

Abstract
Mental retardation (MR) occurs in 2%–3% of the general population. Conventional karyotyping has a resolution of 5–10 million bases and detects chromosomal alterations in ∼5% of individuals with unexplained MR. The frequency of smaller submicroscopic chromosomal alterations in these patients is unknown. Novel molecular karyotyping methods, such as array-based comparative genomic hybridization (array CGH), can detect submicroscopic chromosome alterations at a resolution of 100 kb. In this study, 100 patients with unexplained MR were analyzed using array CGH for DNA copy-number changes by use of a novel tiling-resolution genomewide microarray containing 32,447 bacterial artificial clones. Alterations were validated by fluorescence in situ hybridization and/or multiplex ligation-dependent probe amplification, and parents were tested to determine de novo occurrence. Reproducible DNA copy-number changes were present in 97% of patients. The majority of these alterations were inherited from phenotypically normal parents, which reflects normal large-scale copy-number variation. In 10% of the patients, de novo alterations considered to be clinically relevant were found: seven deletions and three duplications. These alterations varied in size from 540 kb to 12 Mb and were scattered throughout the genome. Our results indicate that the diagnostic yield of this approach in the general population of patients with MR is at least twice as high as that of standard GTG-banded karyotyping.
Introduction
Mental retardation (MR) occurs in 2%–3% of newborns in the general population, but, in most cases, its cause has remained elusive (Roeleveld et al. 1997; Yeargin-Allsopp et al. 1997). Establishing the cause in a mentally retarded individual improves clinical management and facilitates genetic counseling of the family. Chromosome abnormalities are detectable by microscopic analysis of chromosomes isolated from peripheral blood lymphocytes in ∼5% of patients with unexplained MR (Phelan et al. 1996; de Vries et al. 1997; Schinzel 2001). The resolution of microscopic techniques for detection of abnormal karyotypes is typically in the range of 5–10 Mb. Molecular cytogenetic techniques, such as FISH and multiplex ligation-dependent probe amplification (MLPA) (Schouten et al. 2002), have shown that causative submicroscopic rearrangements of the subtelomeric regions can be found in ∼5% of patients with human malformations and MR (Flint et al. 1995; Knight et al. 1999; De Vries et al. 2003; Koolen et al. 2004a). These results for the subtelomeric regions indicate that submicroscopic rearrangements in general may be a more common cause of MR than microscopically visible rearrangements.
Recent technological developments allow the investigation of the human genome at a resolution that is ∼50–100 times higher than that of routine chromosome analysis by karyotyping (Pinkel et al. 1998; Lucito et al. 2003; Ishkanian et al. 2004; Ried 2004). Such DNA-based methods are collectively referred to as “molecular karyotyping.” One such method is array-based comparative genomic hybridization (array CGH) (Solinas-Toldo et al. 1997; Pinkel et al. 1998; Veltman et al. 2002). Pilot studies using intermediate resolution (1-Mb) array CGH have indicated the potential of this technology in diagnosing patients with MR and congenital anomalies (Vissers et al. 2003; Shaw-Smith et al. 2004). Other recent studies have reported the presence of large-scale copy-number variations (LCVs) in healthy individuals (Iafrate et al. 2004; Sebat et al. 2004), which could complicate the clinical interpretation of these genomic alterations.
The goal of the underlying study was to estimate the significance of this whole-genome approach for the general population of patients with MR. Therefore, a recently published, 32,447-clone BAC array, with complete coverage of the entire human genome (Ishkanian et al. 2004), was used for the first time to test 100 mentally retarded patients, with or without additional congenital malformations, for the presence of submicroscopic chromosomal alterations. Novel algorithms were developed and validated for microarray normalization and copy-number detection, and parental samples were used as a control population to distinguish between causative de novo alterations and inherited LCVs. A clinical evaluation of the patient sample used in this study, in comparison with that of the overall patient population seen in our clinical genetics center, allowed us to estimate the importance of this approach in medical practice.
Patients and Methods
Patients
The diagnostic value of the genomewide tiling-resolution array CGH approach was tested by screening 100 patients with unexplained MR (60 males and 40 females; age range 10 mo to 63 years) (table 1). All had normal GTG-banded chromosomes, and all were previously subjected to a specific subtelomeric screen using MLPA, with normal results (Koolen et al. 2004a). All patients were evaluated for additional clinical features by a board-certified clinical geneticist. A clinical scoring system based on the presence of family history of MR, prenatal-onset growth retardation, postnatal growth retardation or advanced growth, facial dysmorphic features, and other congenital abnormalities was applied, with a score from 0 to 10 (table 2) (Baralle 2001; de Vries et al. 2001).
Table 1.
Clinical Characteristics of the 100 Patients with MR
| Characteristic | Valuea |
| Sex: | |
| Male | 60 |
| Female | 40 |
| Age group: | |
| <10 years old | 67 |
| 10–20 years old | 23 |
| >20 years old | 10 |
| Age (years): | |
| Median | 7 |
| Range | 1–63 |
| Level of MR: | |
| Mild | 32 |
| Moderate | 27 |
| Severe | 32 |
| Not assessable | 9 |
| Family history of MR: | |
| Yes: | |
| Compatible with Mendelian inheritance | 13 |
| Incompatible with Mendelian inheritance | 4 |
| No | 83 |
| Prenatal-onset growth retardation: | |
| Yes | 18 |
| No | 82 |
| Postnatal growth abnormalities: | |
| Yes: | |
| Head circumference or stature | 42 |
| Head circumference and stature | 14 |
| No | 44 |
| Facial dysmorphic features: | |
| Yes: | |
| 1 Feature | 19 |
| ⩾2 Features | 73 |
| No | 8 |
| Nonfacial dysmorphism and congenital abnormalities: | |
| Yes: | |
| 1 Abnormality | 35 |
| ⩾2 Abnormalities | 18 |
| No | 47 |
| Total clinical score: | |
| 0 | 0 |
| 1 | 5 |
| 2 | 16 |
| 3 | 32 |
| 4 | 21 |
| 5 | 14 |
| 6 | 7 |
| 7 | 5 |
| 8–10 | 0 |
Data are no. of patients, unless indicated otherwise.
Table 2.
Clinical Checklist
| Trait (Points) | Score |
| Family history of MR: | 1–2 |
| Compatible with Mendelian inheritance (1) | … |
| Incompatible with Mendelian inheritancea (2) | … |
| Prenatal-onset growth retardation | 2 |
| Postnatal growth abnormalitiesb: | 1–2 |
| Microcephaly (1) | … |
| Short stature (1) | … |
| Macrocephaly (1) | … |
| Tall stature (1) | … |
| ⩾2 Facial dysmorphic featuresc | 2 |
| Nonfacial dysmorphism and congenital abnormalitiesd: | 1–2 |
Including discordant phenotypes.
For each abnormality listed, assign 1 point, with a maximum score of 2 points.
Notably, hypertelorism, nasal anomalies, and ear anomalies.
Notably, hand anomaly, heart anomaly, hypospadias with/without undescended testis; for each, assign 1 point, with a maximum score of 2 points.
Genomic DNA was isolated from blood leukocytes by use of routine procedures. For this study, two pools of reference DNA were made—one containing equal amounts of genomic DNA from 10 healthy male blood donors and one containing equal amounts of genomic DNA from 10 healthy female blood donors. Blood was collected for DNA isolation from a total of 72 parents for further analysis. This study was approved by the Medical Ethics Committee of the Radboud University Nijmegen Medical Centre.
Microarray Preparation
We prepared a tiling-resolution microarray consisting of 32,447 overlapping BAC clones, which were selected to cover the entire human genome (Ishkanian et al. 2004; Krzywinski et al. 2004) and are available at the BACPAC Resources Center Web site, using methodology essentially as described elsewhere (Veltman et al. 2004). In brief, genomic target DNAs were isolated from 1-ml bacterial cultures by use of an AutogenPrep 960 (Autogen), in accordance with the instructions of the manufacturer. Degenerate oligonucleotide-primed PCR (DOP-PCR) was performed on 50 ng of DNA from all clones, as described elsewhere (Telenius et al. 1992), with minor modifications (Veltman et al. 2002). DOP-PCR products were dissolved at a concentration of 1 μg/μl in a 30% dimethyl-sulfoxide solution and were spotted onto CMT-ultragaps coated glass slides (Corning) by use of an Omnigrid 100 arrayer (Genomic Solutions). All 32,447 clones were printed once on a single microarray slide.
Isolation of DNA and Microarray-Based Genome Profiling
Isolation of genomic DNA, DNA labeling, hybridization of labeled DNA to the 32,447-BAC array, and spot identification were performed as described elsewhere (Vissers et al. 2003). In brief, genomic DNA from patients and controls was isolated in accordance with standard procedures and was purified using a QIAamp kit (QIAgen), in accordance with the instructions of the manufacturer. Next, 500 ng of genomic DNA from each patient was labeled by random priming with Cy3-dUTP and Cy5-dUTP (Amersham Biosciences) and was hybridized in duplicate with dye-swap against the sex-mismatched reference pool. Parental samples were hybridized once against the same reference pool as their child. Test and reference samples were mixed with 120 μg of human Cot-1 DNA (Roche), were coprecipitated, and were resuspended in 120 μl of a hybridization solution containing 50% formamide, 10% dextran sulfate, 2 × saline sodium citrate (SSC), 4% SDS, and 10 μg/μl of yeast tRNA (Invitrogen). Hybridization and posthybridization washing procedures were performed using a GeneTac Hybridization Station (Genomic Solutions). An 18-h hybridization at 37°C with active circulation of the probe was performed, followed by five posthybridization wash cycles in 50% formamide and 2 × SSC at 45°C and five wash cycles in phosphate buffer at 20°C. Slides were dried by centrifugation and were scanned using a GenePix Autoloader 4200AL laser scanner (Axon Instruments). Spot identification and two-color fluorescence intensity measurements were obtained using the GenePix 5.0 software, and all data were entered into a database for subsequent analysis.
Data Analysis
The log2-transformed test:reference ratio for each clone was normalized by subtracting its local mean log2 test:reference ratio obtained by a weighted median filter (Yin and Yang 1996). This filter averages the log ratios of autosomal clones weighted by a Gaussian distribution with its mean at the spot to be normalized and an SD of 1 physical mm on the array. The normalized ratios were analyzed for loss and gain regions by a standard hidden Markov model (HMM) (Rabiner 1989). This HMM has three hidden states (si) for each clone: normal, loss, and gain. The probability of finding the HMM in a certain state, given normalized log ratios xi, is given by
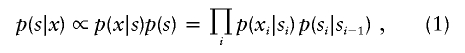 |
where the transition probability is given by
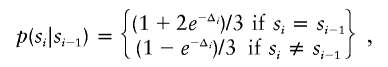 |
where Δi is the distance on the chromosome between the adjacent clones i and i-1. This equation has the following properties: (1) clones with identical chromosomal positions have identical states; (2) clones at an infinite distance from each other (e.g., located on different chromosomes) are noncorrelated; and (3) the transition probability for a distance 2Δi is the square of that for a distance Δi. In this study, we used units of 150 Mb to measure the distances Δi. The observed log ratio xi is assumed to be Gaussian distributed, with SD σ and a mean that depends on its hidden state si, as follows:
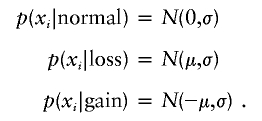 |
The sex-mismatched hybridizations allowed us to use the median of the log ratios of clones mapped to the chromosome X as an internal estimate of a single-copy gain or loss for each experiment and allowed a quick objective general assessment of the quality of individual experiments. For this reason, the sex chromosomes were excluded from further analysis.
