGene Dual SeaPansy Assay System kit; Toyo Ink). Three independent experiments of transfection were performed.
Statistical Analysis
Cases were subdivided by sex, age at onset (<55 years and ⩾55), rheumatoid-factor status, history of orthopedic intervention for RA-related arthropathy, and RA-susceptible HLA-DRB1 status. Controls were also subdivided by sex and were adjusted for sex and for age at sampling (i.e., <30, 30–39, 40–49, 50–59, or ⩾60 years), to fit the case-group distribution. All cases or subgroups of cases were compared with all controls or corresponding subgroups of controls, respectively, with adjustment for sex and age. Alternative hypotheses and null hypotheses for association between cases and controls were evaluated by χ2 tests; in each case, the odds ratio and 95% confidence interval (95%CI) were calculated by Woolf’s method. Haplotype frequencies for pairs of alleles, as well as χ2 values for allele associations, were estimated by the Estimating Haplotype-frequencies software program (Terwilliger and Ott 1994; Web Resources for Genetic Linkage Analysis [Rockefeller University]). LD coefficients D′=D/Dmax were calculated (Devlin and Risch 1995). Hardy-Weinberg equilibrium of alleles at individual loci was assessed by χ2 statistics (Nielsen et al. 1999).
Estimation of Combinatorial Effect of Two Loci, by Use of Genotype Data
Genotype data on eight SNPs in 91 cases and in 91 controls were combined with data on rIL-3-16. The most likely pattern of combinatorial genetic contribution of any two loci was estimated by the maximum-likelihood method (Appendix). Genotype relative risk was defined against the genotype that consisted of homozygotes of nonsusceptibility alleles from both SNPs. The variables used to define a relative risk of combined genotypes were designed such that the relative risk for heterozygotes was either equal to or the square root of the relative risk for homozygotes of susceptibility alleles from both SNPs, or 1, as modeled by Risch and Merikangas (1996). The model represented 10 patterns of distribution of the genotype relative risk, with variation of the relative risk of heterozygotes compared with of homozygotes. One of the 10 patterns was independent, which meant that two loci possessed disease-susceptible alleles on their own and that those alleles increased the risk of RA without mutual interaction The other nine patterns represented combinatorial effects on genotype risk. LOD scores were calculated, for comparison with a hypothesis that no association was present for either SNP; we calculated the difference between the highest LOD score among the nine combinatorial patterns and the LOD score for the independent pattern, to test the likelihood of the combinatorial pattern against the independent pattern. If an independent relationship to RA was estimated as being most likely, then subgrouping of the test population by genotype status of two SNPs was considered inappropriate for testing of associations with the disease; on the other hand, if a combinatorial rather than an independent pattern was estimated as being most likely for two SNPs with a difference between two LOD scores, then subgrouping was considered appropriate. The χ2 test was then performed to detect associations between RA and each SNP, with the entire test population being used for the eight SNPs from unrelated genes and with the subgrouped population being used for an appropriate SNP(s).
Results
A. Data on rIL-3-16, cIL-3-131, and cGM-CSF-23 SNPs, for 254 Cases and 881 Controls
Demographic features and age-at-onset distribution among subjects are summarized in tables 3 and 4. The distribution of HLA-DRB1 types among our cases (table 5) was similar to that in a previous report from Japan (Wakitani et al. 1997). Alleles and genotypes at the rIL-3-16 marker are shown in table 6, along with allele and genotype data on the control group, adjusted according to sex and age distributions. The adjusted data on all control groups deviated more from case data than did nonadjusted control data, suggesting that nonadjusted data are more conservative for detection of association. Therefore, we used nonadjusted data for further discussion.
Table 3.
Characterization of Cases and Controls
|
Cases |
Controls |
|||||
| Overall | Female | Male | Overall | Female | Male | |
| No. | 254 | 206 | 48 | 881 | 525 | 356 |
| Fraction of sample: | ||||||
| Total | .81 | .19 | .60 | .40 | ||
| Positive for: | ||||||
| Rheumatoid factor | .81 | .79 | .88 | … | … | … |
| History of surgical intervention(s) | .47 | .48 | .41 | … | … | … |
| RA-susceptible HLA-DRB1 typea | .69 | .68 | .73 | … | … | … |
| Average age (years): | ||||||
| At sampling | 57.0 | 56.1 | 61.3 | 41.8 | 43.4 | 39.4 |
| At onset | 46.3 | 44.9 | 52.4 | … | … | … |
Includes *0101, *0102, *0401, *0404, *0405, and *1001.
