

Abstract
Cataracts are the leading cause of blindness in most countries. Although most hereditary cases appear to follow an autosomal dominant pattern of inheritance, autosomal recessive inheritance has been clearly documented and is probably underrecognized. We studied a large family—from a relatively isolated geographic region—whose members were affected by autosomal recessive adult-onset pulverulent cataracts. We mapped the disease locus to a 14-cM interval at a novel disease locus, 9q13-q22 (between markers D9S1123 and D9S257), with a LOD score of 4.7. The study of this progressive and age-related cataract phenotype may provide insight into the cause of the more common sporadic form of age-related cataracts.
Cataracts remain a leading cause of blindness worldwide (Thylefors et al. 1995; Lim 1996; Yorston 1998), accounting for 42% of all blindness. The number of cases is expected to double by 2010. Although we are beginning to learn more about lens structure and function, the mechanisms of lens opacification remain poorly understood (Arnold 1998). Segregation analysis and twin studies of populations affected with age-related cataracts suggest that Mendelian inheritance, often autosomal recessive (AR), could account for ∼50% of age-related cataracts (Italian American Cataract Study Group 1991; Heiba et al. 1993, 1995; Group TFOES 1994; Hammond et al. 2000). The study of single-gene hereditary cataracts has been helpful in the identification of ∼20 candidate loci and of 10 human genes, all for autosomal dominant (AD) phenotypes (MIM 604219) (Hejtmancik 1998; Kannabiran et al. 1998; Litt et al. 1998; Mumford et al. 1998; Shiels et al. 1998; Heon et al. 1999; Mackay et al. 1999; Bateman et al. 2000; Kramer et al. 2000; Ren et al. 2000). Only one human locus (Pras et al. 2000) and a few murine loci (Kratochvilova et al. 1988; Chang et al. 1996; Iida et al. 1997; Song et al. 1997) are linked with AR cataracts. Mutations in CRYAA were recently identified in patients with AR cataracts (Pras et al. 2000). For a genetic study, we have recruited a large family that is affected with AR early-onset progressive pulverulent cataracts (ARPCs) and that resides in a relatively isolated region of Switzerland.
This family was documented to have nine members, all in one generation (fig. 1), who were affected with an ARPC (fig. 2). Eight affected individuals and 22 of their children were recruited and examined for this study. None of the offspring (generation IV), whose ages at the time of study were 16–43 years, showed any sign of lens opacity. Because ARPCs progress slowly, the presence of lens opacities can be detected by slit-lamp biomicroscopy several years before the symptoms become manifest. Slit-lamp biomicroscopy was used to examine all participants on two occasions, separated by a 2-year interval. Because no member of the fourth generation showed evidence of lens opacity and because the phenotype affects only one generation, we proposed that the inheritance of this phenotype is AR. In genealogical studies that included the two generations preceding the generation that included affected individuals, we were unable to document any consanguineous link between the two sides of the family. However, consanguineous relationships are documented in both sides of the family (fig. 1), and other consanguineous marriages are documented in that locality. Individuals II:2 and II:3 died during their 70s, at which time they had good functional vision. One sib from each side of the family was examined (II:1 and II:4) and failed to show any cataract phenotype. Furthermore, they were not aware of any vision problem in their respective siblings. In no case was the affection status modified after the second examination.
Figure 1.

Genealogy and summarized haplotype showing the most informative markers. All spouses were examined and found to be normal. Blackened symbols indicate clinically affected individuals; unblackened symbols represent unaffected relatives. Unblackened symbols containing an “N” indicate relatives ⩾30 years old who were examined but who did not show signs of the disease; empty unblackened symbols indicate unaffected relatives who are ⩽30 years old; slashes indicate deceased individuals. The hatched boxes indicate the affected haplotype, whereas the unhatched boxes indicate the unaffected haplotype. “XX” indicates the beginning and end of the disease-gene interval. “?” indicates the genotype was not available.
Figure 2.