The SD of the log ratios of all autosomal clones was used as an estimate for σ. For duplicate experiments, the means and SDs were computed per array, and the probability p(x1i,x2i|si) is the product of the two individual Gaussian distributions. To find the hidden state that best matches the observations, we used a standard Viterbi algorithm that optimizes equation (1) (Forney 1973). This results in a path in which we find regions of consecutive clones that are in the hidden gain or loss state. To quantify the likelihood of a copy-number gain or loss in a certain region, indexed by n, we compared the optimal log likelihood of the Viterbi path, logp*, with the log-likelihood in the absence of the region, logpn. The “certainty” that a region contains a real copy-number change was defined as
The experimental threshold for this certainty was set at 1.5 for the experiments done in duplicate. To exclude individual false-positive results, we furthermore required that a region consist of at least three clone positions. This HMM method was validated by randomly permuting all autosomal data and rerunning the algorithm on the entire set. For single-slide experiments (parental samples), this analysis resulted in the detection of eight aberrations, whereas in the experiments in duplicate (patient samples), only two small aneusomic segments were identified. By randomly permuting all data, we created a database of microarray data for which no spatial correction is expected. Any aberration found can, therefore, be interpreted as a false positive. By doing so, a valid estimate of the expected number of false positives in each experiment is obtained. In addition, an experiment in duplicate was performed in which the male reference pool was hybridized against the female reference pool. Again, the HMM was run, and no significant regions with a copy-number gain or loss were found. From this, we conclude that the reference pools themselves did not contain pronounced gains or losses. From all these analyses, we concluded that the criteria are highly conservative and that, especially for the experiments in duplicate, almost all regions that meet the criteria are excellent candidates for being a true copy-number alteration.
Confirmation Experiments
De novo chromosomal alterations were validated in the individual patients by at least one other method: MLPA (Schouten et al. 2002), with specifically designed synthetic probe sets, and/or FISH. It was similarly confirmed that parental samples contained a normal copy number for these chromosomal regions. For MLPA, we developed one or four uniquely sized probes for each region to be tested, in accordance with a protocol provided by MRC-Holland (all reagents for MLPA reaction and subsequent PCR amplification were purchased from MRC-Holland). Ten probes were combined in one MLPA assay, in combination with four standard control probes. MLPA analysis was performed as described elsewhere, with minor modifications (Koolen et al. 2004a). In brief, 200–400 ng of DNA in a final volume of 8 μl was heated for 5 min at 98°C. Equal amounts of probe mix and SALSA hybridization buffer were added to each sample, followed by heat denaturation for 1 min at 97°C and overnight incubation at 60°C. Next, Ligation-65 mix was added and was incubated for 15 min at 54°C. After heat inactivation, the ligation products were amplified by PCR with the use of the 6-FAM–labeled primer set. Amplification products were identified and quantified by capillary electrophoresis on an ABI 3730 genetic analyzer, using GeneMapper software (Applied Biosystems). Data were normalized by dividing each probe’s signal strength by the average signal strength of the sample. This normalized peak pattern was divided by the average peak pattern of all samples in the same experiment. Values for control wild-type peaks (n=1,667) were centered at 1.0, with an SD of 0.06. The mean ± 5 SD was used as the threshold for copy-number changes (1.3 for gains and 0.7 for losses).
In addition, each de novo DNA copy-number alteration identified by array CGH was validated by FISH analysis on fixed metaphase spreads from the respective patient and from his or her parents. In each region of significant DNA copy-number alteration, four clones—also present on the array—were selected as probes in the FISH validation assay. In brief, BAC DNAs were amplified by DOP-PCR and were labeled with biotin by use of random priming. Subsequent hybridizations were performed overnight at 37°C on an OmniSlide Thermal Cycler System (Thermo Hybaid; Thermo Life Sciences), in accordance with standard protocols. Detection of the biotin-labeled probes was achieved using either streptavidin-Cy3 (Amersham Biosciences) or avidin-FITC followed by goat-a-avidin-FITC (Vector Laboratories). The FISH slides were analyzed using a Leica DM RA fluorescence microscope.
Results
Identification of Chromosomal Alterations by Genomewide Tiling-Resolution Array CGH
Our sample set consisted of 100 patients with unexplained MR, all with normal chromosomes, as determined by routine karyotyping, and with normal subtelomeres, as determined by MLPA. DNA from each patient was labeled and hybridized in duplicate (with label swap) against a sex-mismatched reference pool onto microarrays containing 32,447 human BAC clones, which completely covered the entire human genome. For each patient, fluorescence ratios of the scanned images were quantified and normalized. The two experiments for each patient were merged, clones were ordered according to their position on the human genome sequence, and copy-number states (normal, gain, or loss) were assigned to each clone by use of a validated HMM (see the “Patients and Methods” section). In figure 1, examples are shown of genome profiles obtained using the tiling-resolution BAC arrays. A total of 274 DNA copy-number alterations were identified in the 100 patients, with a range from 0 to 9 alterations per patient (fig. 2). Only three patients in this series did not show any copy-number alterations. Aneusomic segments varied in size from 50 kb to 12 Mb and were covered by 3–118 adjacently mapped clones present on the microarrays.
Figure 1.

Genomic profiles obtained by tiling-resolution array CGH for patients with MR. The arrays contained 32,447 human BAC clones (indicated by small circles representing the log2-transformed and normalized test:reference intensity ratios [“2Log(T/R)”]), ordered from 1pter to Yqter in the two genome profiles and from pter to qter for individual chromosomes, on the basis of physical-mapping positions obtained from the UCSC Genome Browser (May 2004 freeze). A, Examples of de novo alterations, including deletions and duplications, in patients 1, 4, 5, 6, 8, and 9. B, Inherited copy-number variations in patients 117 and 498, including the matching chromosome profiles of one of their parents. The individual chromosome plots show small horizontal lines to indicate the presence of a copy-number alteration (gain shown in green; loss shown in red) detected by HMM analysis. Single-clone changes were ignored.
Figure 2.

Frequency distribution of aneuploid segments per patient
Distinguishing between Inherited and de Novo Alterations
To distinguish clinically relevant copy-number alterations from normal LCVs, 72 clinically normal parents of the 100 patients were tested using the same tiling-resolution microarrays. These parental samples served as a control population and, in addition, provided valuable information on the inheritance of specific copy-number changes. Eleven alterations, detected in 10 individual patients (including two adjacent alterations in patient 9), were present in neither of the parental DNA samples nor in DNA from any of the 70 other unrelated parents tested (table 3). These de novo alterations most likely represent clinically relevant alterations. For five other alterations, de novo occurrence could not be established, as one or both of the patients' parents were not available; however, these alterations were never observed in any of the other parents tested (table 3). For these alterations, it remains unclear whether they are clinically relevant. All 258 remaining alterations detected in this study were shown to occur in at least one of the normal parents tested (table 4). Such inherited alterations most likely represent LCVs that are not related to the disorder under investigation. De novo aneusomic segments ranged in genomic size from 0.54 to 12.37 Mb (median 2.76 Mb), whereas LCVs ranged in size from 0.05 to 2.16 Mb (median 0.43 Mb) (P=.015, by two-sided independent student T test) (fig. 3). This suggests that clinically relevant aberrations are, in general, larger than normal genomic variants. The largest polymorphic variant detected in this study consisted of two adjacently located duplications on chromosome 5p14.3, involving a total of 3.55 Mb.
Table 3.
Patients with de Novo Alterations (Both Parents Tested) or Candidate Alterations (One or Both Parents Unavailable for Testing)[Note]
| Type ofSubmicroscopicAberration andChromosome Band | Patient | GainorLoss | Start(Mb) | End(Mb) | Length(Mb) | No. ofClonesa | Confirmedb | Clinical Findings (Score)c |
| De novo: | ||||||||
| 1p34.3-1p34.2 | 1 | Loss | 39.22 | 43.15 | 3.93 | 42 | Yes | Severe MR, FD, GR, MC, and delayed brain myelinization (7) |
| 2q23.1-2q23.2 | 2 | Loss | 149.17 | 150.09 | .92 | 11 | Yes | Severe MR, FD, GR, MC, BP, and epilepsy (3) |
| 3q27.1-3q29 | 3 | Loss | 184.43 | 196.8 | 12.37 | 124 | Yes | Severe MR, FD, GR, MC, hypogenitalism, hypoplastic kidneys, and deafness (6) |
| 5q35.1 | 4 | Gain | 170.52 | 171.76 | 1.24 | 16 | Yes | Mild MR, semilobular holoprosencephaly, finger-like thumbs, and polydactyly (3) |
| 9q31.1 | 5 | Loss | 99.74 | 102.58 | 2.85 | 30 | Yes | Moderate MR, FD, MC, and transposition of the great vessels (4) |
| 9q33.1 | 6 | Loss | 115.3 | 115.84 | .54 | 6 | Yes | Mild MR, FD, macrocephaly, and autistic spectrum (3) |
| 11q14.1-11q14.2 | 7 | Loss | 78.12 | 85.61 | 7.49 | 64 | Yes | Mild MR and FD (5) |
| 12q24.21-12q24.23 | 8 | Gain | 114.91 | 117.21 | 2.3 | 39 | Yes | Severe MR, FD, GR, MC, and BP (7) |
| 17p13.2-17p13.1 | 9 | Gain | 4.27 | 7.16 | 2.89 | 28 | Not done | Moderate MR and FD (3)d |
| 17p13.1 | 9 | Gain | 7.67 | 9.10 | 1.43 | 14 | Yes | Moderate MR and FD (3)d |
| 17p12 | 9 | Gain | 12.65 | 15.54 | 2.88 | 30 | Yes | Moderate MR and FD (3)d |
| 17p11.2 | 9 | Gain | 18.55 | 20.03 | 1.48 | 24 | Yes | Moderate MR and FD (3)d |
| 22q11.21 | 10 | Loss | 17.1 | 19.75 | 2.66 | 35 | Yes | Mild MR, FD, and GR (3) |
| Candidate: | ||||||||
| 1q21.1 | 11 | Gain | 143.25 | 145.38 | 2.12 | 21 | Yes | Moderate MR, FD, and GR (5) |
| 3p14.1 | 12 | Loss | 67.59 | 68.15 | .56 | 5 | Yes | Moderate MR (1) |
| 7q11.21 | 13 | Loss | 64.23 | 64.58 | .35 | 18 | Not done | Mild MR, FD, and BP (3) |
| 9p24.3 | 14 | Gain | .21 | .45 | .23 | 5 | Yes | Moderate, MR, and FD (2) |
| 15q24.1-15q24.2 | 15 | Loss | 72.21 | 73.86 | 1.65 | 16 | Yes | Mild MR, FD, GR, and MC (6) |
Note.— None of the genomic regions were present in the Database of Genomic Variants (see Web Resources).
