

Abstract
Peptide deformylase (PDF) is a prokaryotic metalloenzyme that is essential for bacterial growth and is a new target for the development of antibacterial agents. All previously reported PDF inhibitors with sufficient antibacterial activity share the structural feature of a 2-substituted alkanoyl at the P1′ site. Using a combination of iterative parallel synthesis and traditional medicinal chemistry, we have identified a new class of PDF inhibitors with N-alkyl urea at the P1′ site. Compounds with MICs of ≤4 μg/ml against gram-positive and gram-negative pathogens, including Staphylococcus aureus, Streptococcus pneumoniae, and Haemophilus influenzae, have been identified. The concentrations needed to inhibit 50% of enzyme activity (IC50s) for Escherichia coli Ni-PDF were ≤0.1 μM, demonstrating the specificity of the inhibitors. In addition, these compounds were very selective for PDF, with IC50s of consistently >200 μM for matrilysin and other mammalian metalloproteases. Structure-activity relationship analysis identified preferred substitutions resulting in improved potency and decreased cytotoxity. One of the compounds (VRC4307) was cocrystallized with PDF, and the enzyme-inhibitor structure was determined at a resolution of 1.7 Å. This structural information indicated that the urea compounds adopt a binding position similar to that previously determined for succinate hydroxamates. Two compounds, VRC4232 and VRC4307, displayed in vivo efficacy in a mouse protection assay, with 50% protective doses of 30.8 and 17.9 mg/kg of body weight, respectively. These N-alkyl urea hydroxamic acids provide a starting point for identifying new PDF inhibitors that can serve as antimicrobial agents.
A largely empirical search for new antibiotics, carried out since the 1940s, has led to an impressive armamentarium of clinically useful antibacterial drugs. However, the actual number of targets interfered with by these agents is very limited. Cross-resistance for antibiotics, resulting from a sharing of the same target, plays an important role in the worsening problem of resistance. It is not surprising that antibacterial drug discovery efforts are now focusing on identifying molecules that block different targets and thus can inhibit the growth of resistant bacteria. Analysis of microbial genomes has revealed an abundance of novel and potentially useful targets (26), but little has so far resulted from this much heralded effort. On the other hand, many bacterial enzymes that have been well characterized hold promise for the discovery of new antibacterial drugs. One such target that has recently received a great deal of attention is peptide deformylase (PDF; EC 3.5.1.31).
Protein synthesis has proven to be a rich source of targets for antibacterial drugs (30). Many of the known antibiotics target one or more steps of this complex process, including the aminoglycosides, macrolides, tetracyclines, and oxazolidinones. Although the protein-synthesizing machineries of bacterial and mammalian cells exhibit overall similarity, there are sufficient differences to allow for selective blocking of the process in bacteria. One significant difference is the transformylation and subsequent deformylation of the initiating methionine in bacterial translation (35, 36). Unlike cytosolic protein synthesis in mammalian cells, protein synthesis in bacteria is initiated with N-formylmethionine (2, 31), which is generated by enzymatic transformylation of methionyl-tRNA. In prokaryotes, the N-formylmethionine of the nascent protein is removed by the sequential action of PDF and a methionine aminopeptidase to afford the mature protein (2, 37). This bacterium-specific requirement for PDF in protein synthesis provides a rational basis for selectivity, making it an attractive drug discovery target. The possible use of PDF as an antimicrobial target was recently reviewed elsewhere (17, 57).
PDF activity was first reported by Adams in 1968 (1). However, further attempts to purify the enzyme from cell lysates failed because the activity was not stable. The enzyme was not characterized further until the early 1990s, when the deformylase gene, def, was cloned (42) and PDF was subsequently overexpressed in Escherichia coli (39, 42).
Bacterial PDF belongs to a new class of metallohydrolases that utilize an Fe2+ ion as the catalytic metal ion (20, 51, 52). The ferrous ion in PDF is very unstable and can be quickly and irreversibly oxidized to the ferric ion, resulting in an inactive enzyme (53). Interestingly, the ferrous ion can be replaced with a nickel ion in vitro, resulting in much greater enzyme stability with little loss of enzyme activity (20).
The three-dimensional structures of various PDF molecules, including structures of enzyme-inhibitor complexes, have been solved and published (6, 10, 13, 14, 20, 21, 38). Although PDF is a ferrous aminopeptidase with a primary sequence very different from those of other metalloproteases, it has been noted that the environment surrounding the catalytic metal ion of PDF appears to be very similar to the active sites of thermolysin and the matrix metalloproteases (MMPs) (10). The catalytic metal ion of PDF is tetrahedrally coordinated with two histidines from the conserved zinc hydrolase sequence, HEXXH, and a conserved cysteine from an EGCLS motif. A water molecule that presumably hydrolyzes the amide bond occupies the fourth position in the tetrahedron.
The fact that PDF is a metalloprotease makes the enzyme a more attractive target for drug discovery. Metalloproteases are among the best studied of the enzyme classes (29), and there are excellent precedents for the mechanism-based design of their inhibitors. In the last few years, several classes of PDF inhibitors have been reported (3, 11-13, 15, 19, 24, 26, 43, 55). While all of these compounds inhibit PDF activity, most of them do not have antibacterial activity, presumably due to weak potency against PDF and/or an inability to penetrate the bacterial cell. It is interesting that among these compounds, those for which the concentrations needed to inhibit 50% of enzyme activity (IC50s) (or Kis) were greater than 1 μM had no antibacterial activity.
Based on mechanistic and structural information, together with an understanding of the general principles of inhibiting metalloproteases, a generic PDF inhibitor structure (Fig. 1) (57) was proposed. In this structure, X represents a chelating pharmacophore that will be the major component to provide binding energy; the n-butyl group mimics the methionine side chain; and P2′ and P3′ are regions of the inhibitor that can provide additional binding energy, selectivity, and favorable pharmacokinetic properties. Recently, actinonin, a naturally occurring antibiotic that was first isolated in 1962 from an actinomycete (18), was shown to be a PDF inhibitor (11). In this study, we report that N-alkyl urea hydroxamic acids, which also fit this generic structure, represent a new class of PDF inhibitors. The structural characterization and biological evaluation of this class of inhibitors are presented here.
FIG. 1.

Generic PDF inhibitor structure derived from the transition state of the deformylation reaction (57).
MATERIALS AND METHODS
Materials.
Actinonin, formate dehydrogenase (FDH), catalase, and NAD+ were obtained from Sigma; E. coli Ni-PDF and Streptococcus pneumoniae Zn-PDF were overproduced and purified as previously described (11, 33). N-formylmethionine-alanine-serine (fMAS) and thio ester peptide Ac-ProLeuGly-S-LeuLeuGly-OC2H5 were obtained from Bachem. Matrilysin (MMP-7) and angiotensin-converting enzyme (ACE) were obtained from Calbiochem. Hydroxypropyl-β-cyclodextrin was purchased from Aldrich. All other chemicals were of the highest commercial grade.
Preparation of N-alkyl urea hydroxamic acids.
The method of preparation of the urea compounds was recently reported (J. Lewis, J. Jacobs, C. Wu, C. Hackbarth, W. Wang, S. Lopez, R. White, J. Trias, Z. Yuan, and D. Patel, Abstr. 41st Intersci. Conf. Antimicrob. Agents Chemother., abstr. 358, 2001) and will be published elsewhere. Briefly, the urea hydroxamic acids (compound 4) were prepared in eight steps from commercially available amino acid precursors as outlined in Fig. 2. The synthetic sequence used Fukuyama-Mitsunobu chemistry followed by thiolytic deprotection to provide N-mono-alkyl-glycine methyl esters (compound 1). Acylation of N-alkyl amino esters with excess phosgene under aqueous reaction conditions provided N-alkyl-N-chlorocarbamoyl-glycine methyl esters (compound 2), which were subsequently acylated with the N-alkyl-N-chlorocarbamoyl-glycine methyl esters in pyridine to produce tetrasubstituted ureas (compound 3). Trifluoroacetic acid (TFA) deprotection followed by coupling of the R2 amine with PyBop peptide coupling reagent and treatment with hydroxylamine provided the final product, urea hydroxamic acid PDF inhibitors (compound 4).
FIG. 2.

Synthesis of N-alkyl urea hydroxamic acids. The letters in the scheme represent the following solvents: a, NsCl, aqueous NaHCO3, DCM; b, R1OH, DIAD, PPh3; c, PhSH, TBDE-PS, DMF; d, COCl2, aqueous NaHCO3, Et2O; e, l-Pro-OtBu, pyridine, DCM; f, TFA, DCM; g, R2NH2, PyBop, DIEA, DMF; and h, aqueous NH2OH, dioxane.
Enzyme assays.
All absorption measurements were obtained with a SpectraMax plate reader (Molecular Devices).