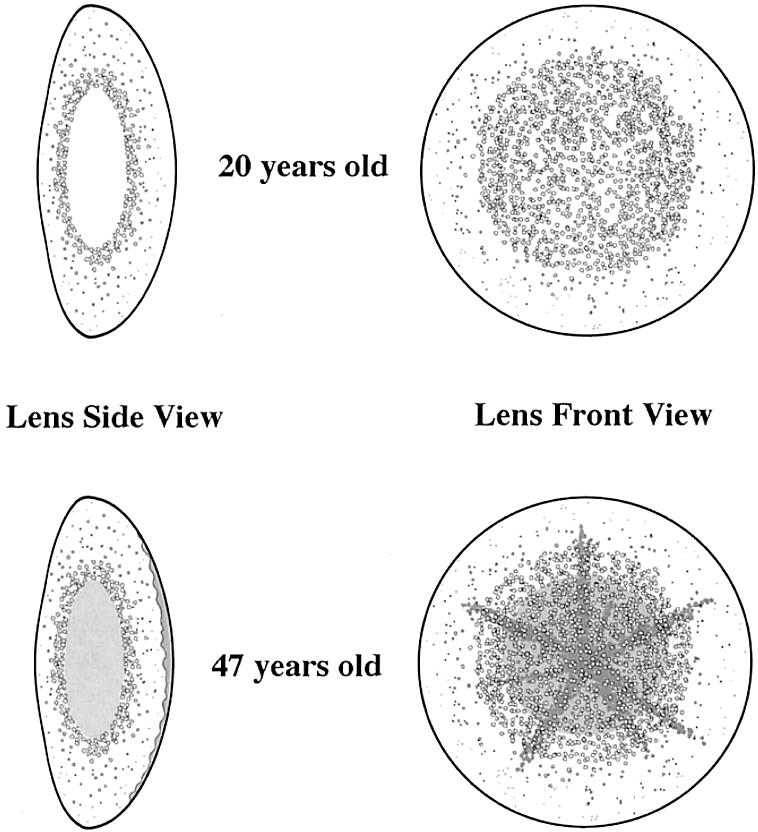
Drawing of the ARPC lens opacity. Top, Individual III:15, at age 20 years, showing pulverulent lamellar and cortical opacity. Bottom, Individual III:5, at age 47 years, showing additional nuclear and posterior subcapsular changes.
The ARPC phenotype did not clearly match any of the cataract phenotypes described to date, because it consisted of a mostly cortical, pulverulent (dustlike) opacity, with occasional nuclear and posterior subcapsular involvement and with early nuclear sclerosis (browning of the nucleus) (fig. 2). The opacification of the nucleus and/or the posterior subcapsular area was progressive and was present to a variable degree (table 1). This cloudy appearance of the lens worsened with age; the affected lens aged prematurely, and, to restore visual function, cataract surgery was usually required by the age of 40 years.
Table 1.
ARPC-Affected Status Clinical Data
| Subject | Vision before Surgery (Age [years])a | Age at Surgery(years) | Other Findingsb |
| III:2 | D .5/.6; N .3/.4 (44) | 55 | Hyperlipidemia |
| III:4 | D .3/.3; N .2/.2 (50) | 53 | |
| III:5 | D .4/.3 | 52 | |
| III:12 | D .3/.3; N .1/.3 (42) | 44 | Hyperthyroid, AS, CS |
| III:14 | D .6/.6; N .2/.2 (30) | 42 | Obesity, hypertension |
| III:15 | D .2/.8 (22) | 40 | Diagnosis at age 20 |
| III:17 | D .4/.4 (37) | 37 | |
| III:20 | D .4/.5 (36) | 36 |
Normal visual acuity has a measurement of 1.0. D = distance vision; N = near vision. Near vision measurements were not available for subjects III:5, III:15, III:17, and III:20.
AS = Albright syndrome; CS = congenital anosmia.
The age at onset of the cataracts in this large family is not entirely clear, because the affected individuals presented with various degrees of visual impairment (table 1). The variation in onset of visual symptoms was influenced by the differing visual needs of individual subjects. The earliest diagnosis was made, at the age of 20 years, in an individual whose vision was decreased by 80% in one eye and by 20% in the other. Lens opacities were most likely present for several years before symptoms were manifest. Affected individuals required cataract surgery at a mean age of 44 years (range 36–55 years), because of progressive visual impairment. At the time of surgery, the mean decrease in vision was 60% for distance vision and 76% for near vision, both decreases being significant.