Table 4.
Distribution of Ages at Onset of RA
|
Fraction |
|||
| Age at Onset(years) | Total Sample | Female | Male |
| <30 | .15 | .17 | .06 |
| 30–39 | .18 | .19 | .09 |
| 40–49 | .26 | .25 | .30 |
| 50–59 | .20 | .19 | .24 |
| ⩾60 | .22 | .20 | .30 |
Table 5.
Distribution of HLA DRB1 Types of among Japanese Patients with RA
Table 6.
Allele and Genotype Data on rIL-3-16 T→C
|
Allele |
Genotype |
|||||
| Major(T) | Minor(C) | Major Homozygotes(TT) | Heterozygotes(TC) | Minor Homozygotes(CC) | Overall | |
| Cases: | ||||||
| Total: | ||||||
| No. (female/male) | 323 (270/53) | 185 (142/43) | 101 (86/15) | 121 (98/23) | 32 (22/10) | 254 (206/48) |
| Fraction (female/male) | .64 (.66/.55) | .36 (.34/.45) | .40 (.42/.31) | .48 (.48/.48) | .13 (.11/.21) | |
| Age at onset <55 years: | ||||||
| No. (female/male) | 167 (146/21) | 75 (60/15) | 55 (48/7) | 57 (50/7) | 9 (5/4) | 121 (103/18) |
| Fraction (female/male) | .69 (.70/.58) | .31 (.29/.42) | .45 (.47/.39) | .47 (.49/.39) | .07 (.05/.22) | |
| Controls: | ||||||
| Total: | ||||||
| No. (female/male) | 954 (559/395) | 808 (491/317) | 252 (140/112) | 450 (279/171) | 179 (106/73) | 881 (525/356) |
| Fraction (female/male) | .54 (.53/.55) | .46 (.47/.45) | .29 (.27/.31) | .51 (.53/.48) | .20 (.20/.21) | |
| Sex adjusted: | ||||||
| No. (female/male) | 907 (731/176) | 855 (699/156) | 221 (177/44) | 465 (377/88) | 195 (161/34) | 881 (715/166) |
| Fraction (female/male) | .51 (.51/.53) | .49 (.49/.47) | .25 (.25/.27) | .53 (.53/.53) | .22 (.23/.21) | |
An association between RA and SNP at rIL-3-16 was statistically significant in a comparison of allele frequencies in total cases versus with those in the controls (P=.0002), as well as in a comparison of female cases versus female controls (P=.00002) and in a comparison of earlier-onset females versus female controls (P=.000004) (table 7). Similar statistical significance was obtained with Fisher’s exact probability test; for example, comparison of the allele frequencies in cases versus those in all controls yielded a two-sided Fisher’s exact probability of .0002. No statistical significance was observed between subgroups of cases classified according to rheumatoid-factor status, history of surgical intervention(s), or RA-susceptible DRB1 status, regardless of sex (P>.3; data not shown). A comparison of male cases versus male controls provided no statistical significance even after adjustment for age (P>.95).
Table 7.
Tests for Association and Odds Ratios, for Genotype Data Regarding the rIL-3-16 Locus
|
Test of Association(χ2a [P] ) |
Odds Ratio [95%CI] |
|||||
| Groups Compared | Allele Frequency(Major Allele vs. Minor Allele) | Genotype Frequency(2×3 table) | Major Homozygotes: Others | Minor Homozygotes: Others | Heterozygotes:Minor Homozygotes | Major:Minor Homozygotes |
| Cases vs. controls | 14.28 [.0002] | 14.88 [.0006] | 11.46 [.0007] | 7.76 [.005] | 1.50 [0.98–2.30] | 2.24 [1.44–3.49] |
| Female cases vs. female controls | 18.22 [.00002] | 19.42 [.00006] | 15.75 [.00007] | 9.27 [.002] | 1.69 [1.01–2.83] | 2.96 [1.74–5.04] |
| Male cases vs. male controls | .003 [1.0] | .003 [1.0] | .0009 [1.0] | .0003 [1.0] | .98 [0.45–2.17] | .98 [0.42–2.29] |
| Earlier-onset cases vs. controls | 19.08 [.00001] | 19.60 [.00006] | 14.21 [.0002] | 11.58 [.0007] | 2.52 [1.22–5.20] | 4.34 [2.09–9.01] |
| Earlier-onset female cases vs. female controls | 21.75 [.000004] | 23.24 [.000009] | 16.32 [.00005] | 13.92 [.0002] | 3.80 [1.48–9.79] | 7.27 [2.80–18.89] |
For χ2 values for genotype frequency (2×3 tables), df =2; for the other χ2 values, df =1.
