


Abstract
Site-selective adenosine (A) to inosine (I) RNA editing by the ADAR enzymes has been found in a variety of metazoan from fly to human. Here we describe a method to detect novel site-selective A to I editing that can be used on various tissues as well as species. We have shown previously that there is a preference for ADAR2-binding to selectively edited sites over non-specific interactions with random sequences of double-stranded RNA. The method utilizes immunoprecipitation (IP) of intrinsic RNA–protein complexes to extract substrates subjected to site-selective editing in vivo, in combination with microarray analyses of the captured RNAs. We show that known single sites of A to I editing can be detected after IP using an antibody against the ADAR2 protein. The RNA substrates were verified by RT–PCR, RNase protection and microarray. Using this method it is possible to uniquely identify novel single sites of selective A to I editing.
INTRODUCTION
Adenosine to inosine (A to I) RNA editing is known to change the sequence of specific pre-mRNAs in metazoans from fly to human. ADAR2, a member of the ADAR (adenosine deaminase that acts on RNA) family, deaminates A to I selectively within double-stranded RNAs (dsRNAs) interrupted by bulges, mismatches or loops [reviewed in (1)]. ADAR editing with low selectivity can also occur on completely dsRNA. This is a type of hyper-editing that has been found within introns and untranslated regions (UTRs) of mRNAs, preferentially in repetitive Alu sequences (2–6). Only a few site-selective ADAR substrates have been detected. In mammals, most selectively edited sites targeted by ADARs have been found in pre-mRNAs expressed in the central nervous system. The most prominent sites of selective editing are in mRNA coding for several subunits of the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole) glutamate receptor (GluR). Editing of subunit B (GluR-B) results in altered receptor properties, changing receptor permeability to Ca2+ and the ability to recover after desensitization (7–9). In exon 11 the Q/R site is edited to nearly 100% giving rise to a codon change from glutamine (Q) to arginine (R). In exon 13 the edited R/G site causes an arginine (R) to glycine (G) codon change that is developmentally regulated. The dsRNA structure required for ADAR editing at these sites is formed by an inverted repeat located in the downstream intron [review by (10)]. Another prominent substrate for site-selective A to I editing is the transcript of the serotonin receptor 5-HT2C. Transcripts encoding the 2C receptor subtype undergo A to I editing at 5 sites: A, B, C′, C and D situated in close proximity to each other (11). Editing alters the coding potential of the second intracellular loop, reducing the efficiency of the interaction between the receptor and the G protein. Most of the selectively edited sites have been found fortuitously as A to G changes when comparing cDNA with genomic sequence, since inosine is seen as guanosine in the process of reverse transcription. However, a significant amount of inosine has been found within the poly(A) fraction of cellular RNA in mouse brain (12).
Co-immunoprecipitation is a powerful tool to precipitate-specific protein complexes. Further, it has been widely used to study RNA–protein interactions. One example is the identification of target RNA for the Nova protein in mouse brain using an ultraviolet cross-linking and immunoprecipitation assay (13). In another more general approach to identify mRNA–protein complexes (mRNPs) called ribonomics, RNA targets were detected using antibodies to RNA-binding proteins followed by genomic arrays (14).
We have shown previously that ADAR binds more preferentially to selectively edited sites than to random sequences of dsRNA (15). Moreover, ADAR2 was shown to bind with a similar affinity to an editing substrate as to the product (16). Based on this knowledge we have developed a method to find novel ADAR substrates by extracting intrinsic ADAR2–RNA substrate complexes from mouse brain by co-immunoprecipitations using an anti-ADAR2 antibody. The specificity of this method has been verified by the detection of known site-selectively edited substrates using RT–PCR, RNase protection and genomic microarray analyses. We present a powerful method with the potential to find novel sites of selective editing in different tissues and organisms.
MATERIALS AND METHODS
Isolation of RNA–protein complex from mouse brain
Three mouse brains were homogenized in HBSS [1× Hank's solution (HBSS GIBCO no. 14185-045)] and 1 M HEPES (pH 7.3) using a glass grinder. The suspension was washed in cold 1× HBSS and the pellet was frozen in liquid nitrogen. The pellet was resuspended in PXL [1× D-phosphate-buffered solution (PBS) (GIBCO no. 14200-67), 0.1% SDS, 0.5% deoxycholate and 0.5% NP-40] and ribonucleoside vanadyl complex (Sigma) on ice. The suspension was sonicated and treated with DNase I RQ1 (SIGMA). After centrifugation at 10 000 g for 20 min, 4°C, the supernatant was used for the immunoprecipitation (IP).