It is the manifestation of the lens opacity at an unusually early age that facilitated detection of the familial pattern. Furthermore, the noncongenital and progressive opacification of the various layers of the lens makes the ARPC phenotype the first human genetic model for the study of age-related cataracts. Cortical, pulverulent cataracts account for a substantial proportion of cataracts, although exact figures are not available (Leske et al. 1991). It is the clear hereditary pattern of these findings that is less commonly documented.
The initial strategy for the genetic study of this family was as follows: (1) candidate-loci screening by linkage analysis, (2) genomewide screening using a DNA-pooling approach, and (3) detailed linkage analysis of the new candidate loci. If we assume an AR mode of inheritance and complete penetrance, the data from our family are powerful enough to detect significant linkage (LOD score >3, odds for linkage ⩾1,000:1 [P=.05]). The power to detect linkage was investigated using SLINK. Two hundred replicates of the pedigree were generated, under the assumption of a completely penetrant AR disease locus with disease-allele frequency = .01 and no phenocopies. Also simulated were data for one marker with five equally frequent alleles at a recombination fraction (θ) of 0. The average LOD score at θ=0 was 3.4 (SD=1.5), and the maximum LOD score was 4.7.
We studied ∼20 loci that are related to genes involved in lens formation, metabolism, or opacification, with an average of four STRP markers per locus (table 2). Two-point and multipoint linkage analyses were performed, considering both AR and dominant inheritance, and no evidence of linkage was detected (data not shown).
Table 2.
Candidate Cataract Loci-Exclusion Data[Note]
| Location | Candidate Gene | MIM | STRP Intervala | Exclusion Regionb(cM) | % of Total Chromosome |
| 1p36 | Unknown | 115665 | D1S468(4)-GATA29A05 | 34 | 12 |
| 116600 | |||||
| 1q21-25 | GJA8 | 116200 | D1S2669(151)-D1S484 | 38.5 | 13 |
| 1q32.2 | PROX-1 | D1S1663(226)-D1S549 | 33.6 | 12 | |
| 2q33-35 | Gamma C | 123660 | D2S155(202)-D2S434 | 13 | 5.3 |
| Gamma D | 115700 | ||||
| Gamma D | 123690 | ||||
| Gamma A–F | 601286 | ||||
| 2q36 | CryBA2 | ||||
| 3p21.1-21.3 | Unknown | D3S2432(58)-D3S1766 | 21 | 9 | |
| 3q21.3-25.2 | BFSP2/phakinin; CRYGS | 603212 | D3S2460(135)-D3S1764 | 28 | 14 |
| 10q23.3-25 | PITX3 | 602669 | D10S1753(113)-D10S1750 | 17 | 9.8 |
| 11q22.3-23.1 | CryAB | D11S898(99)-D11S1998 | 34 | 23 | |
| 12q13 | MP26/LIM1/AQPO | 154050 | D12S368(66)-D12S83 | 30 | 17.5 |
| 13cen-q11 | GJA3 | 601885 | D13S1316(0)-D13S787 | 10 | 9 |
| 16q22.1 | Unknown | 116800 | D16S515(92)-D16S518 | 23 | 17 |
| 17p | Unknown | 601202 | D17S849(.6)-D17S796 | 24 | 19 |
| 17q11.2-q12 | CRYBA1/A3 | 600881 | D17S122(41)-D17S925 | 30 | 24 |
| 17q24 | Galactokinase | 115660 | D17S836(113)-D17S784 | 3.9 | |
| 19q13.3 | IRE/FTL; LIM2/TGFβ1 | 600886 | D19S178(68)-D19S877 | 30 | 29 |
| 21q22.3 | CryAA | 123580 | D21S266(46)-D21S171 | 26 | 45 |
| 22q11.2 | CRYBB2 | 601547 | TOP1P2(19)-D22258 | 13 | 21 |
| 409 |
Note.— Data from the genotyping of the hotspots from the pooling study allow us to exclude 13.8% of the human genome.
From The Center for Medical Genetics, Marshfield Medical Research Foundation. The map position (in parentheses) is expressed in centimorgans.
May include flanking regions.
A genomewide scan consisting of 380 microsatellite markers spaced at ∼10-cM intervals was performed using a pooling strategy (Betard et al. 2000). The protocol was modified for the ABI-3700 DNA analyzer (Applied Biosystems). Initially, the following three DNA pools were genotyped: (1) seven affected individuals, (2) eight spouses, and (3) eight unaffected offspring. We later added a fourth pool, of four unaffected siblings >25 years old.