Odds ratios with 95%CIs are shown in table 7. In the comparison of total cases versus total controls, the odds ratio of genotype TT (major-allele homozygotes) to CC (minor-allele homozygotes) was 2.24 (95%CI 1.44–3.49) and that of TC (heterozygotes) to CC was 1.50 (95%CI 0.98–2.30). When earlier-onset female cases were compared with female controls, those ratios increased to 7.27 (95%CI 2.8–18.89) and 3.8 (95%CI 1.48–9.79), respectively.
Allele and genotype frequencies of rIL-3-16, cIL-3-131, and cGM-CSF-23 are shown in table 8. Almost-identical genotype frequency was seen for cIL-3-131 and rIL-3-16, suggesting that these two loci have the same degree of association with RA. On the other hand, a significant association with RA was not observed for the cGM-CSF-23 SNP locus; this was especially true for the frequency of major-allele homozygote, which showed the strongest association with rIL-3-16. Our analyses of LD, based on the estimated haplotypes of SNPs in the IL-3 vicinity, are summarized in table 9. The rIL-3-16 and cIL-3-131 sites were in complete LD, and rIL-3-16 was linked to cGM-CSF-23 with D′ value of .78. No particular haplotype showed higher statistical significance for association with RA than did rIL-3-16 alone. Hardy-Weinberg equilibrium of the distribution of genotypes for each SNP, for both the case group and the control group, was evaluated by χ2 tests and showed no significant deviation (P>.70; data not shown).
Table 8.
Allele and Genotype Frequencies of rIL-3-16, cIL-3-131, and cGM-CSF-23
|
Frequency |
|||||
| Allele |
Genotype |
||||
| Major | Minor | Major Homozygotes | Heterozygotes | Minor Homozygotes | |
| Cases: | |||||
| rIL-3-16 | .64 | .36 | .40 | .48 | .13 |
| cIL-3-131 | .64 | .36 | .40 | .48 | .13 |
| cGM-CSF-23 | .66 | .34 | .41 | .49 | .11 |
| Controls: | |||||
| rIL-3-16 | .55 | .45 | .29 | .51 | .20 |
| cIL-3-131 | .55 | .45 | .29 | .52 | .19 |
| cGM-CSF-23 | .61 | .39 | .39 | .43 | .18 |
Table 9.
Haplotype-Frequency Estimation and LD, for rIL-3 16-cIL-3 131 and rIL-3 16-cGM-CSF 23
| Group | Frequency of Haplotype | D′ | |||
| T-T |
C-C |
T-C |
C-T |
||
| rIL-3 16-cIL-3 131: | |||||
| Cases (n=254) | .6339 | .3642 | .0020 | .0000 | 1.00 |
| Controls (n=130) | .5577 | .4423 | .0000 | .0000 | 1.00 |
| Total (n=384) | .6081 |
.3906 |
.0013 |
.0000 |
1.00 |
| T-C |
C-T |
C-C |
T-T |
||
| rIL-3 16-cGM-CSF 23: | |||||
| Cases (n=84) | .6050 | .3074 | .0438 | .0438 | .81 |
| Controls (n=91) | .4999 | .3351 | .1100 | .0550 | .75 |
| Total (n=175) | .5504 | .3218 | .0782 | .0496 | .78 |
Thus far, P values have not been corrected for multiple testing; however, even if Bonferroni’s correction were to be applied, our result would still be statistically significant, as is shown below. We did 21 comparisons (three types of comparisons for each of seven pairs of comparison groups): (1) total cases versus total controls, (2) total female cases versus total female controls, (3) total male cases versus total male controls, (4) age-and-sex–adjusted cases versus age-and-sex–adjusted controls, (5) HLA-DRB1–positive group versus HLA-DRB1–negative group, (6) rheumatoid factor–positive group versus rheumatoid factor–negative group, and (7) history-of-surgical-intervention(s)–positive group versus history-of-surgical-intervention(s)–negative group, with all comparisons being done for (i) one type of allele-frequency comparison and (ii) two types of genotype-frequency comparison. Therefore, when Bonferroni’s correction was applied, our most significant P value—that is, .000004, for the comparison of female cases versus total female controls—was corrected to .0001, which is still significant.