Deformylase activity was assayed by a PDF-FDH coupled assay as previously described (11, 28). Briefly, the assay was carried out at room temperature with 5 nM E. coli Ni-PDF or 10 nM S. pneumoniae Zn-PDF (33) in a buffer consisting of 50 mM HEPES (pH 7.2), 10 mM NaCl, and 0.2 mg of bovine serum albumin/ml in half-area 96-well microtiter plates (Corning). The reaction was initiated by the addition of a reaction mixture of 0.5 U of FDH/ml, 1 mM NAD+, and 4 mM fMAS at the desired concentration. To determine the IC50s of the desired compounds, PDF was preincubated for 10 min with various concentrations of test compounds prior to the addition of the reaction mixture. The initial reaction velocity was measured as the initial rate of increase in the absorption at 340 nm.
Matrilysin (MMP-7) activity was assayed as reported previously (56) by using a thio ester peptide as a substrate, with some modifications. Briefly, 0.12 μg of MMP-7/ml was preincubated at room temperature for 10 min with test compounds at various concentrations in a buffer containing 50 mM Tricine (pH 7.5), 0.2 M NaCl, 10 mM CaCl2, and 0.05% Brij. The reaction was initiated by the addition of 0.05 mM thio ester peptide substrate (Ac-ProLeuGly-S-LeuLeuGly-OC2H5) and 0.1 mM 5,5′-dithio-bis(2-nitrobenzoic acid). Reaction progress was monitored by recording the increase in the absorption at 405 nm.
ACE activity was determined with a 96-well format according to the procedure reported by Maclean et al. (32). The hydrolysis product of the enzyme reaction was detected by derivatization with o-phthaldialdehyde reagent (Pierce) by following the manufacturer's protocol. Briefly, 0.03 U of ACE/ml was incubated with test compounds at different concentrations at room temperature for 10 min in a buffer consisting of 25 mM HEPES (pH 8.2) and 0.3 M NaCl. The reaction was initiated when 2 mM substrate (hippuryl-HisLeu) was added to the mixture. The enzyme reaction was carried out at 35°C for 1 h. An equal volume of o-phthaldialdehyde reagent was added, and the reaction signal was detected by recording the change in fluorescence at excitation and emission wavelengths of 360 and 465 nm, respectively.
The IC50 was calculated with the following equation:
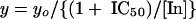 |
(1) |
In this equation, yo is enzyme activity in the absence of inhibitor, and [In] is the inhibitor concentration.
All data fitting was carried out by using nonlinear least-squares regression with the commercial software package DeltaGraph 4.0 (Deltapoint, Inc).
Cytotoxicity assays.
The cytotoxicities of the test compounds were assessed by using human K562 (ATCC CCL-243) and murine P388D1 (ATCC CCL-46) leukemia cell lines. The human cell line K562 was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (Gibco BRL) and 1 mM sodium pyruvate. P388D1 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% bovine calf serum (Gibco BRL). The assays were conducted with 96-well microtiter plates (Corning), and test compounds were serially diluted in 10% dimethyl sulfoxide. A volume of 10 μl of each dilution was added to wells 1 to 11 in each row; well 12, used as a control, contained 10 μl of 10% dimethyl sulfoxide solution without drug; and well 12H contained 0.25 μg of puromycin/ml as a no-growth control. Ninety microliters of log-phase cells (5.5 × 104 cells/ml) was suspended in the appropriate assay medium and added to each well. The cells were exposed to the drug for 3 days at 37°C in the presence of 5% CO2. On day 4, an indicator solution containing 1 mg of XTT/ml and 7.7 μg of phenazine methosulfate (Sigma)/ml in phosphate-buffered saline was added to each well, and the suspension was reincubated for 4 h under the same conditions. The XTT cleavage product was detected by recording the change in the absorption at 450 nm, and cell growth as a percentage of that in the corresponding control well was used to calculate the IC50 with equation 1.
Susceptibility studies and killing curves.
Bacteria used in these studies were stored at −80°C and grown at 35°C in accordance with NCCLS recommendations (44).
For susceptibility tests, MICs were determined by the broth microdilution method in accordance with NCCLS guidelines (44) but with the following modification. Because of the high frequency of resistance previously reported with other PDF inhibitors (4, 13, 34), a modified bacterial inoculum size of 0.5 × 105 to 1.0 × 105 CFU/ml was used to determine the relative activities of the compounds. The organisms used are all part of the Versicor strain collection (11). The MIC was defined as the lowest concentration that yielded no visible growth after 24 h of incubation at 35°C; end points were obtained by measuring the optical density at 600 nm (OD600). Minimum bactericidal concentrations (MBCs) were determined for Haemophilus influenzae (ATCC 31517; VHIN1004) and S. pneumoniae (ATCC 49219; VSPN1001) (45). The MBC was defined as the lowest concentration of antibiotic that resulted in a ≥3-log10 decrease in the bacterial titer.
Time-kill curves were determined by using log-phase cultures of H. influenzae (ATCC 31517), S. pneumoniae (ATCC 49219), and Staphylococcus aureus (ATCC 25923; VSAU1003). The bacteria were exposed to 5 or 10 times the MIC of each compound tested. Cultures were incubated at 35°C, and titers were quantitatively determined at 0, 2, 6, 8, and 24 h of incubation. Results were expressed as log10 CFU per milliliter versus time.
Expression and purification of E. coli PDF used for crystallographic studies.
The E. coli def gene was cloned for overexpression in E. coli BL21(DE3)/pLysS as described previously (11). Fermentation was performed by using a Bioflo 2000 fermentor (New Brunswick Scientific) with an on-demand fed-batch strategy and minimal medium. A 4-liter fermentation was maintained at 37°C until the culture reached an OD600 of ∼8.9. The temperature was then lowered to 18°C, and the culture was induced overnight with isopropyl-β-d-galactopyranoside (IPTG) at a final concentration of 0.4 mM. The bacterial cells (final OD600, ∼40) were harvested by centrifugation and yielded ∼240 g of paste (wet weight). The cells were frozen as pellets and stored at −80°C.
All purification steps were performed at 4°C unless noted otherwise. Aliquots of cells (∼50 g) were thawed briefly at 37°C and resuspended in 250 ml of lysis buffer (20 mM morpholineethanesulfonic acid [MES]-KOH [pH 6.5], 100 mM KCl, 5 mM NiSO4, 5 mM MgSO4, 10 μg of catalase/ml, 100 μM phenylmethylsulfonyl fluoride, 10.0 μg of DNase I/ml, five tablets of complete protease inhibitor cocktail without EDTA [Boehringer Mannheim]). The cells were lysed with four freeze-thaw cycles and then sonicated (four 10-s bursts, full power) with a Virsonic 60 apparatus (Virtus) equipped with a titanium horn. The pellets were collected at 35,000 × g and washed by resuspension in 120 ml of lysis buffer. The supernatants were combined and clarified by centrifugation at 100,000 × g for 30 min. DNA was removed by centrifugation at 35,000 × g following a 15-min treatment with 0.1% polyethyleneimine (final concentration). The supernatant was buffer exchanged with diafiltration against a 5-kDa-cutoff membrane in 20 mM morpholinepropanesulfonic acid (MOPS)-KOH (pH 7.7)-50 mM KCl-1 mM NiSO4 and filtered through a 0.2-μm-pore-size filter. The sample was then loaded at a flow rate of 30 ml/min onto a POROS 20HQ (perfusion chromatography medium; PE Biosystems) column (2 by 16 cm) equilibrated with 20 mM MOPS-KOH (pH 7.7)-1 mM NiSO4 at room temperature. A linear salt gradient, 0 to 300 mM KCl in 20 column volumes, was used to elute bound proteins, and the fractions were immediately placed at 4°C. Fractions containing Ni-PDF were identified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis, pooled, and further purified by size exclusion chromatography on a Superdex 75 HiLoad (16/60) column (Amersham Pharmacia Biotech) equilibrated with 20 mM MOPS-KOH (pH 7.7)-100 mM KCl at a flow rate of 1 ml/min. Proteins were pooled and concentrated to ∼5 to 10 mg/ml. After the addition of glycerol to 10% and 1 mM Tris-(2-carboxyethyl)-phosphine (TCEP) hydrochloride, the enzyme was stored at −80°C. The protein concentration was determined with Bradford reagent (Bio-Rad) and bovine serum albumin as a standard. This highly purified Ni-PDF was used directly for crystallization.
Crystallization and structure determination.
An enzyme-inhibitor complex was formed by incubation of a twofold molar excess of PDF inhibitor VRC4307 with 1.5 mg of PDF/ml in binding buffer (20 mM HEPES [pH 7.5], 100 mM NaCl, 0.5 mM TCEP) for 30 min at room temperature. The complex was buffer exchanged four times in binding buffer with 50 μM compound VRC4307 by using a Millipore concentrator (5,000-molecular-weight cutoff) and finally concentrated to 36 mg/ml. Crystals were grown by the hanging-drop vapor diffusion method by mixing equal volumes of protein solution and well buffer [100 mM HEPES (pH 7.0), 2.0 M (NH4)2SO4] at 17°C. Crystals were cryoprotected at room temperature by a 15-min, 10-step transfer to cryobuffer solution [100 mM HEPES (pH 7.0), 2.4 M (NH4)2SO4, 25% glycerol] with 5 μM compound VRC4307 and immediately flash-frozen in a 100 K nitrogen gas cold stream (model 700 apparatus; Oxford Cryosystems) for data collection. Diffraction data were collected by using an R-AXIS-IIc image-plate system (Molecular Structures Corp. [MSC]) with Osmic Confocal Max-Flux optics (Blue-3 configuration; Rigaku/MSC) and an RU-2HR generator (MSC) operating at 100 mA and 50 kV. A complete data set was collected from a single crystal (0.2 by 0.18 by 0.06 mm) maintained at 100 K. The images were indexed, and integration was performed by using Denzo/Scalepack (50).