Linkage was suggested by the observation of a skew (termed a “hotspot”) in the banding pattern of the affected pool compared with those of the other pools; 22 hotspots were identified. Individual family members were then genotyped with markers from these candidate loci, and significant linkage was observed with markers on chromosome 9q13-q22. Linkage and haplotype analysis confirmed the AR inheritance of the phenotype. A maximum pairwise LOD score of 4.71 at θ=0 was obtained with D9S768 (table 3). Location-score analysis, using SIMWALK2, version 2.60 (Sobel and Lange 1996) (fig. 3), supported the strong evidence for linkage to this region (maximum location score [log10] = 4.70, θ=0, with marker D9S768). With this analysis, the estimate for the most-likely-genetic-descent graph is used as the initial position, and a random walk is then performed on the space-of-descent graphs, using the Metropolis acceptance criterion. Completely typed representative pedigrees are obtained by sampling, in numbers proportional to their true likelihood, from this random walk; and these pedigrees are then used to estimate the location-score curve for the original pedigree. Haplotype analysis showed that recombination events observed with markers D9S1123 and D9S257 defined a 14-cM interval, according to the sex-averaged reference map (fig. 1) (Broman et al. 1998).
Table 3.
Two-Point Linkage Data for ARPC Phenotype and Markers of the 9q13-q22 Region
|
LOD Score at θ = |
|||||||||
| Marker (Distance [cM])a | Location | Heterozygosityb | 0 | .1 | .2 | .3 | .04 | Zmax | θmax |
| D9S301 (66) | 9p21-9q21 | .71 | ∞ | 1.20 | 1.13 | .76 | .27 | 1.23 | .13 |
| D9S1122 (75) | 9pter-qter | .67 | ∞ | 2.90 | 2.32 | 1.52 | .6 | 3.02 | .05 |
| D9S1123 (77) | 9pter-qter | .66 | ∞ | .96 | .84 | .56 | .21 | .96 | .11 |
| D9S153 (79) | 9q13-q22.3 | .68 | 2.00 | 1.63 | 1.23 | .78 | .29 | 2.00 | .00 |
| D9S1867 (79) | 9pter-qter | .71 | 2.00 | 1.62 | 1.21 | .75 | .28 | 2.00 | .00 |
| D9S768 (80) | 9q13-q22.3 | .79 | 4.71 | 3.85 | 2.92 | 1.89 | .76 | 4.71 | .00 |
| D9S167 (83) | 9q13-q22.3 | .83 | 2.30 | 1.94 | 1.49 | .98 | .40 | 2.30 | .00 |
| D9S152 (84) | 9q21-q22 | .72 | 2.00 | 1.38 | .76 | .24 | .02 | 2.00 | .00 |
| D9S1119 (85) | 9pter-qter | .64 | .04 | .02 | .01 | .00 | .00 | .14 | .00 |
| D9S252 (88) | 9q13-q22 | .66 | 2.00 | 1.38 | .76 | .24 | .02 | 2.00 | .00 |
| D9S1812 (90) | 9pter-qter | .57 | 2.30 | 1.93 | 1.48 | .97 | .40 | 2.30 | .00 |
| D9S257 (91) | 9q13-q22 | .84 | ∞ | 1.73 | 1.57 | 1.08 | .42 | 1.74 | .12 |
| D9S283 (94) | 9q13-q22 | .75 | ∞ | .48 | .80 | .67 | .30 | .80 | .21 |
Markers within the critical interval are underlined.
As determined on the basis of family spouses. AR, full-penetrance, and marker-allele frequencies were estimated on the basis of the founders, and disease-allele frequencies of .01 were assumed for the disease locus.
Figure 3.

SIMWALK2 analysis of markers at the 9q13-q22 locus. Simulated location scores for ARPC vs. chromosome 9 markers. The maximum log10 location score was 4.7, with D9S768. The 14-cM disease interval, as defined by haplotype analysis, was between D9S1123 and D9S257.
The ARPC locus represents the second AR cataract locus published and the only one associated with a noncongenital progressive cataract. Also, this new ARPC locus does not correspond to a known candidate cataract locus in mice or humans. Fifteen genes have been identified in this interval. None of these appear to have a role in the maintenance of either lens transparency or lens metabolism. Under the assumption of a common founder for both sides of the family, further genetic mapping will use a combination of recombination and homozygosity mapping. Both the progress of the documentation of the human genome sequence and the recent identification of other families with AR cataracts will assist us in narrowing the disease interval and identifying the ARPC gene.