Number of BAC clones located in the copy-number alteration.
Confirmed by MLPA and/or FISH.
For detailed clinical descriptions, see appendix A (online only). The clinical checklist score is given in parentheses. BP = behavioral problems; FD = facial dysmorphism; GR = growth retardation; MC = microcephaly.
The same patient.
Table 4.
All Polymorphic Variants Found in the 100 Patients
| Patient ID | Certainty | No. of Clones in Region | Genomic Size(Mb) | Chromosome | Start(Mb) | Start Clone | End(Mb) | End Clone | DNA Copy-Number Loss or Gain |
| 498 | 1.54 | 6 | .16 | 1 | 103.87 | RP11-107J8 | 104.03 | RP11-219M14 | Loss |
| 62 | 1.8 | 42 | .85 | 1 | 140.8 | RP11-304H2 | 141.65 | RP11-464H8 | Gain |
| 300 | 1.6 | 38 | .85 | 1 | 140.8 | RP11-304H2 | 141.65 | RP11-464H8 | Loss |
| 1 | 2.05 | 22 | .51 | 2 | 87.28 | CTD-2003O3 | 87.79 | RP11-655P13 | Gain |
| 319 | 2 | 22 | .51 | 2 | 87.28 | CTD-2003O3 | 87.79 | RP11-655P13 | Gain |
| 185 | 1.75 | 20 | .42 | 2 | 87.37 | RP11-223J6 | 87.79 | RP11-655P13 | Gain |
| 49 | 1.69 | 24 | .82 | 2 | 89.06 | RP11-15J7 | 89.88 | RP11-136K15 | Loss |
| 166 | 2.27 | 23 | .82 | 2 | 89.06 | RP11-15J7 | 89.88 | RP11-136K15 | Loss |
| 250 | 1.51 | 22 | .72 | 2 | 89.15 | CTD-2013C24 | 89.88 | RP11-136K15 | Gain |
| 261 | 1.53 | 21 | .72 | 2 | 89.15 | CTD-2013C24 | 89.88 | RP11-136K15 | Loss |
| 298 | 1.82 | 21 | .72 | 2 | 89.15 | CTD-2013C24 | 89.88 | RP11-136K15 | Gain |
| 76 | 1.86 | 28 | 1.76 | 2 | 111.01 | RP11-611D21 | 112.77 | RP11-543D12 | Gain |
| 319 | 1.55 | 10 | .5 | 2 | 111.8 | RP11-803D5 | 112.3 | RP11-726P14 | Gain |
| 39 | 1.85 | 20 | .63 | 2 | 129.48 | RP11-159J24 | 130.12 | RP11-675D6 | Loss |
| 58 | 2.04 | 33 | 2 | 2 | 164.36 | RP11-132J19 | 166.36 | RP11-71B17 | Gain |
| 198 | 1.63 | 18 | .96 | 3 | .72 | RP11-151A4 | 1.67 | RP11-611C4 | Gain |
| 42 | 1.72 | 16 | .44 | 4 | 8.67 | RP11-689P11 | 9.1 | CTD-2205P10 | Gain |
| 79 | 1.83 | 10 | .15 | 4 | 8.67 | RP11-689P11 | 8.81 | RP11-1338A24 | Gain |
| 139 | 1.69 | 10 | .15 | 4 | 8.67 | RP11-689P11 | 8.81 | RP11-1338A24 | Gain |
| 148 | 2.06 | 24 | .66 | 4 | 8.67 | RP11-689P11 | 9.33 | CTD-2067O23 | Loss |
| 350 | 1.71 | 10 | .15 | 4 | 8.67 | RP11-689P11 | 8.81 | RP11-1338A24 | Loss |
| 159 | 1.63 | 14 | .36 | 4 | 8.74 | RP11-637J21 | 9.1 | CTD-2205P10 | Gain |
| 190 | 1.84 | 14 | .36 | 4 | 8.74 | RP11-637J21 | 9.1 | CTD-2205P10 | Loss |
| 298 | 2.08 | 20 | .57 | 4 | 8.75 | RP11-234P23 | 9.33 | CTD-2067O23 | Loss |
| 279 | 1.61 | 18 | .55 | 4 | 8.78 | RP11-612I3 | 9.33 | CTD-2067O23 | Loss |
| 17 | 1.88 | 14 | .26 | 4 | 9.07 | RP13-980J16 | 9.33 | CTD-2067O23 | Loss |
| 32 | 1.67 | 13 | .26 | 4 | 9.07 | RP13-980J16 | 9.33 | CTD-2067O23 | Loss |
| 62 | 1.62 | 26 | .53 | 4 | 48.96 | RP11-749B10 | 49.49 | RP11-259G19 | Gain |
| 198 | 1.9 | 20 | .93 | 5 | .48 | CTD-2228K2 | 1.41 | RP11-600M8 | Gain |
| 498 | 2 | 57 | 1.76 | 5 | 19.31 | RP11-59C4 | 21.07 | RP11-337D24 | Gain |
| 498 | 2.25 | 51 | 1.79 | 5 | 21.57 | RP11-454O18 | 23.36 | RP11-88H16 | Gain |
| 14 | 2.27 | 46 | 1.45 | 5 | 69.16 | RP11-0155O16 | 70.61 | RP11-313J5 | Loss |
| 70 | 1.86 | 44 | 1.23 | 5 | 69.16 | RP11-0155O16 | 70.39 | CTD-2007A12 | Gain |
| 159 | 2.41 | 46 | 1.45 | 5 | 69.16 | RP11-0155O16 | 70.61 | RP11-313J5 | Loss |
| 179 | 1.97 | 45 | 1.45 | 5 | 69.16 | RP11-0155O16 | 70.61 | RP11-313J5 | Loss |
| 194 | 2.27 | 46 | 1.45 | 5 | 69.16 | RP11-0155O16 | 70.61 | RP11-313J5 | Gain |
| 249 | 2.28 | 46 | 1.45 | 5 | 69.16 | RP11-0155O16 | 70.61 | RP11-313J5 | Loss |
| 198 | 1.56 | 40 | .96 | 5 | 69.65 | RP11-89M15 | 70.61 | RP11-313J5 | Loss |
| 1 | 1.57 | 33 | .67 | 5 | 69.72 | RP11-629O23 | 70.38 | CTD-2012J21 | Loss |
| 122 | 2.04 | 36 | .9 | 5 | 69.72 | RP11-629O23 | 70.61 | RP11-313J5 | Gain |
| 169 | 1.59 | 36 | .9 | 5 | 69.72 | RP11-629O23 | 70.61 | RP11-313J5 | Loss |
| 5 | 2.05 | 34 | .79 | 5 | 69.82 | RP11-375P3 | 70.61 | RP11-313J5 | Loss |
| 27 | 2.03 | 33 | .79 | 5 | 69.82 | RP11-375P3 | 70.61 | RP11-313J5 | Gain |
| 73 | 2.09 | 34 | .79 | 5 | 69.82 | RP11-375P3 | 70.61 | RP11-313J5 | Loss |
| 75 | 1.5 | 14 | .22 | 7 | 101.69 | RP11-342G18 | 101.91 | RP11-1385F10 | Gain |
| 319 | 1.83 | 22 | .95 | 7 | 105.55 | CTD-2026C17 | 106.5 | RP11-762B6 | Gain |
| 190 | 1.66 | 8 | .35 | 7 | 143.2 | RP11-703N5 | 143.56 | RP11-714J20 | Gain |
| 76 | 1.51 | 10 | .05 | 8 | .08 | RP11-130K11 | .14 | RP11-809I19 | Loss |
| 128 | 1.63 | 12 | .83 | 8 | 7.13 | RP11-447A6 | 7.96 | CTD-2185F10 | Gain |
| 171 | 1.69 | 12 | .83 | 8 | 7.13 | RP11-447A6 | 7.96 | CTD-2185F10 | Gain |
| 283 | 1.73 | 14 | .98 | 8 | 7.13 | RP11-447A6 | 8.11 | RP11-556O5 | Gain |
| 319 | 1.81 | 10 | .6 | 8 | 7.13 | RP11-447A6 | 7.72 | RP11-739E3 | Loss |
| 185 | 1.61 | 10 | .68 | 8 | 7.28 | RP11-738O21 | 7.96 | CTD-2185F10 | Loss |
| 282 | 1.88 | 15 | .67 | 8 | 47.01 | CTD-2326I15 | 47.69 | CTD-2026P11 | Gain |
| 1 | 2.02 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 2 | 1.58 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 62 | 1.92 | 18 | .31 | 8 | 86.7 | RP11-509F16 | 87.01 | CTD-2067O20 | Loss |
| 76 | 1.54 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 88 | 1.56 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 123 | 1.56 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 148 | 1.71 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Gain |
| 169 | 2.02 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Gain |
| 240 | 1.53 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 258 | 1.67 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 261 | 1.92 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 282 | 1.93 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 498 | 1.69 | 14 | .22 | 8 | 86.7 | RP11-509F16 | 86.92 | RP11-50N4 | Loss |
| 242 | 1.66 | 6 | .4 | 8 | 114.43 | RP11-762L11 | 114.82 | RP11-448E20 | Gain |
| 261 | 1.57 | 10 | .29 | 8 | 131.3 | RP11-531O18 | 131.6 | CTD-2415J03 | Loss |
| 282 | 1.64 | 6 | .17 | 8 | 131.37 | RP11-1292J5 | 131.55 | RP11-1104D13 | Loss |
| 5 | 1.53 | 28 | 2.08 | 9 | 41.43 | RP11-708D13 | 43.51 | CTD-2511E18 | Loss |
| 186 | 2.04 | 12 | .35 | 9 | 80.7 | RP11-72M15 | 81.04 | RP11-411J24 | Loss |
| 117 | 2.04 | 26 | 1.08 | 9 | 130.77 | RP11-83J21 | 131.85 | RP13-195B13 | Loss |
| 240 | 1.67 | 30 | .99 | 10 | 46.17 | CTD-2004H14 | 47.16 | RP11-57L2 | Gain |
| 307 | 1.5 | 20 | .51 | 10 | 46.17 | CTD-2004H14 | 46.68 | RP11-138M8 | Loss |
| 226 | 1.59 | 18 | .47 | 10 | 46.2 | RP11-342C24 | 46.68 | RP11-138M8 | Loss |
| 17 | 1.52 | 12 | .14 | 10 | 47.02 | RP11-30N1 | 47.16 | RP11-57L2 | Loss |
| 282 | 1.75 | 14 | .44 | 11 | 50.19 | RP11-419P14 | 50.63 | RP11-89A1 | Gain |
| 14 | 1.53 | 15 | 1.06 | 14 | 18.33 | CTD-2213M13 | 19.39 | RP11-449I24 | Loss |
| 30 | 1.7 | 16 | 1.06 | 14 | 18.33 | CTD-2213M13 | 19.39 | RP11-449I24 | Loss |
| 57 | 1.98 | 15 | 1.06 | 14 | 18.33 | CTD-2213M13 | 19.39 | RP11-449I24 | Gain |
| 142 | 1.52 | 13 | 1.06 | 14 | 18.33 | CTD-2213M13 | 19.39 | RP11-449I24 | Loss |
| 158 | 1.72 | 14 | 1.06 | 14 | 18.33 | CTD-2213M13 | 19.39 | RP11-449I24 | Loss |
| 282 | 2.04 | 16 | 1.06 | 14 | 18.33 | CTD-2213M13 | 19.39 | RP11-449I24 | Gain |
| 283 | 2 | 14 | .94 | 14 | 18.33 | CTD-2213M13 | 19.27 | RP11-447E14 | Gain |
| 42 | 2.52 | 65 | 1.68 | 15 | 18.44 | RP11-487A3 | 20.11 | RP11-702C12 | Gain |
| 75 | 1.6 | 34 | 1.84 | 15 | 18.44 | RP11-720N23 | 20.11 | RP11-702C12 | Gain |
| 295 | 2.75 | 65 | 1.68 | 15 | 18.44 | RP11-487A3 | 20.11 | RP11-702C12 | Gain |
| 404 | 2.03 | 48 | 1.14 | 15 | 18.44 | RP11-487A3 | 19.58 | RP11-95I9 | Loss |
| 23 | 1.8 | 13 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Loss |
| 42 | 2 | 16 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Gain |
| 51 | 1.74 | 13 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Loss |
| 113 | 1.72 | 14 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Loss |
| 134 | 1.72 | 12 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Loss |
| 193 | 1.61 | 13 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Loss |
| 205 | 1.68 | 14 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Loss |
| 207 | 1.83 | 15 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Loss |
| 313 | 1.5 | 13 | .85 | 14 | 18.53 | RP11-692L20 | 19.39 | RP11-449I24 | Gain |
| 1 | 2.45 | 60 | 1.57 | 15 | 18.55 | RP11-118E23 | 20.11 | RP11-702C12 | Loss |