The structure of the enzyme or enzyme complex was determined by molecular replacement with AMoRe (46) as implemented in the CCP4 (9) package with a structure of E. coli PDF (e.g., 1ICJ) (6) as a search model. The structure was refined with the CNX 2000 (8) package (Accelrys Inc.).
Isolation of resistant mutants of H. influenzae and S. pneumoniae.
Spontaneous resistant mutants were selected as previously described (33, 34). Briefly, 1010 CFU of H. influenzae (ATCC 31517) were plated on chocolate agar plates containing 30 μg of antibiotic/ml. In a second experiment, approximately 1010 CFU of log-phase S. pneumoniae (ATCC 49219) were spread on tryptic soy agar (TSA)-5% sheep blood agar plates containing 30 μg of test compound/ml. The plates were incubated for 48 h at 35°C in a 10% CO2 atmosphere. Colonies were picked and transferred to drug-free plates; stable mutants that survived this passage were subjected to further characterization.
Molecular techniques and sequence analysis.
Primary sequences for the def and fmt homologs of H. influenzae and S. pneumoniae were obtained from public sequence databases and previously published work (16, 34). The def-fmt operon and flanking DNA (230 bp upstream; 60 bp downstream) from wild-type H. influenzae VHIN1004 and a single PDF inhibitor-resistant derivative were amplified by PCR with respective upstream and downstream primers oPV743 (5′-CGCCAGGTTACCAAATACTTATCTAAG-3′) and oPV378 (5′-TTTTCCGCTATAAATTTACCG-3′) for def and oPV177 (5′-GCCAGCGCATTAAAGAAAAGTTG-3′) and oPV179 (5′-CGAAAGTGCGGTCATTTTTACTG-3′) for fmt.
The defA, defB, and fmt genes and flanking DNAs (approximately 120, 350, and 60 bp upstream, respectively; 60 bp downstream for all three) from wild-type S. pneumoniae ATCC 49619 and two PDF inhibitor-resistant derivatives were amplified by PCR with the respective upstream and downstream primers oPV369 (5′-CGTGGCTTGGAAGTAACT-3′) and oPV370 (5′-GATATACCAGGTCGTTGC-3′) for defA, oPV385 (5′-TCCCTTATTGCTCCACTTG-3′) and oPV368 (5′-TATCATTTGTTTTCATGCCTC-3′) for defB, and oPV383 (5′-CCACAACCTCTATCATTACCAG-3′) and oPV384 (5′-ACCGTTCCATAGACCAGC-3′) for fmt.
All PCR products were purified with QiaQuick DNA purification columns (Qiagen), and DNA sequences were determined by the dideoxy chain termination method (Sequetech). In all instances, mutations were confirmed by analysis of a second independent PCR product.
Pharmacokinetic studies.
CD-1 female outbred mice (Charles River Laboratories) were used for pharmacokinetic analyses of selected PDF inhibitors. The compounds were formulated in 20% cyclodextrin (Aldrich) and filter sterilized. After 1 week of acclimation, mice (20 to 25 g each) were dosed either intravenously (i.v.) or orally (p.o.) with antibiotic; at 0.083, 0.25, 0.5, 1, 2, and 4 h after dosing, blood samples were collected from anesthetized mice via cardiac puncture. Groups of four mice were used for each time point. The blood was allowed to clot, and the serum samples were stored immediately at −80°C. Serum was extracted with a mixture of acetonitrile, ethanol, and acetic acid and analyzed by liquid chromatography with triple quadropole mass spectrometry (LC-MS/MS) MS (Phenomenex LUNA-C8, 50 by 2 mm, 3 μm, HP1100, Micromass Quattro LC) with an internal standard. The pharmacokinetic parameters, including Tmax (time to maximum concentration), Cmax (maximum concentration measured), t1/2 (terminal half-life), and AUC (area under the curve), were calculated. The p.o. bioavailability was calculated as the ratio of the AUC for p.o. administration to the AUC for i.v. administration.
Metabolic stability in liver microsomes.
Preparations of hepatic microsomes from male CD-1 mice, male Sprague-Dawley rats, and human donors were obtained from Xenotech LLC. To assess their metabolic stability, test compounds (1 μM) in phosphate buffer (100 mM, pH 7.4) were incubated at 37°C with liver microsomal proteins (0.2 mg/ml) supplemented with NADPH (1 mM), UDP-glucuronic acid (4 mM), and alamethecin (60 μg/mg of protein) for 0, 10, 20, and 30 min. Aliquots were removed at various times, and the reaction was terminated by the addition of acetonitrile. Loss of the parent compound was monitored by LC-MS/MS (PE-SCIEX API3000; triple quadrupole) with reverse-phase high-pressure liquid chromatography separation (Phenomenex LUNA-C18, 2.1 by 30 mm, 3 μm). The viability and activity of the microsomal preparations were separately verified by using a positive metabolic control. Intrinsic clearance was calculated from the slope of the curve of log10 linear concentration versus incubation time.
Metabolite identification.
Mouse serum samples from the pharmacokinetic studies described above were extracted with acetonitrile and analyzed by LC-MS/ MS to identify additional metabolites in the serum. In a separate experiment, test compounds (10 μM) were incubated with mouse, rat, and human liver microsomes as described above at 37 °C for 20 min, and the resulting samples were extracted before being analyzed for metabolites.
Protection from infection in the mouse septicemia model (S. aureus strain Smith).
A standard peritonitis model of infection was used as an initial screen to determine the relative efficacies of the PDF inhibitors (MDS Pharma Services, Bothell, Wash.). Briefly, outbred ICR-derived mice (20 to 25 g each) were inoculated intraperitoneally with a 90 to 100% lethal dose (9.0 × 105 CFU/mouse) of S. aureus strain Smith (ATCC 19636) in 0.5 ml of brain heart infusion broth containing 5% mucin. Compounds were dissolved in 20% cyclodextrin and administered subcutaneously (s.c.) and/or p.o. at 1 and 5 h after infection. Vancomycin was included as a control antibiotic, and animals in this antibiotic regimen were dosed s.c. Groups of five mice were used for each dose level. Mice were monitored daily for 6 days, and cumulative mortality was used to determine the 50% effective dose (ED50) by nonlinear regression with Graph-Pad Prism (Graph Pad Software).
RESULTS
Previously, the alkyl succinate hydroxamic acid VRC3324, (2R)-butyl-N4-hydroxy-N1-[2-methyl-(1S)-(naphthalen-2-ylcarbamoyl)-propyl]-succinamide, was identified as a potent PDF inhibitor with excellent antibacterial activity (C. Hackbarth, S. Lopez, M. Gomez, W. Wang, J. Jacobs, R. Jain, Z.-J. Ni, J. Trias, D. Chen, G. Withers, K. Clark, J. Koehn, B. Weidmann, D. Patel, and Z. Yuan, Abstr. 40th Intersci. Conf. Antimicrob. Agents Chemother., abstr. 2173, 2000). Similar compounds of this chemical class exhibit broad-spectrum antibacterial activity and in vivo activity in a mouse septicemia model (D. Chen, C. Hackbarth, Z.-J. Ni, W. Wang, C. Wu, D. Young, R. White, J. Trias, D. Patel, and Z. Yuan, Abstr. 40th Intersci. Conf. Antimicrob. Agents Chemother., abstr. 2175, 2000). In our efforts to discover new pharmacophores that would improve the biological properties of this class of inhibitors, N-alkyl urea hydroxamic acids were identified. VRC3852 (Table 1) represents an early lead in this new class of PDF inhibitors (Lewis et al., 41st ICAAC). Analogs of VRC3852, prepared in both library format and as individual compounds, were screened to identify those with optimized activity and structure.
TABLE 1.
Structure and activity of N-alkyl urea hydroxamic acidsa

S. pn., S. pneumoniae; S. aur, S. aureus; E. faec, E. faecium; H. influ, H. influenzae; M. cat, M. catarrhalis; E. coli acr, efflux pump mutant of E. coli; C. alb, C. albicans. The number of strains tested is indicated in parentheses. P388, P388D1.
In vitro activity tests.
From more than 200 analogs tested, 21 compounds (Table 1) were purified by high-pressure liquid chromatography and subjected to additional biological characterization. All of these compounds were tested for the inhibition of deformylase activity with the E. coli and S. pneumonia enzymes overproduced and purified from E. coli. In addition, whole-cell activity was assessed with broth microdilution MICs against a panel of 26 organisms, including both gram-positive and -negative bacteria and Candida albicans.