B. IL-3 Promoter Assay
To investigate the possibility that rIL-3-16 SNP plays a role in the promoter activity, we performed a luciferase-activity assay, using two different lengths of genomic fragments with either of the SNP sequences. The ratio of stimulated to unstimulated luciferase activity in assays of the longer segment was 3.63 (SD 0.835) for the T allele and 3.99 (SD 0.746) for the C allele, 48 h after stimulation; the Student’s t value was 1.08 (P=.15). For the short-segment assay at 48 h, the ratios were 2.05 (SD 0.009) and 1.74 (SD 0.052) for the T and C alleles, respectively; Student’s t value was 0.84 (P=.22).
C. Combinatorial Data on Two SNPs
We then investigated a combinatorial effect of two SNPs: the rIL-3-16 and one SNP from a panel of eight other candidate genes. The results of χ2 tests for associations between SNPs in the eight different genes and RA and between rIL-3-16 and RA, as well as the results of maximum-likelihood estimation of relationships between those eight SNPs and rIL-3-16, are shown in table 10. The χ2 values listed in this table were the largest ones observed among comparisons of genotype frequencies for each SNP. The SNPs on the IL-1β, CD44, and TNFR1 genes were considered to have combinatorial effects with IL-3 genotype status, in contributing to RA. The IL-4Rα gene had the same LOD scores for independent and combinatorial patterns, and the other four genes were estimated to be independent of rIL-3-16's genotype status. Of the three genes that appeared to have combinatorial effects with rIL-3-16, IL-1β yielded the largest difference, 0.8 (P=.1), between LOD scores of combinatorial and independent patterns. rIL-3-16 and IL-1β were further assessed for combinatorial effect and association with RA, with the subdivided population.
Table 10.
Results of χ2 Tests for Association between SNPs in Nine Genes and RA, and Estimation of Relationships between Eight SNPs and rIL-3-16
|
LOD Scoreb |
||||
| Gene Containing SNP | Association Test of 91 Cases of RA vs. 91 Controls (χ2a [P]) | Combinatorial Pattern | Independent Pattern | Difference |
| MMP2 | 1.93 [.16] | 1.7 | 1.8 | −.1 |
| TNFR1 | 1.99 [.16] | 1.9 | 1.6 | .3 |
| STAT6 | 2.23 [.14] | 1.6 | 1.7 | −.1 |
| IL−15 | 2.58 [.11] | 1.7 | 1.8 | −.1 |
| IL-1β | 2.91 [.09] | 2.8 | 2.0 | .8 |
| CD44 | 3.92 [.05] | 3.4 | 2.8 | .6 |
| IL-4Rα | 4.85 [.03] | 2.4 | 2.4 | .0 |
| TIMP1 | 5.30 [.02] | 3.3 | 3.4 | -.1 |
| IL-3 (rIL-3-16) | 5.67 [.02] | … | … | … |
Maximum value, among comparisons of genotype frequencies.
Under hypothesis that no association exists between two SNPs and RA.
Our estimates of relative risk for RA with respect to alleles of rIL-3-16 and of the IL-1β gene were as follows: compared with homozygotes for nondisease alleles of both SNPs, homozygotes for disease alleles of both SNPs carried a relative risk of 5.3, whereas persons homozygous for the disease allele of one SNP and heterozygous for the other would carry a relative risk of 2.3; relative risk for heterozygotes for both SNPs were 1.5, and that for the rest was 1.0 (Appendix, pattern 6). This pattern represents the situation in which a disease-susceptible allele of either gene requires the other gene’s disease allele in order to produce its own effect and in which the relative risk for persons homozygous for the disease allele of either SNP is the square of that for heterozygotes. Because pattern 6 was estimated as being most likely, the subgroup of patients with RA who carried either the TT or the TC genotype of rIL-3-16 was selected for further tests of association between the SNP on IL-1β and RA, and the subgroup of those either homozygous or heterozygous for the disease allele of the SNP on IL-1α was selected for testing of association between rIL-3-16 and RA. Eighty-six cases and 72 controls constituted the subgroup of those either homozygous or heterozygous for the disease allele of the SNP on IL-3, and 67 cases and 54 controls constituted the subgroup either homozygous or heterozygous for the disease allele of the SNP on IL-1β. The results are shown in table 11. For rIL-3-16 and the SNP on IL-1β, the statistical significance of associations with RA when these selected subgroups were used were >10 times and >4 times stronger, respectively, than values obtained with association tests comparing 91 cases versus 91 controls.
Table 11.