Immunoprecipitation of RNA–ADAR2 complexes
Anti-human ADAR2 antibody was made from recombinant histidine tagged human ADAR2 (hADAR2) protein, kindly provided by professor Brenda Bass' laboratory. The hADAR2 protein was concentrated using a centricon YM30 (Millipore) run out on 8% SDS–PAGE gel. The band corresponding to hADAR2 was excised and immunized four times into rabbits (Agrisera; Umeå Sweden). The serum was checked for immuno-reactivity and supplemented with 0.05% sodium azide.
To reduce non-specific binding prior to use in IPs the Sepharose A beads were incubated with tRNA (100 µg/ml) and BSA (100 µg/ml) in 1× PBS, washed once in 1× PBS and resuspended in 1 vol of 1× PBS and 0.05% NaN3. The cell lysis extract from one mouse brain was pre-cleared with 50 µl of Sepharose A stock for 30 min at 4°C with rotation. The pre-cleared lysate was incubated with anti-ADAR2 polyclonal antibody or pre-immune serum for 2 h at 4°C with rotation. The lysate-antibody was mixed with 50 µl of prepared Sepharose A stock and incubated for 1 h at 4°C with rotation. The bead–antibody-lysate complex was rinsed three times in wash buffer containing 1× PBS, MgCl2 (2 mM), EDTA (15 mM), NP-40 (1%) and Tween-20 (0.5%) including 1 protease Inhibitor Cocktail tablet/10 ml buffer (Roche) and rinsed once in 1× PBS, and eluted in 1× PBS plus 1% SDS at 65°C for 10 min.
Verification of ADAR2-binding using western blot
The IP eluate (10 µl) was boiled in SDS for 10 min prior to fractionation by electrophoresis on a 4–15% pre-made SDS–PAGE gel (BioRad) and transferred to a PVDF membrane by electroblotting. Anti-hADAR2 was used as primary antibody and anti-rabbit/HRP (DakoCytomation) was used as secondary antibody. The blots were developed using Amershams ECL plus Western Blotting Detection System and developed in a LAS 1000 system (Fujifilm).
Preparation of RNA after immunoprecipitation
The protein fraction was removed from the protein–RNA eluate after the IP by adding 1.8 mg of proteinase K (Roche) and incubated at 37°C for 15 min prior to a phenol/chloroform extraction and precipitation. The RNA was purified using RNeasy according to the manufacturer's instruction (Qiagen).
Microarray preparation
Preparation of labeled cRNA from the immunoprecipitated RNA was done according to Affymetrix Two-Cycle Target Labeling Assay. Labeled cRNA from nine mouse brains were hybridized to each Mouse Genome 430A 2.0 Array (Affymetrix). Scanning was performed after adding streptavidin-phycoerythrin Biotinylated anti-streptavidin antibody (SAPE) according to standard protocols Affymetrix Inc. (Santa Clara, CA).
Verification of known ADAR2 substrates using RT–PCR
The reverse transcription reactions were done with the Sensiscript RT kit (Qiagen) using hexanucleotide mix (Roche). A radioactive PCR using taq polymerase from Qiagen was performed for 25 cycles. Primers mGluRB-R/G-R (5′-GGGGAGTTCTATATTCTACGGC-3′), mGluRB-Q/R-R (5′-GACACCATGAATATCCACTTGAGACC-3′) and serotonin-R (5′-GGCCTTAGTCCGCGAATTGAACCGGC-3′) were radioactively labeled by T4 polykinase (invitrogen) using [γ-32P]ATP (NEN Perkin Elmer). The following non-radioactive primers were also used in the different PCRs: mGluRB-R/G-F (5′-CCCACATTTCTGGCCCTTGTGCC-3′), mGluRB-Q/R-F (5′-TTTGCCTACATTGGGGTCAGTG-3′) and serotonin-F (5′-GTCCATCATGCACCTCTGCG-3′). The result was shown on a native 5% PAGE gel. As negative controls the acidic ribosomal protein P0 (ARPP P0) and GluR-A were amplified using primers ARPP P0-F (5′-GCACTGGAAGTCCAACTACTTC-3′), ARPP P0-R (5′-TGAGGTCCTCCTTGGTGAACAC-3′), mGluRA-F (5′-CCAGAGCTGGTGCTGGTCAGCTCTCG-3′) and mGluRA-R (5′-GAAGTATATACGACCACTGTCATC-3′). All primers were labeled with [γ-32P]ATP as described above. For sequencing the R/G site, primer mGluRB-R/G-seq (5′-GGGCCAGTTCTCAAACTTCTCTGGCCCC-3′) was used.