Twenty of these 21 compounds have IC50s of ≤100 nM for E. coli deformylase. Eighteen of these 20 compounds also have excellent activity (IC50s, ≤75 nM) against S. pneumoniae PDF. These IC50s corresponded to improved activity against S. pneumoniae. In fact, the two compounds with poor IC50s for the S. pneumoniae enzyme, VRC4347 and VRC4390, also lacked whole-cell activity against this organism, providing further evidence that this class of compounds targets deformylase for the inhibition of bacterial growth.
Compared with the MICs of the initial lead in this series, VRC3852, the MICs against the three gram-positive pathogens tested (S. aureus, S. pneumoniae, and Enterococcus faecium) improved for the majority of the compounds listed. One of the compounds that had poor gram-positive activity, VRC4390, nonetheless maintained its activity against H. influenzae. On the other hand, VRC4234 and VRC4305 were more active against S. aureus and S. pneumoniae but less potent against the gram-negative pathogen H. influenzae. Apparently, these compounds readily undergo efflux in E. coli, since none was active against wild-type E. coli but all possessed low MICs against an E. coli acr mutant which lacks the Acr efflux pump (49). None of these compounds was active against C. albicans.
To determine whether this class of compounds inhibited bacterial growth through the inhibition of deformylase, each of the compounds was tested for its ability to inhibit the growth of an actinonin-resistant S. aureus strain, VSAU6011. This strain harbors a loss-of-function mutation in the formyltransferase-encoding gene, bypassing the need for deformylase activity and permitting growth even when PDF activity is inhibited (34). S. aureus VSAU6011 was highly resistant to all of the compounds tested, demonstrating that N-alkyl urea hydroxamic acids, like alkyl succinates (11, 13), inhibit bacterial growth through the inhibition of deformylase.
These compounds were also tested for their potential cytotoxicity and inhibition of matrilysin, a member of the mammalian MMP superfamily which closely resembles PDF (5, 10, 38). Matrilysin activity was not inhibited in the presence of up to and in most instances exceeding 100 μM PDF inhibitor. In addition, the IC50s of VRC4232 and VRC4307 against human ACE, another metalloprotease, were higher than 100 μM. Thus, these N-alkyl urea hydroxamic acids are over 1,000 times more potent against bacterial deformylase than the eukaryotic metalloenzymes tested.
When these compounds were tested against two different mammalian cell lines, the IC50s, determined after 72 h of continuous exposure, were at least 10-fold higher than the MICs for the major bacterial pathogens in the screening panel. A closer examination of the structure-activity relationships of these compounds (see Discussion) suggested that the cytotoxicity of these compounds generally correlated with their hydrophobicity. Given their overall in vitro profiles, VRC4232 and VRC4307 were selected as new leads for additional in vitro and in vivo characterizations.
X-ray analysis of binding interactions.
Key parameters and summary statistics for the refined PDF model are provided in Table 2. After independent rigid-body refinement of the three PDF proteins in the asymmetric unit, the electron density for the three inhibitors was clearly seen in each of the active sites. Several rounds of iterative model building (including the addition of water molecules and correcting loops involved in crystal contacts) with the program O (27) and refinement were conducted before the three inhibitor molecules were added to the model. In complex A, the inhibitor could be positioned unambiguously in the electron density. For complexes B and C, there was a weak, broken electron density for the 4,5-dimethylthiazole ring of the inhibitor. Rfree was monitored while additional water molecules were added. The final model contained three PDF-compound VRC4307 complexes as well as 435 water, 3 sulfate, and 5 glycerol molecules, resulting in an R factor of 18.6% (Rfree = 21.6%) in the resolution range of 30.0 to 1.8 Å.
TABLE 2.
Summary of diffraction data and refinement data for the crystal structure of the E. coli PDF complex
| Parameter | Description or value |
|---|---|
| Space group | C2 |
| Cell | |
| a, b, c (Å) | 138.85, 163.37, 85.96 |
| α, β, γ (°) | 90, 121.39, 90 |
| Asymmetric unit | Three complexes |
| Resolution (Å) | 1.80 (1.86-1.80) |
| No. of unique reflections | 56,820 (5,097) |
| Redundancy | 4.3 (2.7) |
| Rmerge (%) | 5.2 (23.1) |
| Intensity (SE) | 13.7 (2.7) |
| No. of atoms | |
| Protein | 3,977 |
| Solvent | 480 |
| Inhibitor | 93 |
| R (%) | 18.6 (22.5) |
| Rfree (%) (test set of 5%) | 21.9 (25.3) |
| Completeness (F > 1.0σ) (%) | 90.5 (69.4) |
| RMSD from ideal | |
| Bond lengths (Å) | 0.01 |
| Bond angles (°) | 1.4 |
| Dihedral angle | 22.2 |
| Mean B value (Å2) | 24.9 |
The structure of E. coli PDF has already been extensively characterized and will not be elaborated on further here (6, 10, 14, 40). When the PDF structure with bound VRC4307 is examined, the hydroxamic acid is observed to coordinate the active-site Ni2+, as expected; the cyclopentyl ring system of VRC4307 lies in the S1′ pocket of the enzyme (Fig. 3A). The carbonyl oxygen of the proline group of VRC4307 forms a hydrogen bond with the backbone nitrogen of Ile44, maintaining the peptidic interaction observed for the PDF structure with the bound product MAS (6). Details of the metal coordination, hydrogen bonding, and Van der Waals interactions are shown in Fig. 4A. The electron density for most of the compound is clear and unambiguous. The electron density for the amide-dimethylthiazole (Fig. 4A, green atoms) portion of the inhibitor is weak and broken. Apparently, the inhibitor is able to adopt multiple conformations when bound; this hypothesis is illustrated by superimposition of the three complexes (Fig. 3B). Surprisingly, the peptidic interaction with the backbone carbonyl of Glu42 is seen in only one of the three complexes (Fig. 4A).
FIG. 3.

Surface representation of VRC4307 binding to E. coli PDF. PDF is represented as a smooth surface created by GRASP; the catalytic Ni2+ is shown as a magenta sphere. (A) The cyclopentyl ring system is positioned in the S1′ pocket. (B) The inhibitors of three complexes have been superimposed to illustrate relative disorder in the dimethylthiazole rings of the inhibitors.
FIG. 4.

Interactions between E. coli PDF and VRC4307. (A) Schematic representation of key interactions between PDF and VRC4307. (B) Interactions of the hydroxamic acid with the active-site metal. Although the ligand coordination geometry is a distorted trigonal bipyramid, the fifth ligand, the carbonyl oxygen, is positioned too far from the Ni2+ atom for optimal interactions with three complexes (2.7, 2.7, and 3.0 Å). Solid lines indicate proposed interactions; the potential fifth ligand interaction is shown as broken lines.
The hydroxamic acid-metal coordination in the PDF-VRC4307 complex maintains a tetrahedral configuration (Fig. 4B). Although the overall arrangement of the ligand atoms approximates a distorted trigonal bipyrimidal geometry, the carbonyl oxygen of the inhibitor is more distant from the metal than is typically observed in metal-ligand complexes. In all three complexes, the Ni2+ distances are reasonable for the N and O atoms (2.0 to 2.1 Å) and for the S atoms (2.3 Å); however, the carbonyl O atom of VRC4307 is 2.7 to 3.0 Å from the metal. This suboptimal interaction may be offset, in part, by an ideal hydrogen bonding interaction distance between the carbonyl oxygen and the backbone nitrogen of Leu91. The unusual metal-ligand geometry observed appears to be a direct consequence of the relatively planar N compared to an alpha-carbon in substrate/product. The VRC4307 cyclopentyl analog of the methionine side chain is packed on top of His132 of PDF, meaning that the relatively planar configuration of the urea N prevents the hydroxamic acid from fully entering the binding pocket for an optimal interaction with the metal. The favorable hydrogen bond interaction between a carbonyl of VRC4307 and Ile44 is accompanied by a steric clash between the carbonyl and Gly43 (Fig. 4A).
Killing curves and MBCs.
It was previously shown that actinonin, a succinate hydroxamic acid, is bacteriostatic against S. aureus (11). In this study, killing curves for the urea compounds VRC4232 and VRC4307 were determined at 10 times the MICs for S. aureus, S. pneumoniae, and H. influenzae (Fig. 5). Like actinonin, the urea-based compounds are also bacteriostatic against S. aureus, but a killing effect was observed against the other two organisms. For S. pneumoniae, viable counts dropped by greater than 2 log10 units over the initial 12 h of exposure; for H. influenzae, a drop of up to 5 log10 units in viable organisms was observed after 24 h. Both compounds were bacteriostatic over the initial 8 h of exposure; increasing the drug concentration to 20 times the MIC (80 μg/ml) did not improve the initial killing rate (data not shown). These observations were consistent with the MBC (Table 3) determinations for H. influenzae and S. pneumoniae. Unlike S. aureus (data not shown), these two fastidious pathogens are killed by PDF inhibitors, although the reason for the late killing effect is unclear.
FIG. 5.