The leading cause of blindness in most countries is cataracts. The hypothesis that Mendelian inheritance can account for a significant portion of age-related cataracts provides a tremendous incentive to identify as many cataract genes as possible, to better understand the biology of lens opacification. Molecular characterization of AR cataracts is essential to the understanding of the biology of cataracts and to the designing of novel therapies. Although the complete prevention of age-related cataracts is unlikely, the simple ability to delay lens opacification could reduce cataract-related blindness significantly (Wilson 1980).
Acknowledgments
We thank L. Jovanovic and E. Schusselé, for their contribution to patient assessment. A.D.P. is supported by a Medical Research Council (Canada) program grant entitled The Centre for Applied Genomics. We are grateful for the enthusiastic participation of the family, and we thank Corinne Darmond-Zwaig, for her technical assistance in the genome scan. This research was partially supported by grants from the Canadian Genetic Diseases Network, National Networks of Centres of Excellence Program (to E.H. and T.J.H.), and by Swiss Grant fund 32-53750.98 (to E.H., D.F.S., and F.L.M.). T.J.H. is a recipient of a Clinician Scientist Award from the Canadian Institutes of Health Research.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for GJA8 [MIM 116200], Gamma C [MIM 123660], Gamma D [MIM 115700 and MIM 123690], Gamma A–F [MIM 601286], BFSP2 [MIM 603212], PITX3 [MIM 602669], MP26/LIM1/AQPO [MIM 154050], GJA3 [MIM 601885], CRYBA1/A3 [MIM 600881], Galactokinase [MIM 115660], IRE/FTL [MIM 600886], CryAA [MIM 123580], CRYBB2 [MIM 601547], and unspecified/unknown candidate genes [MIM 1115665, MIM 116800, MIM 601202, and MIM 116600])
References
- Arnold J (1998) Global cataract blindness: the unmet challenge. Br J Ophthalmol 82:593–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JB, Johannes M, Flodman P, Geyer DD, Clancy KP, Heinzmann C, Kojis T, Berry R, Sparkes RS, Spence MA (2000) A new locus for autosomal dominant cataract on chromosome 12q13. Invest Ophthalmol Vis Sci 41:2665–2670 [PubMed] [Google Scholar]
- Betard C, Rasquin-Weber A, Brewer C, Drouin E, Clark S, Verner A, Darmond-Zwaig C, Fortin J, Mercier J, Chagnon P, Fujiwara TM, Morgan K, Richter A, Hudson TJ, Mitchell GA (2000) Localization of a recessive gene for North American Indian childhood cirrhosis to chromosome region 16q22—and identification of a shared haplotype. Am J Hum Genet 67:222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman K, Murray J, Sheffield V, White R, Weber J (1998) Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63:861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Smith RS, Heckenlively JR, Davisson MT, Roderick TH (1996) Chromosomal localization of a new mouse lens opacity gene (lop18). Genomics 36:171–173 [DOI] [PubMed] [Google Scholar]
- Group TFOES (1994) Familial aggregation of lens opacities: The Framingham Eye Study and The Framingham Offspring Eye Study. Am J Epidemiol 140:555–564 [PubMed] [Google Scholar]
- Hammond CJ, Snieder H, Spector TD, Gilbert CE (2000) Genetic and environmental factors in age-related nuclear cataracts in monozygotic and dizygotic twins. N Engl J Med 342:1786–1790 [DOI] [PubMed] [Google Scholar]
- Heiba IM, Elston RC, Klein BE, Klein R (1993) Genetic etiology of nuclear cataract: evidence for a major gene. Am J Med Genet 47:1208–1214 [DOI] [PubMed] [Google Scholar]
- ——— (1995) Evidence for a major gene for cortical cataract. Invest Ophthalmol Vis Sci 36:227–235 [PubMed] [Google Scholar]