| 46 | 2.46 | 61 | 1.57 | 15 | 18.55 | RP11-118E23 | 20.11 | RP11-702C12 | Loss |
| 51 | 2.31 | 55 | 1.36 | 15 | 18.55 | RP11-118E23 | 19.9 | RP11-585E3 | Loss |
| 63 | 2.43 | 62 | 1.57 | 15 | 18.55 | RP11-118E23 | 20.11 | RP11-702C12 | Loss |
| 76 | 1.78 | 51 | 1.09 | 15 | 18.55 | RP11-118E23 | 19.63 | RP11-350K22 | Loss |
| 117 | 2.56 | 68 | 1.87 | 15 | 18.55 | RP11-118E23 | 20.41 | RP11-1081C20 | Loss |
| 134 | 2.23 | 60 | 1.57 | 15 | 18.55 | RP11-118E23 | 20.11 | RP11-702C12 | Loss |
| 176 | 1.78 | 42 | 1.04 | 15 | 18.55 | RP11-118E23 | 19.58 | RP11-95I9 | Loss |
| 205 | 2.53 | 59 | 1.57 | 15 | 18.55 | RP11-118E23 | 20.11 | RP11-702C12 | Loss |
| 342 | 2.12 | 60 | 1.57 | 15 | 18.55 | RP11-118E23 | 20.11 | RP11-702C12 | Loss |
| 498 | 1.98 | 62 | 1.57 | 15 | 18.55 | RP11-118E23 | 20.11 | RP11-702C12 | Gain |
| 57 | 1.7 | 17 | .18 | 15 | 18.67 | RP11-452L16 | 18.84 | RP11-509A17 | Gain |
| 142 | 2.21 | 59 | 1.45 | 15 | 18.67 | RP11-452L16 | 20.11 | RP11-702C12 | Loss |
| 282 | 2.57 | 58 | 1.44 | 15 | 18.68 | RP11-717D19 | 20.11 | RP11-702C12 | Gain |
| 17 | 2.12 | 50 | 1.35 | 15 | 18.76 | RP11-631J8 | 20.11 | RP11-702C12 | Gain |
| 54 | 2.5 | 54 | 1.35 | 15 | 18.76 | RP11-631J8 | 20.11 | RP11-702C12 | Loss |
| 140 | 2.04 | 53 | 1.35 | 15 | 18.76 | RP11-631J8 | 20.11 | RP11-702C12 | Loss |
| 179 | 2.17 | 54 | 1.35 | 15 | 18.76 | RP11-631J8 | 20.11 | RP11-702C12 | Loss |
| 207 | 2.26 | 53 | 1.35 | 15 | 18.76 | RP11-631J8 | 20.11 | RP11-702C12 | Loss |
| 96 | 1.53 | 51 | 1.31 | 15 | 18.81 | CTD-2563C10 | 20.11 | RP11-702C12 | Gain |
| 49 | 1.87 | 28 | .94 | 15 | 18.96 | RP11-403L7 | 19.9 | RP11-585E3 | Loss |
| 194 | 1.98 | 28 | .6 | 15 | 19.03 | RP11-773J18 | 19.63 | RP11-350K22 | Gain |
| 198 | 1.8 | 32 | .87 | 15 | 19.03 | RP11-773J18 | 19.9 | RP11-585E3 | Loss |
| 24 | 1.81 | 36 | .83 | 15 | 19.16 | CTD-2155N19 | 19.98 | RP11-603B24 | Loss |
| 28 | 2.12 | 38 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 30 | 1.75 | 36 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 34 | 1.8 | 37 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 39 | 1.65 | 28 | .74 | 15 | 19.16 | CTD-2155N19 | 19.9 | RP11-585E3 | Loss |
| 57 | 2.37 | 39 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Gain |
| 73 | 1.72 | 39 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 89 | 1.83 | 36 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 94 | 1.52 | 20 | .3 | 15 | 19.16 | CTD-2155N19 | 19.45 | RP11-138H15 | Gain |
| 111 | 1.88 | 20 | .3 | 15 | 19.16 | CTD-2155N19 | 19.45 | RP11-138H15 | Loss |
| 113 | 2.23 | 37 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 123 | 2.13 | 40 | 1.12 | 15 | 19.16 | CTD-2155N19 | 20.28 | RP11-561P13 | Gain |
| 238 | 1.77 | 36 | .83 | 15 | 19.16 | CTD-2155N19 | 19.98 | RP11-603B24 | Loss |
| 298 | 2.23 | 32 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 319 | 2 | 37 | .96 | 15 | 19.16 | CTD-2155N19 | 20.11 | RP11-702C12 | Loss |
| 350 | 2 | 35 | .83 | 15 | 19.16 | CTD-2155N19 | 19.98 | RP11-603B24 | Loss |
| 38 | 1.65 | 24 | .73 | 15 | 19.39 | RP11-56N1 | 20.11 | RP11-702C12 | Loss |
| 52 | 1.76 | 26 | .73 | 15 | 19.39 | RP11-56N1 | 20.11 | RP11-702C12 | Loss |
| 93 | 1.58 | 24 | .73 | 15 | 19.39 | RP11-56N1 | 20.11 | RP11-702C12 | Loss |
| 159 | 1.51 | 14 | .35 | 15 | 19.63 | RP11-350K22 | 19.98 | RP11-603B24 | Gain |
| 193 | 1.75 | 14 | .35 | 15 | 19.63 | RP11-350K22 | 19.98 | RP11-603B24 | Loss |
| 189 | 1.61 | 14 | .23 | 15 | 19.89 | RP11-281J20 | 20.11 | RP11-702C12 | Loss |
| 242 | 1.69 | 12 | .1 | 15 | 19.89 | RP11-281J20 | 19.98 | RP11-603B24 | Loss |
| 75 | 1.74 | 16 | .4 | 15 | 21.9 | RP11-350A1 | 22.3 | RP11-110H15 | Loss |
| 117 | 1.75 | 12 | .31 | 15 | 26.27 | RP11-0139P12 | 26.58 | RP11-550A14 | Loss |
| 13 | 1.67 | 24 | .44 | 15 | 30.22 | RP11-624A21 | 30.66 | RP11-720B11 | Loss |
| 111 | 1.52 | 18 | .44 | 16 | 32.52 | RP11-319D11 | 32.96 | CTD-2081G1 | Gain |
| 88 | 1.73 | 28 | 1.43 | 17 | 7.67 | CTD-2306L18 | 9.1 | RP11-342E3 | Gain |
| 88 | 2.35 | 55 | 2.88 | 17 | 12.65 | RP11-333I5 | 15.54 | RP11-640N23 | Gain |
| 80 | 1.7 | 10 | .31 | 17 | 33.31 | CTD-2344A06 | 33.62 | RP11-72I20 | Gain |
| 179 | 1.63 | 10 | .31 | 17 | 33.31 | CTD-2344A06 | 33.62 | RP11-72I20 | Gain |
| 240 | 1.69 | 10 | .31 | 17 | 33.31 | CTD-2344A06 | 33.62 | RP11-72I20 | Gain |
| 319 | 1.73 | 10 | .31 | 17 | 33.31 | CTD-2344A06 | 33.62 | RP11-72I20 | Gain |
| 192 | 1.73 | 7 | .24 | 17 | 33.38 | RP11-121G16 | 33.62 | RP11-72I20 | Loss |
| 205 | 1.74 | 12 | .27 | 17 | 38.56 | RP11-812O5 | 38.83 | CTD-2297L14 | Gain |
| 24 | 1.58 | 10 | .22 | 17 | 38.61 | RP11-1365J18 | 38.83 | CTD-2297L14 | Gain |
| 55 | 1.63 | 12 | .24 | 17 | 38.61 | RP11-1365J18 | 38.85 | RP11-100E5 | Gain |
| 70 | 1.8 | 10 | .22 | 17 | 38.61 | RP11-1365J18 | 38.83 | CTD-2297L14 | Gain |
| 186 | 1.96 | 9 | .22 | 17 | 38.61 | RP11-1365J18 | 38.83 | CTD-2297L14 | Loss |
| 192 | 1.58 | 8 | .22 | 17 | 38.61 | RP11-1365J18 | 38.83 | CTD-2297L14 | Gain |
| 198 | 1.93 | 9 | .22 | 17 | 38.61 | RP11-1365J18 | 38.83 | CTD-2297L14 | Gain |
| 258 | 1.86 | 10 | .22 | 17 | 38.61 | RP11-1365J18 | 38.83 | CTD-2297L14 | Gain |
| 307 | 1.93 | 10 | .22 | 17 | 38.61 | RP11-1365J18 | 38.83 | CTD-2297L14 | Gain |
| 301 | 2.04 | 15 | .5 | 18 | 24.54 | RP11-620F23 | 25.05 | CTD-2024E24 | Loss |
| 282 | 1.56 | 6 | 1.02 | 19 | .25 | RP11-657O13 | 1.27 | CTD-2315C7 | Gain |
| 159 | 1.61 | 16 | 2.16 | 19 | .26 | RP11-9F15 | 2.41 | RP11-148G19 | Gain |
| 226 | 1.78 | 16 | 1.66 | 19 | .54 | CTD-2589F14 | 2.21 | RP11-0333F10 | Gain |
| 205 | 2.07 | 6 | .48 | 19 | .94 | CTD-2563K4 | 1.42 | CTD-2260M22 | Loss |
| 307 | 1.85 | 6 | 1.02 | 19 | .94 | CTD-2563K4 | 1.96 | CTD-2268M1 | Loss |
| 185 | 1.69 | 7 | .69 | 19 | 1.26 | CTD-2295F20 | 1.96 | CTD-2268M1 | Gain |
| 5 | 2.12 | 22 | .33 | 19 | 8.47 | CTD-2509P17 | 8.8 | RP11-1307A5 | Loss |
| 205 | 1.95 | 22 | .33 | 19 | 8.47 | CTD-2509P17 | 8.8 | RP11-1307A5 | Loss |
| 13 | 1.69 | 21 | .17 | 19 | 8.63 | RP11-1077G12 | 8.8 | RP11-1307A5 | Loss |
| 12 | 1.75 | 18 | .16 | 19 | 8.64 | RP11-92E5 | 8.8 | RP11-1307A5 | Loss |
| 34 | 2 | 22 | .16 | 19 | 8.64 | RP11-92E5 | 8.8 | RP11-127L14 | Loss |
| 52 | 2.36 | 21 | .16 | 19 | 8.64 | RP11-92E5 | 8.8 | RP11-1307A5 | Loss |
| 57 | 2.29 | 21 | .16 | 19 | 8.64 | RP11-92E5 | 8.8 | RP11-1307A5 | Loss |
| 111 | 1.9 | 17 | .16 | 19 | 8.64 | RP11-92E5 | 8.8 | RP11-1307A5 | Loss |
| 240 | 1.54 | 21 | .16 | 19 | 8.64 | RP11-92E5 | 8.8 | RP11-1307A5 | Loss |
| 319 | 1.56 | 12 | .1 | 19 | 8.64 | RP11-92E5 | 8.75 | RP11-14H08 | Loss |
| 14 | 1.84 | 16 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 49 | 1.67 | 14 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 54 | 2 | 16 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 89 | 1.8 | 16 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 123 | 1.67 | 10 | .07 | 19 | 8.68 | RP11-743O10 | 8.75 | RP11-14H08 | Loss |
| 136 | 1.82 | 17 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 140 | 2.04 | 18 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 141 | 2.18 | 16 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 142 | 1.55 | 15 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 179 | 1.65 | 18 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Gain |
| 189 | 2.08 | 17 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 190 | 1.84 | 16 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 194 | 1.55 | 15 | .11 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-203K6 | Loss |
| 204 | 1.74 | 21 | .31 | 19 | 8.68 | RP11-743O10 | 8.99 | RP11-478A23 | Gain |
| 208 | 1.54 | 18 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 279 | 1.95 | 12 | .1 | 19 | 8.68 | RP11-743O10 | 8.79 | RP11-598E5 | Loss |
| 301 | 1.53 | 15 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 313 | 1.53 | 14 | .11 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-203K6 | Loss |
| 342 | 2.17 | 13 | .11 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-203K6 | Loss |
| 350 | 2.16 | 17 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 404 | 1.64 | 18 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Gain |
| 498 | 1.85 | 17 | .12 | 19 | 8.68 | RP11-743O10 | 8.8 | RP11-1307A5 | Loss |
| 88 | 1.68 | 10 | .05 | 19 | 8.7 | RP11-1361O8 | 8.75 | RP11-14H08 | Loss |