Bacterial killing curves. Log-phase organisms were exposed to either no drug (▪), 10 times the MIC of VRC4232 (♦), or 10 times the MIC of VRC4307 (•). Results are expressed as log10 CFU per milliliter versus time.
TABLE 3.
MICs and MBCs of PDF inhibitors against H. influenzae VHIN1004 and S. pneumoniae VSPN1001a
| Strain | VRC4232
|
VRC4307
|
||
|---|---|---|---|---|
| MIC | MBC | MIC | MBC | |
| VHIN1004 | 4 | 32 | 4 | 32 |
| VSPN1001 | 4 | >64 | 1 | 32 |
MICs and MBCs are given in micrograms per milliliter.
Resistance and its mechanism.
The in vitro selection of spontaneously resistant H. influenzae and S. pneumoniae mutants occurred at a frequency of 1 in 109 for both VRC4232 and VRC4307 (Table 4). These resistance frequencies are similar to those observed previously for actinonin against S. pneumoniae (33).
TABLE 4.
Antibiotic profiles of H. influenzae and S. pneumoniae mutant strainsa
| Strain tested | Strain selected on | Genotype | DT (min) | MIC (μg/ml) of:
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Act | VRC4232 | VRC4307 | VRC3852 | VRC4349 | VRC4411 | β-Lact | CHL | ERY | TMP | LZD | TET | CIP | ||||
| VHIN1004 | None | 58 | 0.125 | 1 | 0.5 | 1 | 0.5 | 0.125 | 1 | 0.25 | 4 | 0.25 | 8 | 0.25 | 0.008 | |
| VHIN6529 | VRC4232 | fmt (bp 307 deletion) | 117 | 32 | >64 | >64 | >64 | >64 | >64 | 2 | 0.25 | 1 | 1 | 16 | 0.25 | 0.015 |
| VHIN6536 | VRC4307 | ND | ND | 32 | >64 | >64 | >64 | >64 | >64 | 2 | 0.25 | 2 | 0.5 | 16 | 0.25 | 0.015 |
| VSPN1001 | None | 65 | 16 | 2 | 0.25 | 32 | 1 | 2 | 0.5 | 2 | 0.25 | 4 | 1 | 0.25 | 0.5 | |
| VSPN6521 | VRC4232 | defB (G211 → T) | 74 | 4 | 64 | 32 | 256 | 64 | 64 | 0.5 | 2 | 0.25 | 4 | 1 | 0.25 | 0.5 |
| VSPN6522 | VRC4307 | defB (G211 → T) | 82 | 2 | 64 | 8 | 256 | 64 | 16 | 0.5 | 2 | 0.25 | 2 | 2 | 0.25 | 1 |
DT, doubling time. Act, actinonin; β-Lact, ampicillin for H. influenzae and penicillin for S. pneumoniae; CHL, chloramphenicol; ERY, erythromycin; TMP, trimethoprim; LZD, linezolid; TET, tetracycline; CIP, ciprofloxacin. ND, not determined.
Two independent H. influenzae mutants, VHIN6529 and VHIN6536, were selected for further characterization. Both mutants were highly resistant to all PDF inhibitors tested: the MICs of all such compounds, except actinonin, were >64 μg/ml; the MIC of actinonin was 32 μg/ml (Table 4). The antibiotic profile of the mutants is comparable to that of the parent strain, suggesting that there is no cross-resistance with other classes of antibiotics and that this resistance is not due to a defect in a transport mechanism that is shared with other antibiotics. The resistant mutants appear on solid medium as small colonies; they grow slowly both in broth and on agar. When these small colonies are streaked on solid agar in the absence of a drug, large, normally sized colonies frequently appear on the plate. These revertants are again as susceptible to deformylase inhibitors as the parent strain, VHIN1004 (data not shown).
To better define the mechanism of resistance to compounds of the urea series, the def and fmt homologs from H. influenzae mutant VHIN6529 and from three independent revertants were sequenced and compared to those of the wild type. In comparison with the sequences of the genes from parent strain VHIN1004, no change was observed in the def homolog. However, the fmt gene from mutant VHIN6529 (and each revertant) contained a frameshift mutation: the deletion of a C residue following bp 307 of the open reading frame (ORF). The resulting protein is predicted to diverge from the wild-type protein at amino acid 103, terminating four amino acids later (Fig. 6). The def sequences from the three independent revertants were not changed compared to that from the parent strain. However, all three revertants carried, in addition to the original 1-bp deletion in fmt, a T residue inserted following bp 299 of the fmt ORF. This compensatory frameshift mutation restores the fmt reading frame in mutant VHIN6529, such that the revertants express a full-length transformylase with a three-amino-acid substitution at codons 99 through 101 (Fig. 6).
FIG. 6.

Internal DNA sequence and predicted protein sequence (codons 94 to 109) of the H. influenzae fmt ORF from wild-type strain VHIN1004 (top), resistant mutant VHIN6529 (middle), and a susceptible revertant (bottom). Note that the insertion of T at codon 99 restores the fmt reading frame in the revertant. Underlined amino acids indicate divergence from the wild-type formyltransferase protein sequence.
Two independent S. pneumoniae mutants, VSPN6521 and VSPN6522, were further characterized. Morphologically, the resistant mutant strains resemble the parent strain. The doubling times of the mutant strains (74 to 82 min) are slightly longer than that of the corresponding wild type (65 min), a finding that is very similar to what was found with mutants of S. pneumoniae selected with actinonin (33). Like that of the H. influenzae mutants, the overall antibiotic profile of the pneumococcal mutants does not vary from that of the parent strain (Table 4). The defB (PDF), defA (PDF paralog), and fmt (transformylase) genes were amplified from these mutants by PCR, and the DNA sequences were compared to the sequence of the wild-type parent strain. No changes were observed in defA or fmt, but for both mutants, a G-to-T transversion was observed at bp 211 of the defB ORF. This change is predicted to generate a Val (GTT)-to-Phe (TTT) substitution at amino acid 71 of the PDF product, a substitution that is distinct from those observed previously for S. pneumoniae mutants selected with actinonin (33).
One major difference can be observed between the resistant mutants from S. pneumoniae and those from H. influenzae: while H. influenzae mutants selected with urea are cross resistant to both succinate and N-alkyl urea PDF inhibitor classes, mutants from S. pneumoniae are cross resistant to other N-alkyl urea compounds but display increased susceptibility to actinonin, a succinate hydroxamic acid.
Pharmacokinetic studies with mice.
The clearance of VRC4232 and VRC4307 in mouse serum was measured after the compounds were administered i.v. and p.o. The concentrations in serum at different times were used to calculate the corresponding pharmacokinetic parameters presented in Table 5. VRC4232 has a longer half-life (1.1 h) than VRC4307 (0.1 h) after i.v. administration. Both compounds have poor p.o. bioavailability (3.2 and 0.1%, respectively), suggesting that these compounds will not have significant p.o. efficacy.
TABLE 5.
Mouse pharmacokinetic parameters for VRC4232 and VRC4307a
| Compound | Route | Dose (mg/kg) | Tmax (h) | Cmax (ng/ml) | t1/2 (h) | AUC (ng · h/ml) | % Absolute bioavailability |
|---|---|---|---|---|---|---|---|
| VRC4232 | i.v. | 13 | 0.083 | 5,580 | 1.1 | 1,120 | |
| p.o. | 13 | 0.083 | 167 | NC | 35.3 | 3.2b | |
| VRC4307 | i.v. | 3.7 | 0.083 | 1,720 | 0.1 | 413 | |
| p.o. | 3.7 | 0.25b | 1.5 | NC | 0.3 | 0.1b |
NC, not calculated.
Value was not accurate due to an extremely low concentrations of the compound.
In vitro metabolic stability.
The metabolic stability of the compounds was determined in vitro: the compounds were incubated with mouse, rat, and human liver microsomes and then analyzed for loss of the parent compound over time. As summarized in Table 6, both compounds were rapidly metabolized by mouse and rat liver microsomes (t1/2, <3 min) but were much more stable in human liver microsomes (t1/2, >19 min).
TABLE 6.
Comparison of metabolic stability of VRC4232 and VRC4307, as determined in vitro by incubation with liver microsomes from mice, rats, and humans
| Compound |
t1/2 (min) in:
|
Clearance (μl/min/mg) in:
|
||||
|---|---|---|---|---|---|---|
| Mice | Rats | Humans | Mice | Rats | Humans | |
| VRC4232 | 3.1 | 2.6 | 26.8 | 1,100 | 1,309 | 129 |
| VRC4307 | 2.2 | 2.0 | 19.9 | 1,594 | 1,700 | 174 |
Identification of major metabolites for VRC4232.
The serum samples collected from mice after dosing with VRC4232 were analyzed for major metabolites by LC-MS/MS. Similar analyses were also carried out for samples collected after incubation of VRC4232 with liver microsomes. The major metabolites identified from these samples are summarized in Fig. 7. Since the authentic compounds were not available, quantitative analysis was not performed. Although these metabolites were identified in all samples, their relative intensities in the LC-MS spectrum varied. The three major peaks found by LC-MS for these metabolites (in order of decreasing relative peak areas) were E, A, and B for in vivo mouse serum; A, E, and F for mouse liver microsomes; D, E, and A for rat liver microsomes; and C, A, and E for human liver microsomes.