Association between SNPs in either IL-3 or IL-1β and RA, for the Entire Sample and Selected Subgroups
|
Association with RA(χ2 [P]) |
||
| Gene Containing SNP | Entire Sample | Selected Subsamples |
| IL-3 | 5.67 [.02] | 10.01 [.002] |
| IL-1β | 2.91 [.09] | 5.37 [.02] |
Discussion
We have reported here a significant association between a C→T substitution at the −16 position of the IL-3 gene and RA. Although our control group consisted of 3.5 times as many individuals as were in the RA group, application of χ2 tests was statistically appropriate, because similar results were obtained with Fisher’s exact-probability test. Since we intended for the control group to represent the general Japanese population without RA, the distributions of sex and age in the two groups were not the same. However, it seemed clear that sex differences and age differences between the case group and the control group had no significant influence on the statistical analyses, because association tests performed with sex- or age-adjusted controls revealed an even more significant association than was revealed by association tests using the data for control individuals as a whole.
Comparisons between each of the subgroups categorized by female sex and/or earlier onset disclosed strong associations that became even more significant when females with earlier-onset RA were compared with sex- and age-matched controls and with the overall sample of controls. On the other hand, in the male population, no association was observed between the SNP and RA. It seemed that the SNP was associated with RA in females, particularly with females with earlier-onset RA. This result is consistent with epidemiological data indicating that females with earlier-onset RA constitute a distinct subgroup among the total RA population and that, at ages up to the mid-50s, females are more susceptible to RA than are males (Ehrlich et al. 1970; Deal et al. 1985). IL-3 and some other factor(s) specific to reproductive females may have synergistic effects for increasing the risk for RA. This finding is very interesting because it may help to differentiate patients with RA into subgroups with different genetic backgrounds. Other conditions, including rheumatoid-factor status, history of orthopedic surgical intervention, or HLA-DRB1 status, appeared to have no relationship to the effect that IL-3 has on RA; in our experience, the latter association seemed to be, to some extent, independent of HLA-DRB1 and did not exert much influence on either the production of rheumatoid factor or the destructive activity of RA. It is worth noting, too, that the SNP on GM-CSF, the gene lying closest to the IL-3 gene, retained no statistically significant association with disease status, although LD between the two loci was substantial. The rIL-3-16 and cIL-3-131 SNPs, which are in complete LD, might be causative genetic variations—or, at least, might be in closer proximity to a causative genetic alteration than is cGM-CSF-23. Our results support a previous report of association between IL-3 and RA (Heller et al. 1997) and do not contradict two published sib-pair linkage studies of RA (Cornelis et al. 1998; Shiozawa et al. 1998), both of which reported LOD scores close to 2, for D5S422 on 5q32-33. That LOD score is not large enough to allow conclusive identification of a disease locus for RA, but it nonetheless provides evidence supporting our results. In the Results section, we have shown, for the most significant association, the P value corrected with Bonferroni’s correction; however, we have not used Bonferroni's correction for the other P values, because we believe that it would produce a P value that would be too conservative for assessment of our data. The reasons why we believe that Bonferroni’s correction would produce a P value that is too conservative are as follows: (1) we decided to be conservative by avoiding the strongest association found in comparisons between age-sex adjusted groups, and (2) a trend of increase, in association, to explain specific association of a distinct clinical entity and the SNP made our multiple comparisons nonrandom.
With regard to complex genetic traits, each related gene contributes disease susceptibility to some extent, but such genes may also interact with each other. Therefore, in addition to undertaking large-scale, multilocus genotyping studies for identification of genes associated with a multifactorial disease, one must identify loci making small contributions and dissect relationships between multiple loci. However, it is not easy to estimate the interaction of loci, each of which may have only a small effect on disease susceptibility. In the study reported here, we divided cases into subgroups based on sex, age at onset, HLA-DRB1 status, rheumatoid-factor status, and requirement for surgical intervention(s), since previous studies had suggested that these parameters are appropriate for the subgrouping of patients with RA (Gordon and Hastings 1994; Selden et al. 1999). Before we apply genotype data to the effort to subdivide populations for association tests using SNPs in multiple genes, the subgrouping procedure should be justified. For justification of our analyses, we applied two hypotheses to estimate a relationship between any two SNPs; the first hypothesis speculated that there is a functional relationship between two genes, and the other hypothesis justified the use of subdivided populations by expecting that genotype information for one SNP would strengthen our power to detect other associated loci. The maximum difference between LOD scores for independent patterns and LOD scores for combinatorial patterns was 0.8, for the relationship between IL-3 and IL-1β. Statistical significance should be obtained by approximating a distribution of a doubled value of the difference between LOD scores, to a 50:50 mixture of a χ2distribution with 1 df and a probability mass at 0, because, in the number of variables used, the difference between independent and combinatorial models was 1 and because, to maintain the intensity of relative risk of homozygosity of the susceptibility allele, heterozygosity, and homozygosity of the nonsusceptibility allele, in this order, significance was declared in one direction. The P value for the 0.8 difference between LOD scores is .1, which is not statistically significant. The reason why statistical significance is not large is that the sample size that we used for our preliminary estimation is small. However, sample size should be larger for future studies, to evaluate relationship among genes. Therefore, statistical significance has been simulated for larger sample sizes, as is shown below; for example, if, instead of 91, the sample size were 1,000, for both the case group and the control group, and if the same genotype frequency were obtained as was seen in our preliminary observation of IL-3 and IL-1β, then the difference between LOD scores would be 8.8 instead of 0.8, and the P value would be .000014, which would seem to be significant enough to justify subgrouping.