Verification of known ADAR2 substrates using RNase protection
The RNase protection assay was done using Ribonuclease Protection Assay kit (RPA III no. 1414) from Ambion. Template RNA was immunoprecipitated from five mouse brains. To make the probe, the GluR-B was amplified by PCR using the mGluRB-R/G-F and mGluRB-R/G-R primers on genomic DNA from N2 cells, and the PCR product was ligated into the pGEM-T Easy vector (Promega). The vector (insert) was cut with HpaI (10 U, Invitrogen) and a uniformly labeled mGluRB-R/G probe was transcribed using SP6 RNA polymerase (30 U, invitrogen) in the buffer supplied by the provider in the presence of [α-32P]UTP (NEN Perkin Elmer). The 225 nt long radioactive probe (GTTAACTCTTTGTATTCCTATTTTGTTGTTTGTTTATTTTTTAGTGGAGTCACATTCAAGACACTGTATTTGTTTGTTGTGGATGTGAGTACATTGCCGTAGAATATAGAACTCCCCA) is complementary to 118 nt of the GluR situated 698 nt downstream of the R/G site. The probe was purified on a 8% PAGE plus 7 M Urea gel. The assay was performed according to the manufacturer's instructions (Ambion).
RESULTS
Specific enrichment of targets for site-selective editing
A method was developed to detect novel site-selective A to I editing in vivo (Figure 1). To identify ADAR2 associated mRNAs, cell lysate from mouse brain was incubated with anti-human ADAR2 polyclonal antibody. ADAR2–RNA complexes were pulled down using protein A–Sepharose beads. The co-purified pre-mRNAs were identified by probing of microarrays after removal of the proteins. ADAR proteins are known to bind tightly to dsRNA of any sequence (16,17,18). However, from previous studies we know that ADAR2 preferentially binds single sites of selective editing over a random sequence of completely dsRNA (15). This might be due to a higher affinity to site-selectively edited substrates. We therefore hypothesized that this method would specifically enrich RNA transcripts subjected to single sites of selective editing.
Figure 1.

Illustration of the IP-array method to find novel substrates for A to I editing. Cell lysis extract was prepared from mouse brain. The extract was immunoprecipitated using an ADAR2-specific polyclonal antibody. Target RNA was extracted from the mRNP complexes upon protein removal. The RNA was amplified, labeled and further hybridized to a mouse genomic oligo array.
ADAR2 co-immunoprecipitation using mouse brain
Using this method it is important to retain intact RNA–protein complexes. Therefore, the cell extracts were treated with a ribonucleoside vanadyl complex to prevent RNA degradation prior to being used as load in the IP. DNA was also removed before further extractions to minimize non-specific background. The specificity of the RNA–protein interaction was optimized by washing the immunoprecipitate three times in 1× PBS, MgCl2 (2 mM), EDTA (15 mM), NP-40 (1%) and Tween-20 (0.5%) in presence of protease inhibitor. After SDS treatment the specificity of the IP for ADAR2 was determined by western blot (Figure 2). An enrichment of ADAR2 was seen when the anti-ADAR2 antibody was used in the IP compared with precipitation using pre-immune serum.
Figure 2.
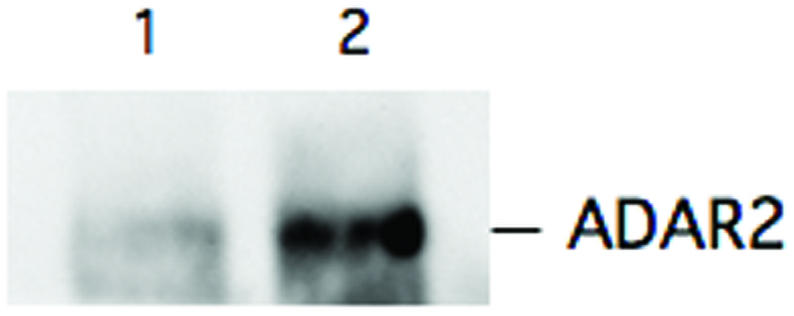
Western blot analysis using anti-human ADAR2 antibody on IP eluates. Three mouse brains were used for ADAR2-specific and pre-immune serum IP, respectively. One-tenth of the IP eluate was used for western blot. Lane 1 represents the pre-immune serum IP and lane 2 shows the amount of ADAR2 in the ADAR2-specific IP.