| 96 | 1.94 | 17 | .1 | 19 | 8.7 | RP11-1361O8 | 8.8 | RP11-1307A5 | Loss |
| 76 | 1.72 | 17 | .63 | 19 | 32.77 | CTD-2544G22 | 33.4 | RP11-722H20 | Gain |
| 342 | 1.8 | 14 | .69 | 19 | 47.86 | RP11-653D16 | 48.55 | RP11-160A19 | Loss |
| 136 | 1.56 | 16 | .26 | 21 | 9.86 | RP11-430M17 | 10.12 | RP11-149I22 | Gain |
| 283 | 1.5 | 11 | .13 | 21 | 9.99 | RP11-282F14 | 10.12 | RP11-149I22 | Gain |
| 242 | 1.99 | 6 | .09 | 21 | 20.89 | RP11-580L2 | 20.98 | RP11-47C12 | Gain |
| 1 | 1.84 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Gain |
| 2 | 1.68 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 13 | 1.86 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 14 | 2.12 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 23 | 2.17 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 30 | 2.25 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 42 | 2.07 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 49 | 1.95 | 21 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 51 | 2.31 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 52 | 1.84 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 57 | 2.34 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 58 | 1.89 | 23 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 96 | 1.69 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 111 | 1.62 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 113 | 1.83 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 117 | 2 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 122 | 1.88 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 134 | 1.83 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 142 | 1.9 | 21 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 158 | 2.26 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 185 | 1.78 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Gain |
| 189 | 1.78 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 190 | 1.98 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 193 | 1.72 | 21 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 205 | 1.88 | 23 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 207 | 2.17 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 226 | 1.73 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 238 | 1.58 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 242 | 1.96 | 23 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 279 | 2.11 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Loss |
| 282 | 2.38 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 283 | 2.45 | 24 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 307 | 1.8 | 22 | .36 | 22 | 14.38 | RP11-586C14 | 14.74 | RP11-532J16 | Loss |
| 313 | 2.02 | 23 | .43 | 22 | 14.38 | RP11-586C14 | 14.81 | RP11-561P7 | Gain |
| 38 | 2.02 | 22 | .33 | 22 | 14.49 | RP11-790P12 | 14.81 | RP11-561P7 | Gain |
| 79 | 1.88 | 20 | .26 | 22 | 14.49 | RP11-790P12 | 14.74 | RP11-532J16 | Loss |
| 30 | 1.76 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Loss |
| 55 | 1.51 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Loss |
| 62 | 1.65 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Gain |
| 94 | 1.51 | 12 | .28 | 22 | 17.1 | CTD-2280L11 | 17.37 | CTD-2522F24 | Loss |
| 128 | 1.52 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Gain |
| 169 | 1.9 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Gain |
| 192 | 1.86 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Loss |
| 261 | 1.57 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Loss |
| 282 | 1.59 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Loss |
| 298 | 1.85 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Loss |
| 306 | 1.53 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Gain |
| 498 | 1.71 | 8 | .19 | 22 | 17.1 | CTD-2280L11 | 17.29 | RP11-690P21 | Loss |
Figure 3.

Size distribution of chromosomal aberrations, divided into de novo alterations (n=10) and polymorphic variants (n=258). Black horizontal bars indicate median size (de novo alteration median 2.76 Mb; genomic variant median 0.43 Mb).
Confirmation Studies and Molecular Description of Clinically Relevant Aberrations
All de novo chromosomal alterations were validated in the individual patients by at least one other method: FISH and/or MLPA. Parental samples were similarly checked to confirm that they contained a normal copy-number for these chromosomal regions (table 2 and figs. A1–A10 in appendix A [online only]). In addition, the presence of a copy-number alteration was confirmed in four patient-parents trios with inherited copy-number variations, in addition to four alterations detected in patients for whom no parents were available for testing. In all cases, these MLPA and FISH studies confirmed the aberrations predicted by array CGH analysis.
Figure A1.

Details on patient 1, showing the genomewide array CGH profile, with the 3.93-Mb deletion on 1p34 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 1 (deletion indicated [red arrow in panel II]), confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III), and confirmation by FISH (panel IV) using clone RP11-708H15 (green) and a centromere probe as the control (red).
Figure A10.

Details on patient 10, showing the genomewide array CGH profile, with the 2.66-Mb deletion on 22q11 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 22 (deletion indicated [red arrow in panel II]), and confirmation of the de novo deletion by FISH (panel IV) using the LSI Tuple (Vysis) probe (red) and a centromere probe as the control (green).
DNA from 10 patients contained validated de novo segmental aneusomies: seven deletions and three duplications. The aneusomic segments identified varied in genomic size from 540 kb to 12 Mb, and they were scattered throughout the genome. In all but one patient, the de novo alteration involved a single genomic segment of adjacent DNA clones. DNA from patient 9 contained a complex duplication on the short arm of chromosome 17, consisting of four interspersed regions covering a total of 8.93 Mb (table 3 and figs. 1 and A9 [online only]). For this patient, one of the regions involved encompasses a known microduplication of the PMP22 gene associated with Charcot-Marie-tooth disease (CMT [MIM 118220]). Another known microdeletion region was observed in patient 10 at 22q11, the region associated with classical DiGeorge syndrome (MIM 188400) and velocardiofacial syndrome (VCFS [MIM 192430]). The other chromosomal alterations did not map to known microdeletion/duplication regions. Two of these alterations had a genomic size <1 Mb—one deletion of 920 kb on 2q23.1-q23.2 (in patient 2) and one deletion of 540 kb on 9q33.1 (in patient 6).
Figure A9.

Details on patient 9, showing the genomewide array CGH profile, with the interspersed 8.68-Mb duplication on 17p11-17p13 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 17 (duplication indicated [red arrow in panel II]), and confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III).
Clinical Implications of Tiling-Resolution Copy-Number Screening
Clinical details of the individual patients are presented in appendix A (online only) and are summarized in table 3. The study group included 32 patients with mild MR, 27 with moderate MR, and 32 with severe MR. In nine patients, the level of MR was not accurately assessable, mainly because of young age. The distribution of the level of MR did not differ significantly between the group of patients with a de novo copy-number alteration (n=10) and the patients with reliable cognitive assessment (n=81) (P=1.00 by Mann-Whitney U test). In addition to MR, all patients were evaluated for other clinical features by use of a clinical checklist (tables 1 and 2). The 10 patients with de novo alterations had a mean score of 4.4 ± 1.7, which was not significantly different from that of the remaining 90 patients without detectable de novo aneusomic segments (mean score 3.6 ± 1.4) (P=.17 by Mann-Whitney U test). This implies that, on average, the patients with de novo alterations did not have more clinical features suggestive of a chromosomal disorder than did patients without such an alteration. The distribution of the clinical scores of the 100 patients is shown in table 5.