The effect that the use of subdivided populations has on tests of association has been assessed as described below. As shown in table 10, in the tests for association with RA that do not use subdivided population, IL-1β shows a smaller χ2 value than does either IL-4Rα or TIMP1. In the estimation of its relationship with IL-3, IL-1β showed larger LOD scores than did either IL-4Rα or TIMP1. When the subdivided population was used, the χ2 value in the test for association with RA increased from 2.91 to 5.37. The increased value, 5.37, was more than the χ2 value for IL-4Rα, 4.85, and was closer to that of TIMP1, 5.30. This change suggests that the estimation may be useful in screening for modifier genes that, by themselves, do not affect the pathogenesis of the disease strongly enough to be identified by examination of the association between a disease and a single gene. In addition, the relationship between IL-3 and IL-1β has been supported by biological studies. IL-1β has been reported to up-regulate IL-3 in the mast cell (Hultner 2000), suggesting that, in combination, the SNP causing amino acid substitution in the IL-1β gene and the SNP in the promoter region of IL-3 may affect the expression level of IL-3. IL-1β and IL-3 also have been shown to regulate hyaluronan binding to the monocyte CD44 molecule, a key event in the initiation of inflammation (Levesque and Haynes 1997). These biological observations, combined with our preliminary genetic estimation, indicate that there is a network including these three molecules that regulates inflammation and that the change of this network by these SNPs will be one of the main events in the pathogenesis of RA.
Acknowledgments
We thank Toyokazu Seki, Koji Suematsu, Maiko Minami, Norihiro Kushida, Koji Jinno, Hiroto Kawakami, and Eri Tatsu for their contribution to the completion of our study.
Appendix
Relative Risk of Genotypes
Posit SNP A and SNP B, which are not linked physically. Disease-susceptibility alleles of SNP A and B are denoted as “a1” and “b1,” and nonsusceptibility alleles are denoted as “a2” and “b2.” Let Gijkl be the relative risk of genotype aiajbkbl, compared with the risk of genotype a2a2b2b2, when each of i, j, k, and l is either 1 or 2.
Define Gijkl by five parameters—G, r1, r2, r3, and r4 (G>1, ri=0, .5, 1):
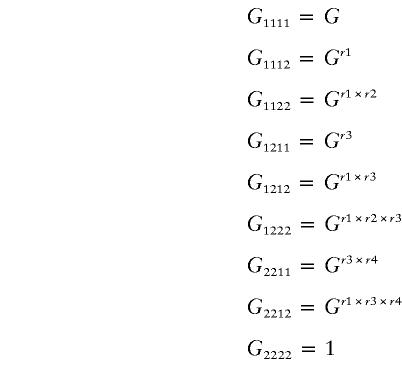 |
Arrange Gijkl in a 3×3 matrix:
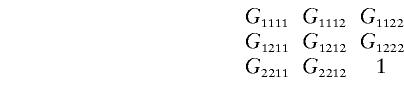 |
By this definition, distribution patterns of relative risk are modeled as below. Note that g takes either G or the square root of G, as defined above.