Specific amplification of known A to I editing substrates
GluR-B is a transcript that is A to I edited site-selectively at two sites (Q/R and R/G) within the coding sequence [reviewed in (10)]. Although some other receptor subunits are subjected to editing, no editing has been detected in the subunit A (GluR-A) transcript. Another well-known substrate for A to I editing is the transcript of the serotonin receptor 5-HT2C. This transcript has been shown to be site-selectively edited at several sites (A, B, C, C′ and D) (11). The specificity of the IP for these known RNA targets was analyzed by semi-quantitative RT–PCR (Figure 3a and b). An enrichment of target substrates was observed in the ADAR2 IP when primers specific for the edited sites in the GluR-B and 5-HT2C transcripts were used (Figure 3a). The pre-immune IP did not show an enrichment of target RNA. When primers specific for GluR-A were used for amplification no product could be detected during the 25 cycles of PCR considered to give a semi-quantitative product (Figure 3a). During an extended PCR to 30 cycles a product of equal amount could be detected in the ADAR2 and pre-immune IP (data not shown). As an additional negative control primers specific for the mRNA of the ribosomal phosphoprotein P0 that is not edited were used for amplification. No enrichment of this product could be detected as the level of transcripts appears to be equal in the ADAR2 and pre-immune IP elutes (Figure 3b). Editing at the Q/R and R/G sites of the target RNAs was verified by sequencing a population from RT–PCR (Figure 3c). Although a mixed population of edited and non-edited products was seen at the R/G site the Q/R site was edited to 100%. These data are in line with previous results showing the extent of GluR-B editing in the mammalian brain [reviewed in (10)]. From the sequencing analysis we can also verify that both pre-mRNA and mRNA of the GluR-B transcript is present in the specific IP. The GluR-B R/G site was amplified using primers specific for the pre-mRNA while the Q/R site was amplified from primers situated in the exons, giving a product from the spliced mRNA. The specificity for the GluR-B transcript in the ADAR2 IP was also verified by an RNase protection assay detecting an RNA from the ADAR2 IP but not from pre-immune IP (Figure 3d). Our data confirm that an RNA that is edited can be specifically enriched from a mammalian brain tissue using an anti-ADAR2 antibody in IPs.
Figure 3.

Detection of known substrates for A to I editing using ADAR2-specific IP. (a) Semi-quantitative RT–PCR on GluR-B at the R/G and Q/R site, GluR-A and the serotonin receptor (5-HT2C) using radioactively labeled primers. Lane 1 shows the amplification of the R/G site from an ADAR2-specific IP, with an estimated size of 314 bp. Lane 2 shows a product amplified from the R/G site from an IP using pre-immune serum. Lane 3 shows the amplification of the Q/R site from an ADAR2-specific IP, with an estimated size of 253 bp. Lane 4 shows product amplification of the Q/R site from an IP using pre-immune serum. RT–PCR on GluR-A, lacking sites for A to I editing, shows no detectable amplification from an ADAR2-specific IP (lane 5), or a pre-immune serum IP (lane 6). Product using total RNA is shown in lane 7 and the estimated size is 203 bp. RT–PCR on 5-HT2C shows the amplification from an ADAR2-specific IP (lane 8), the estimated size is 94 bp. Lane 9 shows a product amplified from the 5-HT2C transcript from an IP using pre-immune serum. Lane M is a size marker with bands of sizes as indicated. (b) RT–PCR on the ribosomal phosphoprotein P0, lacking sites for A to I editing. No enrichment could be detected in the ADAR2 IP (lane 1) compared with the pre-immune serum IP (lane 2). The estimated size is 265 bp. Lane M is a size marker with bands of sizes as indicated. (c) The product from the RT–PCR-specific for the R/G and the Q/R sites were DNA sequenced to determine the editing efficiency. At the R/G site a forward primer was used to give a dual A and G peak at the R/G site. At the Q/R site a reverse primer was used so that an edited site is a C in the sequence. Edited nucleotides are indicated with an arrow in the chromatogram. (d) An RNase protection assay was used to confirm the enrichment of GluR-B in the presence of anti-ADAR2 antibody. A 225 nt long α-32P-labeled probe, 118 nt complementary downstream of the R/G site, was hybridized to RNA from an IP using pre-immune serum (lane 1) and to RNA from an IP using anti-ADAR2 antibody (lane 2).