FIG. 7.

Modification of VRC4232 in vivo (mice) and in vitro (liver microsomes) and identification of its major metabolites (A to F).
The majority of the major metabolites correspond to modifications of the hydroxamic acid, including hydrolysis (metabolites A, B, and C in Fig. 7) and glucuronic acid conjugation (metabolites E and F in Fig. 7). All of these modifications would result in loss of the chelating activity of the parent compound, an effect which should render the compounds devoid of PDF inhibitory activity. Other major modifications identified include hydroxylation of the proline group (metabolite C in Fig. 7) and oxidation of dimethylthiazole (metabolites B, D, and F in Fig. 7).
Mouse S. aureus septicemia model.
VRC4232 and VRC4307 both have MICs of 0.015 μg/ml against S. aureus strain Smith, used in the septicemia model. After s.c. administration of VRC4232, four of five mice survived at 32 mg/kg of body weight and none of five survived at 16 mg/kg, resulting in an ED50 of 29.7 mg/kg. VRC4307 was more efficacious in this model. The survival of three, three, one, and none of five mice was observed after s.c. doses of 32, 16, 8, and 4 mg/kg, respectively, resulting in an ED50 of 17.9 mg/kg. VRC4307 was also tested for its efficacy after p.o. administration; no protection was observed at the highest tested dose of 30 mg/kg (i.e., none of five mice survived). The ED50 for the control antibiotic, vancomycin, was 2.4 mg/kg after s.c. dosing.
DISCUSSION
N-alkyl urea hydroxamic acids represent a new class of PDF inhibitors. All previously published PDF inhibitors with reasonable antibacterial activity share the structural feature of a 2-substituted alkanoyl, particularly 2-substituted hexanoyl, at the P1′ site (57). In this study, it was shown that bacterial PDF can accommodate N-alkyl urea at the P1′ site. The tested compounds also can effectively inhibit the growth of both gram-positive and gram-negative bacteria. The N-alkyl urea structure allows easy preparation of analogs with different P1′ and P3′ substitutions to explore the combination of moieties at either site that might provide the best antibacterial activity.
Various alkyl groups at the P1′ site were explored in a combinatorial library format: branched and cyclic aliphatic groups such as N-cyclopentylethyl and N-isopentyl showed the best antibacterial activity (J. Lewis, unpublished data). Compounds combining these two P1′ substitutions with different P3′ groups were prepared as purified compounds and tested in a panel of in vitro assays (Table 1). The data in Table 1 demonstrate that the N-alkyl urea compounds can very effectively inhibit bacterial PDFs with IC50s well below 1 μM. In general, compounds with substituted thiazoles have excellent activity against S. aureus and S. pneumoniae. Overall, all of the N-alkyl urea compounds prepared resulted in moderate inhibition of H. influenzae growth.
A comparison of VRC4390, VRC4156, VRC5157, and VRC4234, all of which share the P1′ N-isopentyl substitution, suggests that increasing hydrophobicity at the P3′ site improves enzymatic and whole-cell activities for this class of compounds. The trend is particularly obvious when a methyl group is incorporated in the P3′ site, a substitution which results in a dramatic loss of inhibition of S. pneumoniae PDF but which has only a slight effect on E. coli PDF inhibition. The methyl substitution also renders the compound inactive against S. aureus and S. pneumoniae in whole-cell assays, while activity against H. influenzae and efflux-deficient E. coli is maintained. These differences suggest that VRC4390 lacks activity against S. aureus and S. pneumoniae because of a species-specific loss of activity against the corresponding PDF. This observation further suggests that deformylases from different bacterial species may have different preferences at their S3′ sites.
The crystal structure of the complex of VRC4307 with E. coli PDF revealed the interactions between the urea hydroxamate and the enzyme. Surprisingly, the hydroxamate did not adopt the expected bidentate interaction that is common with MMP inhibitors (7) and that was observed in the recently published PDF-actinonin structure (13). The relatively planar N (relative to an alpha-C atom of a substrate or actinonin) coupled with the P2′ proline limits the ability of the inhibitor to fully reach the active-site metal. The result is a single metal-ligand interaction with the hydroxamate, while the remaining oxygen atom forms a hydrogen bond interaction with the backbone amide of Leu91. In order to achieve these interactions and maintain the P1′ interactions, the peptidic interaction between the carbonyl of the inhibitor and the backbone amide of Ile44 is also distorted relative to the actinonin complex. A good hydrogen bonding distance is achieved, and close contact with the carbonyl and the alpha-carbon of Gly43 results (2.8 Å).
Due to the structural resemblance of the key catalytic residues between PDFs and MMPs, the 2-substituted alkanoyl can fit very well into the S1′ site of the active center of both enzymes (10, 41). This similarity could result in the inhibition of MMPs by many deformylase inhibitors. In this study, we show that bacterial PDF can accommodate N-alkyl urea in the P1′ site. In addition to structural novelty, this alternative P1′ substitution also provides excellent selectivity, as demonstrated by the absence of MMP inhibition. The data in Table 1 indicate that none of the compounds exhibits inhibitory activity against MMP-7 at 200 μM; analogous compounds with a 2-substituted alkanoyl substitution at the P1′ site usually display complete inhibition of MMP-7 at this concentration (data not shown). Based on the crystal structure, it appears that the selectivity may require both the N-alkyl urea hydroxamate and a P2′ proline group. The conformation adopted by the urea hydroxamate places its P2′ proline ring at a sterically unfavorable distance from the MMP carbonyl of Pro238 (matrilysin numbering). This proline is conserved in the MMP family, and the orientation of its carbonyl is quite different from that of Gly89, which occupies the equivalent position in E. coli PDF. This structural property of the N-alkyl urea hydroxamic acids should avoid the potential toxicity of hydroxamic acids that is due to the inhibition of MMPs.
The urea hydroxamates appear to be bacteriostatic against S. aureus, as was observed previously for other PDF inhibitors against both S. aureus and E. coli (4, 13, 34). Huntington et al. (25) reported the lysis of Bacillus subtilis by a peptide thiol PDF inhibitor, although the compound appeared to be bacteriostatic when tested with log-phase organisms. In contrast to the situation with S. aureus, VRC4232 and VRC4307 both had a killing effect on S. pneumoniae and H. influenzae, two pathogens that cause upper respiratory tract infections. Notably, neither compound displayed a significant degree of killing of H. influenzae over the initial hours of the experiments, but there was significant death of this species by 24 h of drug exposure. The basis for this delayed killing effect is not clear, but these data and the characterization of the resistant mutants indicate that the inability to remove the N-formyl group from proteins is toxic in H. influenzae. Hypothetically, several cycles of growth in the presence of deformylase inhibitors would deplete the pool of normally processed polypeptides, while N-formylated proteins would accumulate. If, at that late stage, one or more enzymes are required but are nonfunctional due to N formylation (e.g., improper folding), then cell death will ensue.
The frequency of resistance to the urea-based inhibitors in both H. influenzae and S. pneumoniae is approximately 10−9, as was previously reported for actinonin, a PDF inhibitor of the succinate hydroxamic acid series. This value is comparable to what was found for a peptide thiol PDF inhibitor against B. subtilis (25) and is 2 to 3 orders of magnitude smaller that what has been reported for the spontaneous selection of resistance to other deformylase inhibitors in S. aureus (13, 34) and E. coli (4, 13). Two mechanisms of resistance to succinyl hydroxamate-based inhibitors of bacterial PDF have been reported. In E. coli, S. aureus, Moraxella catarrhalis, and H. influenzae, loss-of-function mutations in fmt have been identified (4, 13, 33; P. Margolis, C. Hackbarth, M. Maniar, S. Lopez, W. Wang, R. White, Z. Yuan, and J. Trias, Abstr. 40th Intersci. Conf. Antimicrob. Agents Chemother., abstr. 2174, 2000). The data presented here indicate that resistance to urea-based PDF inhibitors in H. influenzae occurs through a mechanism similar to that seen for succinyl hydroxamate-based compounds in the same species. As was observed previously for actinonin resistance mutations, resistance to VRC4232 and VRC4307 in H. influenzae apparently is derived from a frameshift mutation that results in a truncated (and presumably nonfunctional) formyltransferase protein. Consistent with previous observations made for E. coli, S. aureus, and H. influenzae, H. influenzae fmt mutant strain VHIN6529 grew more slowly than the wild-type parent strain. Revertants of strain VHIN6529 simultaneously displayed an improved growth phenotype, increased susceptibility to PDF inhibitors, and a compensatory frameshift mutation that restored the fmt reading frame. This correlation strongly supports a model whereby resistance to urea-based PDF inhibitors can occur through bypassing of the formylation cycle.