- Pattern 1—heterozygosity or homozygosity of susceptibility allele of either SNP is adequate to raise risk:

- Pattern 2—heterozygosity or homozygosity of susceptibility allele of SNP A is necessary to raise risk:

- Pattern 3—homozygosity of susceptibility allele of SNP A is necessary to raise risk:

- Pattern 4—heterozygosity or homozygosity of susceptibility allele of SNP B is necessary to raise risk:

- Pattern 5—homozygosity of susceptibility allele of one SNP B is necessary to raise risk:

- Pattern 6—heterozygosity or homozygosity of susceptibility allele of both SNPs is necessary to raise risk:

- Pattern 7—heterozygosity or homozygosity of susceptibility allele of SNP A and homozygosity of susceptibility allele of SNP B are necessary to raise risk:

- Pattern 8—homozygosity of susceptibility allele of SNP A and heterozygosity or homozygosity of susceptibility allele of SNP B is necessary to raise risk:

- Pattern 9—homozygosity of susceptibility allele of both SNPs is necessary to raise risk:

One more important relationship, not modeled in the equations above, was that in which two loci contribute independently, regardless of the partner gene’s genotype status. For this relationship, we introduced a different definition: parameters ga and gb represent a homozygote’s relative risk of SNP A and SNP B, respectively. Assume parameters fa and fb to be 0, .5, or 1, to control the heterozygote’s relative risk according to ga and gb.
Independent Pattern of Genotypes
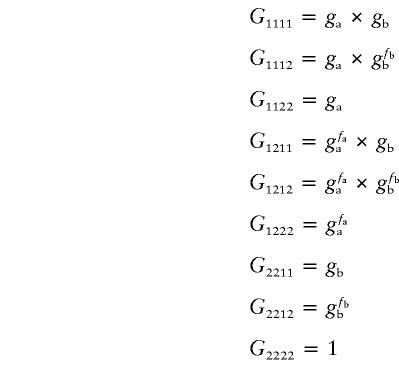 |
Proportion of Genotypes among Population and Case or Control Group
The test population was in Hardy-Weinberg equilibrium. Disease prevalence d was extrapolated from epidemiological studies; in our text, we have assumed that d=.01 for RA. Frequencies of disease-susceptibility alleles of either SNP A or SNP B are pa or pb, respectively.
Let Paiajbkbl, Caiajbkbl, and Naiajbkbl denote a proportion of people with genotype aiajbkbl, in the whole population, in the case population, and in the control population, respectively. The following equalities are present:
 |
Maximum-Likelihood Model and Likelihood Ratio
Consider a random case-control sampling from A sufficiently large population; that is, on in which sampling an individual would not affect conditions of successive sampling. Let Qaiajbkbl and Raiajbkbl denote the observed data on cases and controls, respectively, with genotype aiajbkbl. The conditional probabilities for sampling a case and a control with genotype aiajbkbl are, respectively, Caiajbkbl/d and Naiajbkbl/(1-d). Then the logarithm of likelihood (L) of the observed data is
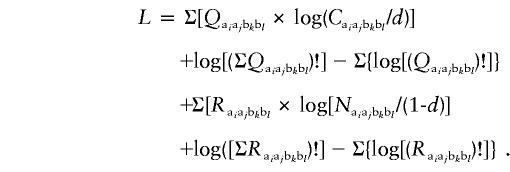 |
The most likely estimate of parameters is obtained, and the most-fit pattern of relative-risk distribution is estimated by comparison of the values of L.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for RA [MIM 180300])
- Web Resources of Genetic Linkage Analysis, http://linkage.rockefeller.edu/
References
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, Medsger TA Jr, Mitchell DM, Neustadt DH, Pinals RS, Schaller JG, Sharp JT, Wilder RL, Hunder GG (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31:315–324 [DOI] [PubMed] [Google Scholar]
- Cornelis F, Faure S, Martinez M, Prud’homme JF, Fritz P, Dib C, Alves H, et al (1998) New susceptibility locus for rheumatoid arthritis suggested by a genome-wide linkage study. Proc Natl Acad Sci USA 95:10746–10750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deal CL, Meenan RF, Goldenberg DL, Anderson JJ, Sack B, Pastan RS, Cohen AS (1985) The clinical features of elderly-onset rheumatoid arthritis: a comparison with younger-onset disease of similar duration. Arthritis Rheum 28:987–994 [DOI] [PubMed] [Google Scholar]
- Devlin B, Risch N (1995) A comparison of linkage disequilibrium measures for fine-scale mapping. Genomics 29:311–322 [DOI] [PubMed] [Google Scholar]
- Ehrlich GE, Katz WA, Cohen SH (1970) Rheumatoid arthritis in the aged. Geriatrics 25: 103–113 [PubMed] [Google Scholar]