Detection of A to I editing targets using microarray
After protein removal from the IP using proteinase K treatment and phenol/chloroform extraction the RNA was amplified and labeled according to Two-Cycle Target Labeling assay (Affymetrix). The cRNA was hybridized to a mouse genome array 430A 2.0 (Affymetrix) to detect enriched ADAR2–RNA targets compared with IPs using pre-immune serum. Three arrays from three independent target extractions were done. The diagram in Figure 4 illustrates the extent of enrichment of the 200 genes that are significantly enriched in all three arrays. The GluR-B transcript was significantly amplified in the three arrays and is indicated in red. Other known A to I substrates enriched in the specific IPs are specified in Table 1. Although present, the GluR-A transcript did not show an increase in the microarray (Table 1). This is in agreement with the presented data from RT–PCR and RNase protection. These results indicate that the method specifically amplifies known selectively edited targets that can be detected by microarray.
Figure 4.

Genes enriched in ADAR2 IP compared with pre-immune serum IP. 200 genes were significantly increased in all three different arrays. The mean value for the three arrays is shown as fold increase 2x. The GluR-B, marked in red, is ranked 25 of the 200 enriched genes.
Table 1.
Enriched known editing targets and non-edited transcripts verified by microarray
| Transcript | Mean (2x-fold increase) | SD |
|---|---|---|
| Known editing targets | ||
| GluR-B | 1.37 | 0.23 |
| 5-HT2C | 1.03 | 1.63 |
| Ednrb | 0.97 | 1.02 |
| Igfbp | 0.50 | 0.10 |
| Blcap | 0.33 | 0.12 |
| ADAR2 | 0.33 | 1.20 |
| Non-edited transcripts | ||
| GluR-A | −0.17 | 0.11 |
| ARPP P0 | −0.50 | 0.20 |
The following abbreviations are used: GluR-B, Glutamate receptor subunit B; 5-HT2C, serotonin receptor subtype 2C; Ednrb, Endothelin receptor type B; Igfbp, insulin-like growth factor-binding protein 2; Blcap, bladder cancer associated protein; ADAR2, adenosine deaminase that acts on RNA 2; GluR-A, Glutamate receptor subunit A; ARPP P0, acidic ribosomal phophoprotein P0.
DISCUSSION
During the past decade several methods have been developed to find new ADAR substrates. By computational analysis a vast amount of edited sites have been detected in 5′- and 3′-UTRs within Alu repetitive elements that are hyper-edited at multiple sites, but very few sites were found in coding sequences (4–6). Although editing of Alu repeats might be important, no function has so far been proposed.
We have developed a method to detect single sites of A to I editing in vivo and have chosen mouse brain in our initial experiments. The mouse genome contains fewer repetitive elements than the human genome and lacks the Alu repeats. By choosing ADAR2 and mouse material we can focus on single sites of editing in coding sequences, with the potential of creating alternative isoforms of the protein. Mouse is therefore a good model organism to avoid extensive A to I hyper-editing of non-coding sequence.
Most dsRNA-binding proteins (dsRBPs) interact with the RNA by sequence-specific structural features rather than base-specific interactions [reviewed in (19)]. A dsRNA-binding motif makes at least two structure-specific interactions with the RNA double-helix. These interactions have been proven to occur in a sequence-independent manner (20,21). However, it has been proposed by us and others that the mismatch opposing the R/G site in GluR-B serves as a structural feature in concert with the neighboring nucleotides to direct site-selective editing (18,22). Further, studies on other dsRBPs indicate that there are regions in the RNA-binding motif that interact with RNA loop structures in the vicinity of the helical structure (23–26). These results are in line with our previous result indicating that the ADAR2 enzyme discriminate between a completely dsRNA structure and a selectively edited substrate interrupted by bulges and loops, possibly with a slower off rate on the latter sites (15). To minimize the background binding to dsRNA we exclude any form of cross-linking between RNA and protein prior to the IP.