It is not evident why the loss-of-function mutations found in H. influenzae fmt occur at a lower frequency (10−8) than those found with succinate inhibitors in E. coli or S. aureus (10−6). One hypothesis is that the difference in resistance rates may be due to the bactericidal effects of these compounds on H. influenzae, which may lead to a decreased number of viable mutants able to survive the selection process. Alternatively, rather than a bypass mechanism, perhaps the truncated Fmt created by the frameshift mutation remains partly functional, and fmt is essential in H. influenzae. In S. pneumoniae, a second mechanism has been identified; resistance to actinonin results from missense mutations in the defB gene. The distinction in resistance mechanisms observed in different species presumably reflects the fact that fmt is essential in pneumococci (33). Resistance to the urea-based compounds VRC4232 and VRC4307 in S. pneumoniae also appears to be mediated by an alteration in the target, but the location of the missense mutation in defB (V71F) is distinct from those of two other pneumococcal defB mutations, A123D and Q172K, that were identified previously (33). The V71F mutation seen in strains VSPN6521 and VSPN6522 is located in the highly conserved box 1 (70GVGLAAPQ77) of S. pneumoniae PDF. As a comparison with the PDF consensus sequence shows, Val71 does not correspond to a strictly conserved residue; an Ile-to-Ala substitution of the equivalent residue in the E. coli enzyme is well tolerated (41). However, this residue does lie between Gly residues known to be critical for enzyme activity in the E. coli enzyme.
It also appears that the defB (V71F) mutants VSPN6521 and VSPN6522 are able to distinguish between inhibitors of the urea and succinate hydroxamate series. These mutants are more susceptible to actinonin (Table 4) as well as to other succinate inhibitors (data not shown), although they are highly resistant to all urea-series PDF inhibitors tested. In contrast, mutants selected with actinonin contain distinct defB mutations (33) and are resistant to both classes of PDF inhibitors (data not shown). A crystal structure of the enzyme containing this V71F mutation is not available; however, the crystal structure of E. coli PDF complexed with VRC4307 can be used to infer the effect of this mutation. The two most common rotamers for Phe71 would place it in either the S1′ or the S3′ pocket. Given that the V71F mutant is still active, the more likely position would be in the large S3′ pocket, which could accommodate this large side chain with only minor perturbations. The urea hydroxamate inhibitors are less likely to tolerate any movement of Phe71 (and the adjacent Gly70) into the active site due to the close contact between Gly70 and the inhibitor. These observations may reflect a species-specific structural aspect of the pneumococcal PDF enzyme.
The metabolic instability of the urea-series compounds seen in rodent microsomes (Table 6) is likely a factor in the rapid clearance of the compounds in this species (Table 5). However, the rate of clearance of both compounds in human liver microsomes is approximately 10-fold slower than that in rodent microsomes, suggesting that this class of compounds should have longer half-lives in humans. The predictive value of these in vitro models has been reported by others (22, 23, 47, 48, 54) and is borne out by the similar spectra of the metabolic products observed in vitro (mouse liver microsomes) and in vivo (mouse serum). Two major modifications, hydroxamic acid hydrolysis and glucuronic acid conjugation, were prominent in both instances, and the sites of modification were also comparable.
VRC4232 and VRC4307 were selected for further in vivo studies. In an S. aureus septicemia model, both compounds showed moderate protective activity after s.c. administration, establishing N-alkyl urea hydroxamic acids as potential antibacterial agents. However, when the more potent compound VRC4307 was tested after p.o. administration in the same model, no protective effect was observed up to 30 mg/kg. This lack of p.o. efficacy presumably reflects the poor p.o. bioavailability of VRC4307, as demonstrated by the in vivo pharmacokinetic analysis (absolute p.o. bioavailability, 0.1%). These data suggest the need to incorporate bioavailability tests at early stages of compound screening by using Caco-2 cell in vitro testing or by using cassette dosing.
In summary, we have identified N-alkyl urea hydroxamic acids as a new class of PDF inhibitors. This class of compounds has potent whole-cell activity against both gram-positive and gram-negative bacteria but is devoid of MMP inhibition. The potential use of this class of compounds as antibacterial agents is supported by their protective activity in an in vivo infection model.
REFERENCES
- 1.Adams, J. M. 1968. On the release of the formyl group from nascent protein. J. Mol. Biol. 33:571-589. [DOI] [PubMed] [Google Scholar]
- 2.Adams, J. M., and M. R. Capecchi. 1966. N-formylmethionyl-sRNA as the initiator of protein synthesis. Proc. Natl. Acad. Sci. USA 55:147-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Apfel, C., D. W. Banner, D. Bur, M. Dietz, T. Hirata, C. Hubschwerlen, H. Locher, M. G. Page, W. Pirson, G. Rosse, and J. L. Specklin. 2000. Hydroxamic acid derivatives as potent peptide deformylase inhibitors and antibacterial agents. J. Med. Chem. 43:2324-2331. [DOI] [PubMed] [Google Scholar]
- 4.Apfel, C. M., H. Locher, S. Evers, B. Takacs, C. Hubschwerlen, W. Pirson, M. G. Page, and W. Keck. 2001. Peptide deformylase as an antibacterial drug target: target validation and resistance development. Antimicrob. Agents Chemother. 45:1058-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker, A., I. Schlichting, W. Kabsch, D. Groche, S. Schultz, and A. F. Wagner. 1998. Iron center, substrate recognition and mechanism of peptide deformylase. Nat. Struct. Biol. 5:1053-1058. [DOI] [PubMed] [Google Scholar]
- 6.Becker, A., I. Schlichting, W. Kabsch, S. Schultz, and A. F. Wagner. 1998. Structure of peptide deformylase and identification of the substrate binding site. J. Biol. Chem. 273:11413-11416. [DOI] [PubMed] [Google Scholar]
- 7.Browner, M. F., W. W. Smith, and A. L. Castelhano. 1995. Matrilysin-inhibitor complexes: common themes among metalloproteases. Biochemistry 34:6602-6610. [DOI] [PubMed] [Google Scholar]
- 8.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D 54:905-921. [DOI] [PubMed] [Google Scholar]
- 9.CCP4. 1994. The CCP4 Suite: programs for protein crystallography. Acta Crystallogr. Sect. D 50:760-763. [DOI] [PubMed] [Google Scholar]
- 10.Chan, M. K., W. Gong, P. T. Rajagopalan, B. Hao, C. M. Tsai, and D. Pei. 1997. Crystal structure of the Escherichia coli peptide deformylase. Biochemistry 36:13904-13909. (Erratum, 37:13042, 1998.) [DOI] [PubMed] [Google Scholar]
- 11.Chen, D. Z., D. V. Patel, C. J. Hackbarth, W. Wang, G. Dreyer, D. C. Young, P. S. Margolis, C. Wu, Z. J. Ni, J. Trias, R. J. White, and Z. Yuan. 2000. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry 39:1256-1262. [DOI] [PubMed] [Google Scholar]
- 12.Chu, M., R. Mierzwa, L. He, L. Xu, F. Gentile, J. Terracciano, M. Patel, L. Miesel, S. Boanon, C. kravec, C. Cramer, T. O. Fischman, A. Hruza, L. Ramanathan, P. Shipkova, and T. Chan. 2001. Isolation and structure elucidation of two novel deformylase inhibitors produced by Streptomyces sp. Tetrahedron Lett. 42:3549-3551. [Google Scholar]
- 13.Clements, J. M., R. P. Beckett, A. Brown, G. Catlin, M. Lobell, S. Palan, W. Thomas, M. Whittaker, S. Wood, S. Salama, P. J. Baker, H. F. Rodgers, V. Barynin, D. W. Rice, and M. G. Hunter. 2001. Antibiotic activity and characterization of BB-3497, a novel peptide deformylase inhibitor. Antimicrob. Agents Chemother. 45:563-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dardel, F., S. Ragusa, C. Lazennec, S. Blanquet, and T. Meinnel. 1998. Solution structure of nickel-peptide deformylase. J. Mol. Biol. 280:501-513. [DOI] [PubMed] [Google Scholar]
- 15.Durand, D. J., B. Gordon Green, J. F. O'Connell, and S. K. Grant. 1999. Peptide aldehyde inhibitors of bacterial peptide deformylases. Arch. Biochem. Biophys. 367:297-302. [DOI] [PubMed] [Google Scholar]
- 16.Fleischmann, R. D., M. D. Adams, O. White, R. A. Clayton, E. F. Kirkness, A. R. Kerlavage, C. J. Bult, J. F. Tomb, B. A. Dougherty, J. M. Merrick, et al. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496-512. [DOI] [PubMed] [Google Scholar]