- Hejtmancik JF (1998) The genetics of cataract: our vision becomes clearer. Am J Hum Genet 62:520–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heon E, Priston M, Schorderet DF, Billingsley GD, Girard PO, Lubsen N, Munier FL (1999) The gamma-crystallins and human cataracts: a puzzle made clearer. Am J Hum Genet 65:1261–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida F, Matsushima Y, Hiai H, Uga S, Honda Y (1997) Rupture of lens cataract: a novel hereditary recessive cataract model in the mouse. Exp Eye Res 64:107–113 [DOI] [PubMed] [Google Scholar]
- Italian-American Cataract Study Group (1991) Risk factors for age-related cortical, nuclear, and posterior subcapsular cataracts: The Italian-American Cataract Study Group. Am J Epidemiol 133:541–553 [PubMed] [Google Scholar]
- Kannabiran C, Rogan PK, Olmos L, Basti S, Rao GN, Kaiser-Kupfer M, Hejtmancik JF (1998) Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the βA3/A1-crystallin gene. Mol Vis 4:21 [PubMed] [Google Scholar]
- Kramer PL, LaMorticella D, Schilling K, Billingslea AM, Weleber RG, Litt M (2000) A new locus for autosomal dominant congenital cataracts maps to chromosome 3. Invest Ophthalmol Vis Sci 41:36–39 [PubMed] [Google Scholar]
- Kratochvilova J, Favor J, Neuhauser-Klaus A (1988) Dominant cataract and recessive specific-locus mutations detected in offspring of procarbazine-treated male mice. Mutat Res 198:295–301 [DOI] [PubMed] [Google Scholar]
- Leske MC, Chylack LT Jr, Wu SY (1991) The Lens Opacities Case-Control Study: risk factors for cataract. Arch Ophthalmol 109:244–251 [DOI] [PubMed] [Google Scholar]
- Lim AS (1996) Vision for the world. Eur J Ophthalmol 6:95 [DOI] [PubMed] [Google Scholar]
- Litt M, Kramer P, LaMorticella DM, Murphey W, Lovrien EW, Weleber RG (1998) Autosomal dominant congenital cataract associated with a missense mutation in the human αcrystallin gene CRYAA. Hum Mol Genet 7:471–474 [DOI] [PubMed] [Google Scholar]
- Mackay D, Ionides A, Kibar Z, Rouleau G, Berry V, Moore A, Shiels A, Bhattacharya S (1999) Connexin46 mutations in autosomal dominant congenital cataract. Am J Hum Genet 64:1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumford A, Vulliamy T, Lindsay J, Watson A (1998) ereditary hyperferritinemia-cataract syndrome: two novel mutations in the l-ferritin iron-responsive element. Blood 6:665–668 [PubMed]
- Pras E, Frydman M, Levy-Nissenbaum E, Bakhan T, Raz J, Assia EI, Goldman B (2000) A nonsense mutation (W9X) in CRYAA causes autosomal recessive cataract in an inbred Jewish Persian family. Invest Ophthalmol Vis Sci 41:3511–3515 [PubMed] [Google Scholar]
- Ren Z, Li A, Shastry BS, Padma T, Ayyagari R, Scott MH, Parks MM, Kaiser-Kupfer MI, Hejtmancik JF (2000) A 5-base insertion in the γC-crystallin gene is associated with autosomal dominant variable zonular pulverulent cataract. Hum Genet 106:531–537 [DOI] [PubMed] [Google Scholar]
- Shiels A, Mackay D, Ionides A, Berry V, Moore A, Bhattacharya S (1998) A missense mutation in the human connexin50 gene (GJA8) underlies autosomal dominant “zonular pulverulent” cataract, on chromosome 1q. Am J Hum Genet 62:526–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337 [PMC free article] [PubMed] [Google Scholar]
- Song CW, Okumoto M, Mori N, Kim JS, Han SS, Esaki K (1997) Mapping of new recessive cataract gene (lr2) in the mouse. Mamm Genome 8:927–931 [DOI] [PubMed] [Google Scholar]
- Thylefors B, Négrel A, Pararajasegaram R, Dadzie K (1995) Global data on blindness. Bull World Health Organ 73:115–121 [PMC free article] [PubMed] [Google Scholar]
- Wilson J (1980) The six main causes of blindness. In: Wilson J (ed) World blindness and its prevention. Oxford University Press, London, pp 7–9 [Google Scholar]
- Yorston D (1998) Are intraocular lenses the solution to cataract blindness in Africa? Br J Ophthalmol 82:469–471 [DOI] [PMC free article] [PubMed] [Google Scholar]