Table 5.
Estimated Frequencies of Submicroscopic Aberrations for Each Clinical Score, in the Study Cohorts and an Unselected Cohortof Patients with MR[Note]
|
Study Cohorts |
Unselected Cohort (n=120) |
||||||||
| Array CGH Tested(n=100) |
MLPA Tested(n=297) |
||||||||
| ClinicalScore | No. ofPatients | No. ofAberrations | Prevalence | No. ofPatients | No. ofAberrations | Prevalence | CombinedPrevalence | No. ofPatients | EstimatedFrequency ofAberrations |
| 0 | 0 | 0 | .00 | 1 | 0 | .00 | .00 | 20 | .0 |
| 1 | 5 | 0 | .00 | 36 | 1 | .03 | .03 | 25 | .7 |
| 2 | 16 | 0 | .00 | 58 | 1 | .02 | .02 | 33 | .6 |
| 3 | 32 | 5 | .16 | 86 | 5 | .06 | .21 | 17 | 3.5 |
| 4 | 21 | 1 | .05 | 62 | 1 | .02 | .06 | 14 | .9 |
| 5 | 14 | 1 | .07 | 32 | 4 | .13 | .19 | 4 | .8 |
| 6 | 7 | 1 | .14 | 15 | 1 | .07 | .20 | 5 | 1.0 |
| 7 | 5 | 2 | .40 | 6 | 0 | .00 | .40 | 1 | .4 |
| 8 | 0 | 0 | .00 | 1 | 1 | 1.00 | 1.00 | 1 | 1.0 |
| 9 | 0 | 0 | .00 | 0 | 0 | .00 | .00 | 0 | .0 |
| 10 | 0 |
0 |
.00 | 0 |
0 |
.00 | .00 | 0 |
.0 |
| Total | 100 | 10 | .10 | 297 | 14 | .05 | .14 | 120 | 8.8 |
Note.— The estimations are based on the combined prevalences of the submicroscopic anomalies in the study cohorts.
The 100 patients tested by array CGH in the present study were part of a series of 297 mentally retarded patients previously tested for subtelomeric abnormalities by use of MLPA. A subtelomeric aberration was found in 14 (4.7%) of 297 patients (table 5). To ascertain the representativeness of the current sample set for all patients with MR who were referred to our clinical genetics center for cytogenetic analysis, we gathered information on the 710 patients with unexplained MR (excluding Down syndrome) who were referred for karyotyping during a 2-year period (September 2002 to September 2004). Within this group, 34 patients (4.8%) had a microscopically visible chromosomal abnormality. A total of 120 unselected patients from the remaining 676 were clinically scored (table 5). Their mean score of 2.2 ± 1.7 was significantly less than that of the 100 patients tested in the current study (P<.0001 by Mann-Whitney U test). Thus, the current detection rate of 10% in our sample partly reflects clinical preselection based on phenotypes with additional dysmorphic features and/or growth abnormalities. The use of this genomewide tiling-resolution array CGH approach in an unselected sample of patients with MR will likely have a lower yield of de novo microdeletions/duplications. To estimate the expected yield of submicroscopic alterations over the entire genome, including the subtelomeric regions, within an unselected population of mentally retarded patients, we calculated the frequencies of submicroscopic abnormalities for each clinical score among the group of 100 individuals tested by array CGH and the group of 297 tested by MLPA. These frequencies were then used to estimate the expected frequency of the submicroscopic aberrations for each clinical score among 120 unselected patients with MR. On the basis of bootstrapping with 1,500 replications, the overall estimated frequency of microdeletions/duplications in this unselected series of mentally retarded patients who were referred to our department for karyotyping is 7.3% (95% CI 4.2%–10.3%). This is considerably higher than the rate of microscopically visible deletions and duplications in this patient group (4.8%).
Discussion
In this study, we demonstrate that submicroscopic interstitial chromosomal abnormalities are a frequent cause of MR in patients with additional phenotypic features. In a set of 100 patients who have unexplained MR with or without congenital anomalies, 10 de novo aneusomic segments were detected by use of a tiling-resolution genomewide BAC array. For an additional five alterations detected, it is unknown whether the chromosome abnormality identified was de novo, because one or both parents were unavailable for testing. The 10 validated de novo copy-number alterations detected in this study by genomewide tiling-resolution array CGH were not detected by routine chromosome analysis or by quantitative analysis of the subtelomeric regions with the use of MLPA. This can be explained by the fact that all de novo alterations were interstitial and that 7 of the 10 abnormalities were <4 Mb (2 were <1 Mb). The tiling-resolution array CGH approach used in this study has a 10-fold increase in coverage over that of routinely used 1-Mb resolution BAC arrays with ∼3,000 clones (Vissers et al. 2003; Shaw-Smith et al. 2004; Rosenberg et al. 2005). Therefore, it can be expected that the application of this approach leads to an increase in diagnostic yield for MR. Although, indeed, many more aberrations were detected by this approach (on average, three per patient), only two de novo aberrations <1 Mb were detected. This result might suggest that a reliable 1-Mb copy-number screening could identify the majority of submicroscopic anomalies related to MR. However, additional data are needed before the percentage of deletions and duplications <1 Mb in patients with MR can be reliably assessed. One added value of tiling-path resolution arrays, moreover, lies in the robustness of the detection. A 1-Mb duplication, for instance, will be detected by 10 clones present on the tiling-path resolution array, compared with only 1 clone on a 1-Mb resolution array. This results in a significantly lower false-positive rate. In fact, no false positives were detected in this study. Furthermore, a tiling-path array approach results in a more precise mapping of the start and endpoints of each rearrangement, thus allowing detailed genotype-phenotype correlation assessments.
Conventional GTG-banded karyotyping detects chromosomal alterations in ∼5% of individuals with unexplained MR. Recent studies involving techniques such as telomeric FISH and MLPA have revealed that small subtelomeric deletions and duplications occur in ∼5% of clinically preselected individuals with unexplained MR whose routine chromosome analysis was normal (Biesecker 2002; de Vries et al. 2003; Koolen et al. 2004a). Combined, these studies suggest that chromosomal alterations, both microscopic and submicroscopic and interstitial as well as subtelomeric, are responsible for 20% of cases of unexplained MR. However, we need to take into account that our series of 100 patients is not representative of all patients with unexplained MR who are referred to our clinical genetics department. Correction for this clinical preselection suggests an overall incidence of ∼12% of chromosomal alterations in this patient group, of which ∼7% are submicroscopic (interstitial and subtelomeric combined). We conclude that molecular karyotyping by tiling-resolution array CGH will increase the diagnostic yield of chromosomal abnormalities among mentally retarded individuals by approximately twofold. This is probably a conservative estimate, since aberrations on the X chromosome could not be analyzed for the presence of genomic rearrangements because of the sex-mismatched hybridizations needed for quality control (see the “Patients and Methods” section). At present, the robustness of the tiling-resolution array CGH procedure is such that these internal quality assessments are no longer required, thus allowing sex-matched hybridizations in a routine diagnostic setting.
The clinical consequences of submicroscopic de novo copy-number alteration are determined by the kind of alteration (deletion or duplication) and by the number and function of genes affected by the dosage alteration. Three patients had a de novo duplication varying in size from 1.2 to 8.9 Mb, leading to trisomy of 7–50 genes. These patients were mildly to severely mentally retarded, and two had brain abnormalities, which varied from microcephaly to semilobar holoprosencephaly. The duplication on chromosome 17p11.2-p13.2 (patient 9) was complex, since four separate regions were involved, including the CMT critical region (Suter and Scherer 2003). At age 6 years, this moderately retarded girl had not yet developed signs of CMT. The seven de novo deletions varied in size from 540 kb to 12.4 Mb. The gene content of the affected chromosomal areas varied from monosomy of 1 gene in patient 6 to monosomy of >70 genes in patient 3. The microdeletions at 2q23.1-q23.2 (patient 2) and 22q11.21 (patient 10) have been described previously. The latter deletion is involved in the DiGeorge syndrome and VCFS, which is a well-known, common, and clinically recognizable syndrome (Lindsay 2001). The 4-year-old boy with this deletion had mild MR, short stature, and some suggestive facial features but lacked diagnostic VCFS features, such as cardiac anomalies, cleft palate, long/thin fingers, hypocalcemia, or thymic hypoplasia. This shows that array CGH also aids in diagnosing atypical clinical presentation of common microdeletion syndromes. The 920-kb microdeletion on 2q23.1-q23.2 (in patient 2) partly overlaps a microdeletion previously reported by our group and determined using a 1-Mb resolution BAC array (Vissers et al. 2003; Koolen et al. 2004b). Both patients had severe MR, postnatal growth retardation, microcephaly, coarse facies, and epilepsy. The overlap in clinical features between the two patients with these microdeletions may point to a new syndrome. Three genes are located in the overlapping deletion region—MBD5, EPC2, and KIF5C. Mice lacking the gene for KIF5C, a neuronal kinesin enriched in motor neurons, show a smaller brain size and a loss of peripheral neurons (Kanai et al. 2000). MBD5 is a member of the methyl CpG-binding-domain protein family specifically expressed in the brain (Nagase et al. 2000; Roloff et al. 2003).
In addition to clinically relevant DNA copy-number alterations, we detected 258 LCVs in our series of 100 patients. All these variations were also observed in one or more normal parental samples. This finding confirms and extends recent reports of the frequency of LCVs in normal individuals (Iafrate et al. 2004; Sebat et al. 2004). Such variation represents a novel class of polymorphisms within the human genome whose exact frequency in different ethnic groups remains to be established. In general, normal genomic variants were smaller than clinically relevant copy-number alterations. It is essential to rule out such submicroscopic variation, by studying parental samples, before drawing any conclusions about whether an aneusomic segment is causative for the MR. In addition, it is important to check whether such an alteration has been described previously in normal individuals, since this will reduce the likelihood that it is causative. Moreover, clinical comparison with previously described patients who have overlapping alterations will aid in the clinical interpretation. Systematic collection of information on the phenotypic effects of rare submicroscopic alterations is urgently needed, now that molecular karyotyping is becoming part of clinical patient evaluation (see ECARUCA–European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations Web site).
In conclusion, this study demonstrates, for the first time, the use of a tiling-resolution BAC array in the diagnosis of unexplained MR in patients. We were able to distinguish de novo alterations from inherited DNA copy-number alterations, and we confirmed all findings identified by the array CGH method. After correction for clinical preselection, our data show that the percentage of submicroscopic rearrangements among individuals with unexplained MR exceeds that of microscopically visible alterations. Offering molecular karyotyping by array CGH to patients who have unexplained MR, with or without congenital anomalies, will therefore significantly improve diagnostic yield, leading to accurate diagnosis and genetic counseling.