- Firestein GS, Xu WD, Townsend K, Broide D, Alvaro-Gracia J, Glasebrook A, Zvaifler N (1988) Cytokines in chronic inflammatory arthritis. I. Failure to detect T cell lymphokines (interleukin 2 and interleukin 3) and presence of macrophage colony-stimulating factor (CSF-1) and a novel mast cell growth factor in rheumatoid synovitis. J Exp Med 168:1573–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DA, Hastings DE (1994) Rheumatoid arthritis: clinical features: early, progressive and late disease. In: Klippel JH, Dieppe PA (eds) Rheumatology. Mosby, London, section 3, pp 4.1–4.14 [Google Scholar]
- Harant H, Lindley IJD (1998) Interleukin 3. In: Mire-Sluis A, Thorpe R, Page C (eds) Cytokines. Academic Press, San Diego, pp 35–52 [Google Scholar]
- Heller RA, Schena M, Chai A, Shalon D, Bedilion T, Gilmore J, Woolley DE, Davis RW (1997) Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci USA 94:2150–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultner L, Kolsch S, Stassen M, Kaspers U, Kremer JP, Mailhammer R, Moeller J, Broszeit H, Schmitt E (2000) In activated mast cells, IL-1 up-regulates the production of several Th2-related cytokines including IL-9. J Immunol 164:5556–5563 [DOI] [PubMed] [Google Scholar]
- Jeong MC, Navani A, Oksenberg JR (1998) Limited allelic polymorphism in the human interleukin-3 gene. Mol Cell Probes 12:49–53 [DOI] [PubMed] [Google Scholar]
- Levesque MC, Haynes BF (1997) Cytokine induction of the ability of human monocyte CD44 to bind hyaluronan is mediated primarily by TNF-alpha and is inhibited by IL-4 and IL-13. J Immunol 159:6184–6194 [PubMed] [Google Scholar]
- Levesque MC, Heinly CS, Whichard LP, Patel DD (1998) Cytokine-regulated expression of activated leukocyte cell adhesion molecule (CD166) on monocyte-lineage cells and in rheumatoid arthritis synovium. Arthritis Rheum 41:2221–2229 [DOI] [PubMed] [Google Scholar]
- Nepom GT (1998) Major histocompatibility complex-directed susceptibility to rheumatoid arthritis. Adv Immunol 68:315–332 [DOI] [PubMed] [Google Scholar]
- Nielsen DM, Ehm MG, Weir BS (1998) Detecting marker-disease association by testing for Hardy-Weinberg disequilibrium at a marker locus. Am J Hum Genet 63:1531–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Selden MF, Amos CI, Ward R, Gregerson PK (1999) The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum 42:1071–1079 [DOI] [PubMed] [Google Scholar]
- Shiozawa S, Hayashi S, Tsukamoto Y, Goto H, Kawasaki H, Wada T, Shimizu K, Yasuda N, Kamatani N, Takasugi K, Tanaka Y, Shiozawa K, Imura S (1998) Identification of the gene loci that predispose to rheumatoid arthritis. Int Immunol 10:1891–1895 [DOI] [PubMed] [Google Scholar]
- Terwilliger J, Ott J (1994) Handbook of human genetic linkage. Johns Hopkins University Press, Baltimore [Google Scholar]
- Waalen K, Sioud M, Natvig JB, Forre O (1992) Spontaneous in vivo gene transcription of interleukin-2, interleukin-3, interleukin-4, interleukin-6, interferon-gamma, interleukin-2 receptor (CD25) and proto-oncogene c-myc by rheumatoid synovial T lymphocytes. Scand J Immunol 36:865–873 [DOI] [PubMed] [Google Scholar]
- Wakitani S, Murata N, Toda Y, Ogawa R, Kaneshige T, Nishimura Y, Ochi T (1997) The relationship between HLA-DRB1 alleles and disease subsets of rheumatoid arthritis in Japanese. Br J Rheumatol 36:630–636 [DOI] [PubMed] [Google Scholar]
- Yamada R, Tanaka T, Ohnishi Y, Suematsu K, Minami M, Seki T Yukioka M, Maeda A, Murata N, Saiki O, Teshima R, Kudo O, Ishikawa K, Ueyoshi A, Tateishi H, Inaba M, Goto H, Nishizawa Y, Tohma S, Ochi T, Yamamoto K, Nakamura Y (2000) Identification of 142 single nucleotide polymorphisms in 41 candidate genes for rheumatoid arthritis in the Japanese population. Hum Genet 106:293–297 [DOI] [PubMed] [Google Scholar]
- Yang Y-C, Kovacic S Kriz R Wolf S, Clark SC Wellems TE, Nienhuis A, Epstein N (1988) The human genes for GM-CSF and IL-3 are closely linked in tandem on chromosome 5. Blood 71:958–961 [PubMed] [Google Scholar]