- 17.Giglione, C., M. Pierre, and T. Meinnel. 2000. Peptide deformylase as a target for new generation, broad spectrum antimicrobial agents. Mol. Microbiol. 36:1197-1205. [DOI] [PubMed] [Google Scholar]
- 18.Gordon, J. J., B. K. Kelly, and G. A. Miller. 1962. Actinonin: an antibiotic substance produced by an actinomycete. Nature 195:701-702. [DOI] [PubMed] [Google Scholar]
- 19.Green, B. G., J. H. Toney, J. W. Kozarich, and S. K. Grant. 2000. Inhibition of bacterial peptide deformylase by biaryl acid analogs. Arch. Biochem. Biophys. 375:355-358. [DOI] [PubMed] [Google Scholar]
- 20.Groche, D., A. Becker, I. Schlichting, W. Kabsch, S. Schultz, and A. F. Wagner. 1998. Isolation and crystallization of functionally competent Escherichia coli peptide deformylase forms containing either iron or nickel in the active site. Biochem. Biophys. Res. Commun. 246:342-346. [DOI] [PubMed] [Google Scholar]
- 21.Hao, B., W. Gong, P. T. Rajagopalan, Y. Zhou, D. Pei, and M. K. Chan. 1999. Structural basis for the design of antibiotics targeting peptide deformylase. Biochemistry 38:4712-4719. [DOI] [PubMed] [Google Scholar]
- 22.Houston, J. B. 1994. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharmacol. 47:1469-1479. [DOI] [PubMed] [Google Scholar]
- 23.Houston, J. B., and D. J. Carlile. 1997. Prediction of hepatic clearance from microsomes, hepatocytes, and liver slices. Drug Metab. Rev. 29:891-922. [DOI] [PubMed] [Google Scholar]
- 24.Hu, Y. J., P. T. Rajagopalan, and D. Pei. 1998. H-phosphonate derivatives as novel peptide deformylase inhibitors. Bioorg. Med. Chem. Lett. 8:2479-2482. [DOI] [PubMed] [Google Scholar]
- 25.Huntington, K. M., T. Yi, Y. Wei, and D. Pei. 2000. Synthesis and antibacterial activity of peptide deformylase inhibitors. Biochemistry 39:4543-4551. [DOI] [PubMed] [Google Scholar]
- 26.Hutchison, C. A., S. N. Peterson, S. R. Gill, R. T. Cline, O. White, C. M. Fraser, H. O. Smith, and J. C. Venter. 1999. Global transposon mutagenesis and a minimal Mycoplasma genome. Science 286:2165-2169. [DOI] [PubMed] [Google Scholar]
- 27.Jones, T. A., J. Y. Zou, S. W. Cowen, and M. Kjeldgaard. 1991. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 28.Lazennec, C., and T. Meinnel. 1997. Formate dehydrogenase-coupled spectrophotometric assay of peptide deformylase. Anal. Biochem. 244:180-182. [DOI] [PubMed] [Google Scholar]
- 29.Leung, D., G. Abbenante, and D. P. Fairlie. 1999. Protease Inhibitors: current status and future prospects. J. Med. Chem. 43:305-341. [DOI] [PubMed] [Google Scholar]
- 30.Leviton, I. 1999. Inhibitors of protein synthesis. Cancer Investig. 17:87-92. [PubMed] [Google Scholar]
- 31.Lucchini, G., and R. Bianchetti. 1980. Initiation of protein synthesis in isolated mitochondria and chloroplasts. Biochim. Biophys. Acta 608:54-61. [DOI] [PubMed] [Google Scholar]
- 32.Maclean, D., J. R. Schullek, M. M. Murphy, Z. J. Ni, E. M. Gordon, and M. A. Gallop. 1997. Encoded combinatorial chemistry: synthesis and screening of a library of highly functionalized pyrrolidines. Proc. Natl. Acad. Sci. USA 94:2805-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margolis, P., C. Hackbarth, S. Lopez, M. Maniar, W. Wang, Z. Yuan, R. White, and J. Trias. 2001. Resistance of Streptococcus pneumoniae to deformylase inhibitors is due to mutations in defB. Antimicrob. Agents Chemother. 45:2432-2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margolis, P. S., C. J. Hackbarth, D. C. Young, W. Wang, D. Chen, Z. Yuan, R. White, and J. Trias. 2000. Peptide deformylase in Staphylococcus aureus: resistance to inhibition is mediated by mutations in the formyltransferase gene. Antimicrob. Agents Chemother. 44:1825-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazel, D., E. Coic, S. Blanchard, W. Saurin, and P. Marliere. 1997. A survey of polypeptide deformylase function throughout the eubacterial lineage. J. Mol. Biol. 266:939-949. [DOI] [PubMed] [Google Scholar]
- 36.Mazel, D., S. Pochet, and P. Marliere. 1994. Genetic characterization of polypeptide deformylase, a distinctive enzyme of eubacterial translation. EMBO J. 13:914-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meinnel, T., and S. Blanquet. 1993. Evidence that peptide deformylase and methionyl-tRNAfMet formyltransferase are encoded within the same operon in Escherichia coli. J. Bacteriol. 175:7737-7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meinnel, T., S. Blanquet, and F. Dardel. 1996. A new subclass of the zinc metalloproteases superfamily revealed by the solution structure of peptide deformylase. J. Mol. Biol. 262:375-386. [DOI] [PubMed] [Google Scholar]
- 39.Meinnel, T., C. Lazennec, and S. Blanquet. 1995. Mapping of the active site zinc ligands of peptide deformylase. J. Mol. Biol. 254:175-183. [DOI] [PubMed] [Google Scholar]
- 40.Meinnel, T., C. Lazennec, F. Dardel, J. M. Schmitter, and S. Blanquet. 1996. The C-terminal domain of peptide deformylase is disordered and dispensable for activity. FEBS Lett. 385:91-95. [DOI] [PubMed] [Google Scholar]
- 41.Meinnel, T., C. Lazennec, S. Villoing, and S. Blanquet. 1997. Structure-function relationships within the peptide deformylase family. Evidence for a conserved architecture of the active site involving three conserved motifs and a metal ion. J. Mol. Biol. 267:749-761. [DOI] [PubMed] [Google Scholar]
- 42.Meinnel, T., Y. Mechulam, and S. Blanquet. 1993. Methionine as translation start signal: a review of the enzymes of the pathway in Escherichia coli. Biochimie 75:1061-1075. [DOI] [PubMed] [Google Scholar]
- 43.Meinnel, T., L. Patiny, S. Ragusa, and S. Blanquet. 1999. Design and synthesis of substrate analogue inhibitors of peptide deformylase. Biochemistry 38:4287-4295. [DOI] [PubMed] [Google Scholar]
- 44.National Committee for Clinical Laboratory Standards. 2000. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 4th ed. Approved standard M7-A5. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 45.National Committee for Clinical Laboratory Standards. 1999. Methods for determining bactericidal activity of antimicrobial agents. Approved guideline M26-A. National Committee for Clinical Laboratory Standards, Wayne, Pa.
- 46.Navaza, J. 1994. AMoRe: an automated package for molecular replacement. Acta Crystallogr. Sect. A 50:157-163. [Google Scholar]
- 47.Obach, R. S. 1999. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 27:1350-1359. [PubMed] [Google Scholar]
- 48.Obach, R. S., J. G. Baxter, T. E. Liston, B. M. Silber, B. C. Jones, F. MacIntyre, D. J. Rance, and P. Wastall. 1997. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 283:46-58. [PubMed] [Google Scholar]
- 49.Okusu, H., D. Ma, and H. Nikaido. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 178:306-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otwinowski, Z., and W. Minor (ed.). 1997. Macromolecular crystallography, part A. Methods Enzymol. 276. [DOI] [PubMed]
- 51.Ragusa, S., S. Blanquet, and T. Meinnel. 1998. Control of peptide deformylase activity by metal cations. J. Mol. Biol. 280:515-523. [DOI] [PubMed] [Google Scholar]
- 52.Rajagopalan, P. T., A. Datta, and D. Pei. 1997. Purification, characterization, and inhibition of peptide deformylase from Escherichia coli. Biochemistry 36:13910-13918. [DOI] [PubMed] [Google Scholar]
- 53.Rajagopalan, P. T., and D. Pei. 1998. Oxygen-mediated inactivation of peptide deformylase. J. Biol. Chem. 273:22305-22310. [DOI] [PubMed] [Google Scholar]
- 54.Rane, A., G. R. Wilkinson, and D. G. Shand. 1977. Prediction of hepatic extraction ratio from in vitro measurement of intrinsic clearance. J. Pharmacol. Exp. Ther. 200:420-424. [PubMed] [Google Scholar]
- 55.Thorarensen, A., M. R. Douglas, Jr., D. C. Rohrer, A. F. Vosters, A. W. Yem, V. D. Marshall, J. C. Lynn, M. J. Bohanon, P. K. Tomich, G. E. Zurenko, M. T. Sweeney, R. M. Jensen, J. W. Nielsen, E. P. Seest, and L. A. Dolak. 2001. Identification of novel potent hydroxamic acid inhibitors of peptidyl deformylase and the importance of the hydroxamic acid functionality on inhibition. Bioorg. Med. Chem. Lett. 11:1355-1358. [DOI] [PubMed] [Google Scholar]
- 56.Weingarten, H., and J. Feder. 1985. Spectrophotometric assay for vertebrate collagenase. Anal. Biochem. 147:437-440. [DOI] [PubMed] [Google Scholar]
- 57.Yuan, Z., J. Trias, and R. J. White. 2001. Deformylase as a novel antibacterial target. Drug Discovery Today 6:954-961. [DOI] [PubMed] [Google Scholar]