Note added in proof.—The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE3191.
Acknowledgments
We thank Walter van der Vliet, Hanneke Mieloo, Martin Elferink, Gerard Merkx, Judith Derks-Willemen, Diederick Passchier, and Martina Versteeg, for expert technical assistance; Nine van Slobbe-Knoers, Ben Hamel, Ernie Bongers, and Carlo Marcelis, for referral of patients; and the patients and their parents, for participation in this study. Kazutoyo Osoegawa, Pieter de Jong, Marco Marra, and Martin Kryzwinski are acknowledged for their contribution in establishing the tiling-resolution BAC set. This work was supported by grants from the Netherlands Organisation for Health Research and Development (ZonMW 907-00-058 [to B.B.A.d.V.], ZonMW 920-03-338 [to D.A.K.], and ZonMW 912-04-047 [to H.G.B. and J.A.V.]).
Appendix A: Description of Patients
Patient 1
This 2-year-old, severely mentally retarded boy was born by Caesarean section at 37 wk and 2 d of gestation, with a birth weight of 2,000 g. There was intrauterine growth retardation and a breach presentation. At age 1 year, his height was 71 cm (−2.5 SD), and his head circumference was 41.5 cm (−4 SD). He had hypertelorism, blepharophimosis, epicanthic folds, long philtrum with thin upper lip, micrognathia, prominent ear ridges, and repositionable club feet. He could not roll over and was very hypotonic. At age 10 mo, MRI of the brain showed a generally delayed myelinization. The patient's mother had a mentally retarded brother with hydrocephaly and epilepsy who died at age 2 years.
Results of routine chromosomal analysis were normal, including normal subtelomeric regions determined using MLPA. Array CGH revealed a de novo deletion on chromosome 1p34 (3.93 Mb), which was confirmed by FISH (fig. A1)
Patient 2
This 3-year-old, severely mentally retarded girl was born after an uneventful pregnancy at 41 wk, with a birth weight of 3,750 g. Her development was delayed from birth onward, with sitting at age 1 year and no walking or words at age 3 years and 5 mo. At age 3 years and 5 mo, she had a height of 92.5 cm (2nd percentile) and a head circumference of 45.5 cm (−2.5 SD). Her facial features included a high broad forehead, full round face, light-blue irises with a dark rim, small ears with large uplifted ear lobules, a broad mouth with a short philtrum and full lips, and a broad chin. Her hands were short and broad, with tapering fingers and clinodactyly of the fifth fingers, and her feet were broad, with short toes and hypoplasia of the toenails. Her behavior was abnormal, with frequent picking of the eyes, periods of hyperpnea, and the putting of full hands into the mouth. She was given treatment for epilepsy.
Extensive studies—which included analysis of chromosomes with the subtelomeric regions; DNA studies for fragile X, Angelman, Rett, and Smith-Magenis syndromes; and cerebral MRI—did not reveal any abnormalities. Array CGH revealed a de novo deletion on chromosome 2q23.1q23.2 (0.92 Mb), which was confirmed by FISH (fig. A2).
Figure A2.

Details on patient 2, showing the genomewide array CGH profile, with the 0.92-Mb deletion on 2q23 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 2 (deletion indicated [red arrow in panel II]), confirmation of the de novo deletion by MLPA of patient DNA (panel III), and confirmation by FISH (panel IV) using clone RP11-469G04 (green).
Patient 3
This severely mentally retarded, 22-year-old male was born at 38 wk of gestation, with a low birth weight of 1,620 g (<3rd percentile) and a short length of 39 cm (<3rd percentile). Facial dysmorphism—including downward-slanting palpebral fissures; low-set, simply formed ears; low nasal bridge; and small mouth—and hypogenitalism with a bifid scrotum were noted at birth. In the years that followed, both the growth retardation and the MR of the patient became more obvious, with a height of 119 cm (−4 SD) and a head circumference of 46 cm (−4 SD) at age 12 years. In addition to the dysmorphism, he had a double row of teeth, hypoplastic kidneys, and mixed conductive/perceptive deafness.
Array CGH revealed a de novo deletion on chromosome 3q27.1-q29 (12.37 Mb), which was confirmed by FISH (fig. A3)
Figure A3.

Details on patient 3, showing the genomewide array CGH profile, with the 12.37-Mb deletion on 3q27-3q29 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 3 (deletion indicated [red arrow in panel II]), confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III), and confirmation by FISH (panel IV) using clone RP11-469G04 (red).
Patient 4
This 15-year-old, mildly mentally retarded boy was born at term after a normal gestation, with a normal birth weight of 3,000 g. He had an asymmetric crying face and preaxial polydactyly on the right hand. Although he had delayed motor development (sitting at age 9 mo and walking at age 2 years), his speech developed normally. He developed epilepsy at age 6 years; subsequently, an MRI of the brain showed a semilobular holoprosencephaly. A small right kidney and a normal left kidney were detected. At age 15 years, the patient was small in stature (height 1.57 m; <3rd percentile), with a normal head circumference of 57 cm (75th percentile), an asymmetric face (left < right), synophrys, upslanting palpebral fissures, finger-like thumbs, remnants of the polydactyly near the right thumb, and a sandal gap between the first and second toes.
Array CGH revealed a de novo duplication on chromosome 5q35.1 (1.24 Mb), which was confirmed by MLPA (fig. A4).
Figure A4.

Details on patient 4, showing the genomewide array CGH profile, with the 1.24-Mb duplication on 5q35 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 5 (duplication indicated [red arrow in panel II]), and confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III).
Patient 5
This 3-year-old, moderately mentally retarded girl was born at term, with a birth weight of 3,000 g and a transposition of the great vessels. At 2.5 years of age, she had a normal height but a small head (circumference 44.5 cm; <2.5 SD) with a plagiocephalic shape. She had a coarse facies with hypertelorism, upward-slanting palpebral fissures, broad mouth, and low-set ears and had short and broad hands and feet, with short fifth fingers. MRI of the brain showed frontal brain atrophy.
Array CGH revealed a de novo deletion on chromosome 9q31.1 (2.85 Mb), which was confirmed by FISH (fig. A5).
Figure A5.

Details on patient 5, showing the genomewide array CGH profile, with the 2.85-Mb deletion on 9q31 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 9 (deletion indicated [red arrow in panel II]), confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III), and confirmation by FISH (panel IV) using clone RP11-177I08 (green) and a centromere probe as the control (red).
Patient 6
This mildly mentally retarded, 16-year-old boy was born prematurely at 33 wk and 5 d, with a normal birth weight of 2,030 g and a relatively large head circumference of 34.6 cm. His motor milestones were mildly delayed (sitting at age 13 mo and walking at age 18 mo), and his IQ score was 66 points at age 5 years. He had a dorsal fusion of the second and third cervical vertebrae, whereas his mother’s sister had full Klippel-Feil syndrome. He had one epileptic seizure at age 15 years. His behavior was within the autistic spectrum. At age 16 years, he had a normal height of 1.71 m (15th percentile), with macrocephaly (head circumference 61.5 cm; +3 SD), minor facial asymmetry (right < left), slightly everted ears, long fingers, clinodactyly of the fourth and fifth toes, and striae on the lower back. Other family members on the mother’s side (notably, the mother's father and sister) also had large head circumferences.
Array CGH revealed a de novo deletion on chromosome 9q33.1 (0.54 Mb), which was confirmed by FISH (fig. A6).
Figure A6.

Details on patient 6, showing the genomewide array CGH profile, with the 0.54-Mb deletion on 9q33 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 9 (deletion indicated [red arrow in panel II]), confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III), and confirmation by FISH (panel IV) using clone RP11-668H03 (red).
Patient 7
This 11-year-old, mildly mentally retarded boy was born at 42 wk, after an uneventful pregnancy, with a birth weight of 3,490 g and a head circumference of 35 cm. His motor development was mildly delayed, with walking at age 18 mo. His IQ was 68 points at age 5 years.
At age 6 years, he had a normal height of 117 cm (15th percentile) and a normal head circumference of 50 cm (15th percentile). He had upward-slanting palpebral fissures; epicanthus inversus; mild ptosis; a broad nasal bridge; a thin upper lip; micrognathia; short hands and fingers, with clinodactyly of the fifth fingers and distal brachydactyly of all fingers; and a shawl scrotum. Perthes disease of the left hip was diagnosed. MRI of the brain revealed a partial temporal agenesis. Results of routine chromosomal analysis of lymphocytes and fibroblasts were normal, including normal subtelomeric regions determined using the MLPA technique.
Array CGH revealed a de novo deletion on chromosome 11q14.1-q14.2 (7.49 Mb), which was confirmed by FISH (fig. A7).
Figure A7.

Details on patient 7, showing the genomewide array CGH profile, with the 7.49-Mb deletion on 11q14 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 11 (deletion indicated [red arrow in panel II]), confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III), and confirmation by FISH (panel IV) using clone RP11-118L16 (green) and a centromere probe as the control (red).
Patient 8
This severely mentally retarded girl was born after 42 wk of gestation, with a normal birth weight of 2,565 g and a normal head circumference of 34.2 cm. At age 4 mo, delayed development and lack of (eye) contact were noted. In the years that followed, further studies—including chromosome analysis of lymphocytes and fibroblasts, MRI of the brain, metabolic studies, MECP2 analysis, and methylation studies of the Angelman region (15q11-q13)—revealed no abnormalities, except for a small cerebrum, which fit the development of brachymicrocephaly. Her motor developments were delayed, with sitting at age 2 years and walking at age 3 years. She lacked any speech and developed behavioral disturbances (i.e., aggressiveness). At 7.5 years of age, she was short in stature (1.13 m; −2.5 SD) and microcephalic (head circumference 48 cm; −2 SD), with hypertelorism; epicanthic folds; arched eyebrows; normally shaped but low-set ears; a short philtrum with an open mouth appearance and full lips, with everted lower lip; broad gums with a flattened palate; clinodactyly of the fifth fingers; distal brachydactyly of the toes; and two café au lait spots on the right leg.
Array CGH revealed a de novo duplication on chromosome 12q24.22 (2.3 Mb), which was confirmed by MLPA (fig. A8).
Figure A8.

Details on patient 8, showing the genomewide array CGH profile, with the 2.3-Mb duplication on 12q24 (red arrow in panel I), the detailed array CGH profile of the affected chromosome 12 (duplication indicated [red arrow in panel II]), and confirmation of the de novo deletion by MLPA of both patient and parental DNA (panel III).
Patient 9
This 6-year-old, moderately mentally retarded girl was born after an uneventful pregnancy at 40 wk, with a normal birth weight of 3,400 g. She had a slow start, with Apgar scores of 3, 5, and 6 at 1 min, 5 min, and 10 min, respectively. From birth onward, feeding problems were present. Her development appeared to be mildly delayed, with walking at age 18 mo and speaking her first words at age 2 years. Her IQ was 50 points (Snijders-Oomen Non-Verbal Intelligence Tests–Revised) at age 4 years and 9 mo.