Using our approach we can collect potential ADAR substrates in vivo and enrich for selectively edited sites. Using microarray analysis as the method to detect potential targets allows us to tolerate a certain amount of background but also to detect products of relatively low abundance since the material is amplified prior to the array. However it should be noted that the microarray is limited in its detection of enriched transcripts. Table 1 shows the enrichment of known edited substrates. Most of the known transcripts subjected to A to I editing show a significant enrichment in the microarray after the ADAR2-specific IP. However, in order to get a better statistical value on the microarray an increased number of independent array analyses are required. The enrichment of edited substrates in the specific IP was verified by semi-quantitative RT–PCR on a selective set of RNAs using primers specific for the GluR-B R/G site, the GluR-B Q/R site and the A–D sites in the 5-HT2C transcript. Using this technique we could detect an enrichment of RNA containing all of these sites but not for GluR-A and ARPP P0 transcripts that are not edited. We are therefore confident that edited substrates indeed are enriched in the specific IP. From sequencing analysis of the PCR products from the amplified edited transcripts we can detect both pre-mRNA and mRNA. Detection of spliced transcripts indicate that splicing has occurred subsequent to binding during the IP. Since ADAR2 has been shown to bind to the inosine containing product with almost the same affinity as to the substrate (16), we expect that edited as well as non-edited A to I substrates are extracted using this assay. Approximately 200 genes showed a significant increase in all three arrays compared with microarrays based on RNA form an IP using pre-immune serum (Figure 4). We are using computational analysis to identify the position of editing sites in the candidate genes. When a computational search on an entire genome is used as the sole method to identify A to I editing it is hard to detect single sites of selective editing in the background of single nucleotide polymorphisms, sequencing errors and mis-alignments. Since we utilize the candidates identified in the experimental setup as the input the computational search can be more general. Three main criteria are used to get a high score on editing probability: (i) A/G mismatches between genomic and cDNA sequence, (ii) phylogenetic conservation of the A/G mismatch between mammals and (iii) inverted repeats with acceptance of mismatches and internal loops (M. Ensterö, B.-M. Sjöberg and M. Öhman, manuscript in preparation). Each criterium is scored individually and high score candidates are verified experimentally. This unique combination of experimental and bioinformatical analysis has the potential to detect novel sites of selective editing that have previously been foreseen using the methods separately. We have detected several new candidates of A to I editing substrates in mouse brain using this strategy (J. Ohlson, M. Ensterö, B.-M. Sjöberg and M. Öhman, manuscript in preparation).
Our approach has numerous applications, it can be used to find novel editing substrates in different tissues as well as to identify editing discrepancies between different species. It is also possible to apply this method on other ADAR protein family members like ADAR1 but also ADAR3, so far without known targets, as well as on other dsRBPs. A to I editing is an essential event for normal brain function (27). Several diseases with altered brain functions have been shown to have an effect on specific sites of editing (28,29). Our method has a potential to give a more general overview of the editing events in a normal brain compared with a diseased one.
Acknowledgments
We thank Lars Wieslander for helpful discussions and comments on the manuscript. We are grateful to Ann-Kristin Östlund Farrants and Patrick Asp and the Wennergren Institute, WGI, Stockholm University for technical assistance. We also thank the Affymetrix core facility at the Karolinska Institute, Novum. This work was supported by grants from Wallenberg consortium North. Funding to pay the Open Access publication charges for this article was provided by Wallenberg consortium North.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bass B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morse D.P., Bass B.L. Long RNA hairpins that contain inosine are present in Caenorhabditis elegans poly(A)+ RNA. Proc. Natl Acad. Sci. USA. 1999;96:6048–6053. doi: 10.1073/pnas.96.11.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morse D.P., Aruscavage P.J., Bass B.L. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proc. Natl Acad. Sci. USA. 2002;99:7906–7911. doi: 10.1073/pnas.112704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levanon E.Y., Eisenberg E., Yelin R., Nemzer S., Hallegger M., Shemesh R., Fligelman Z.Y., Shoshan A., Pollock S.R., Sztybel D., et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 5.Blow M., Futreal P.A., Wooster R., Stratton M.R. A survey of RNA editing in human brain. Genome Res. 2004;14:2379–2387. doi: 10.1101/gr.2951204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Athanasiadis A., Rich A., Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hume R.I., Dingledine R., Heinemann S.F. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- 8.Burnashev N., Monyer H., Seeburg P.H., Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 9.Lomeli H., Mosbacher J., Melcher T., Hoger T., Geiger J.R., Kuner T., Monyer H., Higuchi M., Bach A., Seeburg P.H. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 10.Seeburg P.H., Higuchi M., Sprengel R. RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res. Brain. Res. Rev. 1998;26:217–229. doi: 10.1016/s0165-0173(97)00062-3. [DOI] [PubMed] [Google Scholar]
- 11.Burns C.M., Chu H., Rueter S.M., Hutchinson L.K., Canton H., Sanders-Bush E., Emeson R.B. Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature. 1997;387:303–308. doi: 10.1038/387303a0. [DOI] [PubMed] [Google Scholar]
- 12.Paul M.S., Bass B.L. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 1998;17:1120–1127. doi: 10.1093/emboj/17.4.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ule J., Jensen K.B., Ruggiu M., Mele A., Ule A., Darnell R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 14.Tenenbaum S.A., Lager P.J., Carson C.C., Keene J.D. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods. 2002;26:191–198. doi: 10.1016/S1046-2023(02)00022-1. [DOI] [PubMed] [Google Scholar]
- 15.Klaue Y., Källman A.M., Bonin M., Nellen W., Öhman M. Biochemical analysis and scanning force microscopy reveal productive and non-productive ADAR2 binding to RNA substrates. RNA. 2003;9:839–846. doi: 10.1261/rna.2167603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Öhman M., Källman A.M., Bass B.L. In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA. 2000;6:687–697. doi: 10.1017/s1355838200000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai F., Drakas R., Nishikura K. Mutagenic analysis of double-stranded RNA adenosine deaminase, a candidate enzyme for RNA editing of glutamate-gated ion channel transcripts. J. Biol. Chem. 1995;270:17098–17105. doi: 10.1074/jbc.270.29.17098. [DOI] [PubMed] [Google Scholar]
- 18.Stephens O.M., Yi-Brunozzi H.Y., Beal P.A. Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry. 2000;39:12243–12251. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 19.Fierro-Monti I., Mathews M.B. Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem. Sci. 2000;25:241–246. doi: 10.1016/s0968-0004(00)01580-2. [DOI] [PubMed] [Google Scholar]
- 20.Nanduri S., Carpick B.W., Yang Y., Williams B.R., Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryter J.M., Schultz S.C. Molecular basis of double-stranded RNA–protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Källman A.M., Sahlin M., Öhman M. ADAR2 A→I editing: site selectivity and editing efficiency are separate events. Nucleic Acids Res. 2003;31:4874–4881. doi: 10.1093/nar/gkg681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chanfreau G., Buckle M., Jacquier A. Recognition of a conserved class of RNA tetraloops by Saccharomyces cerevisiae RNase III. Proc. Natl Acad. Sci. USA. 2000;97:3142–3147. doi: 10.1073/pnas.070043997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramos A., Grunert S., Adams J., Micklem D.R., Proctor M.R., Freund S., Bycroft M., St Johnston D., Varani G. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 2000;19:997–1009. doi: 10.1093/emboj/19.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagel R., Ares M., Jr Substrate recognition by a eukaryotic RNase III: the double-stranded RNA-binding domain of Rnt1p selectively binds RNA containing a 5′-AGNN-3′ tetraloop. RNA. 2000;6:1142–1156. doi: 10.1017/s1355838200000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leulliot N., Quevillon-Cheruel S., Graille M., Van Tilbeurgh H., Leeper T.C., Godin K.S., Edwards T.E., Sigurdsson S.T., Rozenkrants N., Nagel R.J., et al. A new alpha-helical extension promotes RNA binding by the dsRBD of Rnt1p RNAse III. EMBO J. 2004;23:2468–2477. doi: 10.1038/sj.emboj.7600260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higuchi M., Maas S., Single F.N., Hartner J., Rozov A., Burnashev N., Feldmeyer D., Sprengel R., Seeburg P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 28.Akbarian S., Smith M.A., Jones E.G. Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer's disease, Huntington's disease and schizophrenia. Brain Res. 1995;699:297–304. doi: 10.1016/0006-8993(95)00922-d. [DOI] [PubMed] [Google Scholar]
- 29.Sodhi M.S., Burnet P.W., Makoff A.J., Kerwin R.W., Harrison P.J. RNA editing of the 5-HT(2C) receptor is reduced in schizophrenia. Mol. Psychiatry. 2001;6:373–379. doi: 10.1038/sj.mp.4000920. [DOI] [PubMed] [Google Scholar]