At age 6 years and 10 mo, she presented with a small stature of 110 cm (<3rd percentile) and a head circumference of 49 cm (10th percentile). Facial features observed were synophrys, upward slanting of the palpebral fissures, flattened upper-ear helices with large ear lobules, upturned nasal tip, and a broad mouth with a long philtrum and full lower lip. She had a cardiac murmur; short broad hands, with clinodactyly of the fifth fingers and absent distal interphalangeal joint creases; and short broad feet, with short toes and a slight skin syndactyly between the second and third toes bilaterally. She had an outgoing personality and could easily approach strangers.
Results of routine chromosomal analysis were normal, including normal subtelomeric regions determined using the MLPA technique. Array CGH revealed four duplicated regions on chromosome 17p: p13.2p13.1 (2.89 Mb), p13.1p13.1 (1.43 Mb), p12p12 (2.88 Mb), and p11.2p11.2 (1.48 Mb). The duplications were de novo (fig. A9).
Patient 10
This 4-year-old, mildly mentally retarded boy was born at term after an uneventful pregnancy, with a normal birth weight of 3,450 g. At age 3 years, he had a small stature (88 cm; −2.5 SD) and a normal head circumference (49 cm; 15th percentile). He had long eyelashes, upward-slanting palpebral fissures, periorbital fullness, small low-set ears, a normal palate, sharply edged upper-canine teeth, a thin upper lip, and a simian crease of the right hand, with tapering fingers.
Array CGH revealed a de novo deletion on chromosome 22q11 (2.66 Mb), which was confirmed by FISH (fig. A10).
Web Resources
The URLs for data presented herein are as follows:
- BACPAC Resources Center, http://bacpac.chori.org/
- Database of Genomic Variants, http://projects.tcag.ca/variation/
- ECARUCA–European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations, http://www.ecaruca.net/ [DOI] [PubMed]
- MRC-Holland, http://www.mlpa.com/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for CMT, DiGeorge syndrome, and VCFS)
References
- Baralle D (2001) Chromosomal aberrations, subtelomeric defects, and mental retardation. Lancet 358:7–8 10.1016/S0140-6736(00)05300-9 [DOI] [PubMed] [Google Scholar]
- Biesecker LG (2002) The end of the beginning of chromosome ends. Am J Med Genet 107:263–266 10.1002/ajmg.10160 [DOI] [PubMed] [Google Scholar]
- de Vries BB, van den Ouweland AM, Mohkamsing S, Duivenvoorden HJ, Mol E, Gelsema K, van Rijn M, Halley DJ, Sandkuijl LA, Oostra BA, Tibben A, Niermeijer MF, for the Collaborative Fragile X Study Group (1997) Screening and diagnosis for the fragile X syndrome among the mentally retarded: an epidemiological and psychological survey. Am J Hum Genet 61:660–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries BB, White SM, Knight SJ, Regan R, Homfray T, Young ID, Super M, McKeown C, Splitt M, Quarrell OW, Trainer AH, Niermeijer MF, Malcolm S, Flint J, Hurst JA, Winter RM (2001) Clinical studies on submicroscopic subtelomeric rearrangements: a checklist. J Med Genet 38:145–150 10.1136/jmg.38.3.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries BB, Winter R, Schinzel A, Ravenswaaij-Arts C (2003) Telomeres: a diagnosis at the end of the chromosomes. J Med Genet 40:385–398 10.1136/jmg.40.6.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint J, Wilkie AO, Buckle VJ, Winter RM, Holland AJ, McDermid HE (1995) The detection of subtelomeric chromosomal rearrangements in idiopathic mental retardation. Nat Genet 9:132–140 10.1038/ng0295-132 [DOI] [PubMed] [Google Scholar]
- Forney GD (1973) The Viterbi algorithm. Proc IEEE Inst Electr Electron Eng 61:268–278 [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large-scale variation in the human genome. Nat Genet 36:949–951 10.1038/ng1416 [DOI] [PubMed] [Google Scholar]
- Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, Snijders A, Albertson DG, Pinkel D, Marra MA, Ling V, MacAulay C, Lam WL (2004) A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet 36:299–303 10.1038/ng1307 [DOI] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N (2000) KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci 20:6374–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SJ, Regan R, Nicod A, Horsley SW, Kearney L, Homfray T, Winter RM, Bolton P, Flint J (1999) Subtle chromosomal rearrangements in children with unexplained mental retardation. Lancet 354:1676–1681 10.1016/S0140-6736(99)03070-6 [DOI] [PubMed] [Google Scholar]
- Koolen DA, Nillesen WM, Versteeg MH, Merkx GF, Knoers NV, Kets M, Vermeer S, van Ravenswaaij CM, de Kovel CG, Brunner HG, Smeets D, de Vries BB, Sistermans EA (2004a) Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet 41:892–899 10.1136/jmg.2004.023671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Vissers LE, Nillesen W, Smeets D, van Ravenswaaij CM, Sistermans EA, Veltman JA, de Vries BB (2004b) A novel microdeletion, del(2)(q22.3q23.3), in a mentally retarded patient, detected by array-based comparative genomic hybridization. Clin Genet 65:429–432 10.1111/j.0009-9163.2004.00245.x [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Bosdet I, Smailus D, Chiu R, Mathewson C, Wye N, Barber S, Brown-John M, Chan S, Chand S, Cloutier A, Girn N, Lee D, Masson A, Mayo M, Olson T, Pandoh P, Prabhu AL, Schoenmakers E, Tsai M, Albertson D, Lam W, Choy CO, Osoegawa K, Zhao S, de Jong PJ, Schein J, Jones S, Marra MA (2004) A set of BAC clones spanning the human genome. Nucleic Acids Res 32:3651–3660 10.1093/nar/gkh700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay EA (2001) Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genet 2:858–868 10.1038/35098574 [DOI] [PubMed] [Google Scholar]
- Lucito R, Healy J, Alexander J, Reiner A, Esposito D, Chi M, Rodgers L, Brady A, Sebat J, Troge J, West JA, Rostan S, Nguyen KC, Powers S, Ye KQ, Olshen A, Venkatraman E, Norton L, Wigler M (2003) Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res 13:2291–2305 10.1101/gr.1349003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase T, Kikuno R, Ishikawa K, Hirosawa M, Ohara O (2000) Prediction of the coding sequences of unidentified human genes. XVII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res 7:143–150 [DOI] [PubMed] [Google Scholar]
- Phelan MC, Crawford EC, Bealer DM (1996) Mental retardation in South Carolina. In: Saul RA, Phelan MC (eds) Proceedings of the Greenwood Genetic Center. Greenwood Genetic Center, Greenwood, SC, pp 45–60 [Google Scholar]
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, Dairkee SH, Ljung BM, Gray JW, Albertson DG (1998) High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 20:207–211 10.1038/2524 [DOI] [PubMed] [Google Scholar]
- Rabiner LR (1989) A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE Inst Electr Electron Eng 77:257–286 [Google Scholar]
- Ried T (2004) Cytogenetics—in color and digitized. N Engl J Med 350:1597–1600 10.1056/NEJMp038242 [DOI] [PubMed] [Google Scholar]
- Roeleveld N, Zielhuis GA, Gabreels F (1997) The prevalence of mental retardation: a critical review of recent literature. Dev Med Child Neurol 39:125–132 [DOI] [PubMed] [Google Scholar]
- Roloff TC, Ropers HH, Nuber UA (2003) Comparative study of methyl-CpG-binding domain proteins. BMC Genomics 4:1 10.1186/1471-2164-4-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg C, Knijnenburg J, Bakker E, Vianna-Morgante A, Sloos WC, Otto PA, Kriek M, Hansson K, Krepisch-Santos AC, Fiegler H, Carter NP, Bijlsma EK, van Haeringen A, Szuhai K, Tanke HJ (2005) Array-CGH detection of micro rearrangements in mentally retarded individuals: clinical significance of imbalances present both in affected children and normal parents. J Med Genet (http://jmg.bmjjournals.com/cgi/rapidpdf/jmg.2005.032268v1) (electronically published June 24, 2005; accessed August 22, 2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel A (2001) Catalogue of unbalanced chromosome aberrations in man. 2nd ed. Walter de Gruyter, Berlin [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G (2002) Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 30:e57 10.1093/nar/gnf056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M (2004) Large-scale copy number polymorphism in the human genome. Science 305:525–528 10.1126/science.1098918 [DOI] [PubMed] [Google Scholar]
- Shaw-Smith C, Redon R, Rickman L, Rio M, Willatt L, Fiegler H, Firth H, Sanlaville D, Winter R, Colleaux L, Bobrow M, Carter NP (2004) Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet 41:241–248 10.1136/jmg.2003.017731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Dohner H, Cremer T, Lichter P (1997) Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20:399–407 [DOI] [PubMed] [Google Scholar]
- Suter U, Scherer SS (2003) Disease mechanisms in inherited neuropathies. Nat Rev Neurosci 4:714–726 10.1038/nrn1196 [DOI] [PubMed] [Google Scholar]
- Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A (1992) Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13:718–725 10.1016/0888-7543(92)90147-K [DOI] [PubMed] [Google Scholar]
- Veltman JA, Schoenmakers EF, Eussen BH, Janssen I, Merkx G, van Cleef B, van Ravenswaaij CM, Brunner HG, Smeets D, Geurts van Kessel A (2002) High-throughput analysis of subtelomeric chromosome rearrangements by use of array-based comparative genomic hybridization. Am J Hum Genet 70:1269–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Yntema HG, Lugtenberg D, Arts H, Briault S, Huys EH, Osoegawa K, de Jong P, Brunner HG, Geurts van Kessel A, van Bokhoven H, Schoenmakers EF (2004) High resolution profiling of X chromosomal aberrations by array comparative genomic hybridisation. J Med Genet 41:425–432 10.1136/jmg.2004.018531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LELM, de Vries BBA, Osoegawa K, Janssen IM, Feuth T, Choy CO, Straatman H, van der Vliet W, Huys EHLPG, van Rijk A, Smeets D, van Ravenswaaij-Arts CMA, Knoers NV, van de Burgt I, de Jong PJ, Brunner HG, Geurts van Kessel A, Schoenmakers EFPM, Veltman JA (2003) Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am J Hum Genet 73:1261–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeargin-Allsopp M, Murphy CC, Cordero JF, Decoufle P, Hollowell JG (1997) Reported biomedical causes and associated medical conditions for mental retardation among 10-year-old children, metropolitan Atlanta, 1985 to 1987. Dev Med Child Neurol 39:142–149 [DOI] [PubMed] [Google Scholar]
- Yin L, Yang R (1996) Weighted median filters: a tutorial. IEEE Trans Circuits Syst 43:157–192 10.1109/82.486465 [DOI] [Google Scholar]