

Abstract
Studies of genetic contributions to risk can be family-based, such as the case-parents design, or population-based, such as the case-control design. Both provide powerful inference regarding associations between genetic variants and risks, but both have limitations. The case-control design requires identifying and recruiting appropriate controls, but it has the advantage that nongenetic risk factors like exposures can be assessed. For a condition with an onset early in life, such as a birth defect, one should also genotype the mothers of cases and the mothers of controls to avoid potential confounding due to maternally mediated genetic effects acting on the fetus during gestation. The case-parents approach is less vulnerable than the case-mother/control-mother approach to biases due to population structure and self-selection. The case-parents approach also allows access to epigenetic phenomena like imprinting, but it cannot evaluate the role of nongenetic cofactors like exposures. We propose a hybrid design based on augmenting a set of affected individuals and their parents with a set of unaffected, unrelated individuals and their parents. The affected individuals and their parents are all genotyped, whereas only the parents of unaffected individuals are genotyped, although exposures are ascertained for both affected and unaffected offspring. The proposed hybrid design, through log-linear, likelihood-based analysis, allows estimation of the relative risk parameters, can provide more power than either the case-parents approach or the case-mother/control-mother approach, permits straightforward likelihood-ratio tests for bias due to mating asymmetry or population stratification, and admits valid alternative analyses when mating is asymmetric or when population stratification is detected.
Introduction
Diseases with an onset early in life, such as asthma, pregnancy complications, schizophrenia, and birth defects, arise through complex etiologies involving both genetic and environmental components. The genetic component itself might be complex, involving both the affected individual’s inherited genotype and maternally mediated mechanisms—that is, the effects of the mother’s genotype on her own phenotype, which can perturb the development of her fetus during gestation. An example is maternal phenylketonuria (PKU), an autosomal recessive metabolic disease that can lead to congenital heart defects in the offspring (Lee et al. 2005).
To elucidate the etiology of early-life conditions, epidemiologists typically use the case-control design. This design is population-based, because it assumes cases and controls are randomly sampled from the population that gave rise to the cases. Epidemiologists compare the genotypes and exposure histories of the population controls with those of the cases (Rothman and Greenland 1998). Geneticists, however, worry about population structure with case-control studies, because bias can arise if mating is assortative and a subpopulation has both a higher prevalence of the allele under study and a higher baseline incidence of the disease. Some have argued that bias due to genetic population structure is negligible in most settings, even in an incompletely mixed population like that of the United States (Wacholder et al. 2002), but concern about confounding due to population structure continues to fuel a preference among geneticists for family-based inference and to stimulate the development of protective analytic strategies, such as the use of unlinked ethnicity markers (Pritchard and Rosenberg 1999; Devlin et al. 2001).
Falk and Rubinstein (1987) suggested the case-parents approach, applying it to the problem of assessing effects of human leukocyte antigen (HLA) genotypes on risk of diabetes. With the family-based design, one, in effect, uses the nontransmitted parental alleles in parents of affected individuals as their genetic controls. The case and both parents are genotyped, and one looks for a pattern in which a particular allele was transmitted more than half the time from heterozygous parents to affected offspring. Such apparent distortions give evidence that the allele either is itself causal or is in linkage disequilibrium with another allele involved in the etiology of the condition. This insight led to the well-known transmission/disequilibrium test (TDT) (Spielman et al. 1993).
Analyses of case-parents data that condition on the parental genotype—for example, the TDT (Spielman and Ewens 1996) or likelihood-based approaches (Self et al. 1991; Schaid and Sommer 1993; Weinberg et al. 1998)—are inherently resistant to biases due to population structure, because conditioning on the parental genotype ensures that the inference is based purely on the apparent transmission rate from heterozygous parents to affected offspring. Likelihood-based methods allow estimation of relative risks associated with one and two copies of the variant allele in the offspring and, also, the relative risks associated with the maternal genotype, permitting one to disentangle the offspring-mediated genetic effects from those that act through the mother’s genotype/phenotype during gestation (Wilcox et al. 1998).
Besides its ability to resist potential biases from genetic population structure, the case-parents approach has practical advantages over the case-control approach. For example, parents of a baby born with a birth defect are usually available and willing to help investigate its causes. Furthermore, the power for a case-parents design when there are only offspring-mediated effects, and not maternally mediated effects, appears to be comparable to that for a case-control design with the same number of cases (Wilcox et al. 1998).
The major drawback of the case-parents design is that, although one can assess gene-by-environment interaction in relation to a multiplicative null model, one cannot estimate the “main effects” of an exposure (Umbach and Weinberg 2000). Thus, for example, one might be able to infer that the relative risk associated with an exposure is higher in one genotype group than in another, but not even be able to tell whether the relative risk conferred by the exposure is >1 (deleterious) or <1 (protective) in any of the genotype groups. In addition, inferences rely on key assumptions that cannot readily be checked with case-parents data only.
The case-control approach has its own shortcomings. Although they are not usually included in case-control studies involving genetic contributions to childhood diseases, mothers should be studied for many loci potentially influencing susceptibility to conditions with onset early in life, because the most important potential confounder for the offspring genotype is the maternal genotype. Thus, rather than studying case children and a sample of control children, one would prefer to study case-mother pairs as the fundamental unit of analysis and to compare those pairs with control-mother pairs, adjusting the model for maternal genotype when looking at the effects of the offspring genotype, and adjusting for the offspring genotype when looking at the effect of the maternal genotype (Mitchell 2003). The relative advantages and disadvantages of these family-based versus population-based approaches are summarized as follows:
Case-Control Design Strengths
-
•
Inference can include both main effects of exposure and main effects of genotypes.
-
•
Nonmultiplicative models can be fit for joint effects of genetic and environmental factors.
-
•
Non-Mendelian inheritance (e.g., due to gene-related attrition during gestation) will not bias the results.
-
•
If mothers are studied too, the design allows maternally mediated effects to be distinguished from offspring-mediated effects.
Case-Control Design Vulnerabilities
-
•
Population structure and self-selection can potentially cause bias, as can recall, if differential by case/control status.
-
•
To avoid confounding, mothers should also be studied (i.e., two individuals need to be genotyped for each case and each control).
-
•
Inference related to maternal effects is ambiguous, because imprinting cannot be studied.
-
•
Suitable controls may be hard to identify and harder to recruit; low participation rates weaken the validity of the inferences.
Case-Parents Design Strengths
-
•
Robust against hidden genetic population structure. Parents provide ideal controls for genetic effects.
-
•
Parents are easy to recruit for studies of diseases in their children.
-
•
Self-selection is not a serious issue, because one studies transmission within families, conditioning on the parental genotypes.
-
•
The design allows maternally mediated effects to be distinguished from offspring-mediated effects and can also be used to study imprinting effects.
Case-Parents Design Vulnerabilities
-
•
Problematic for diseases with onset in later life, when availability of parents is low, and may be genetically selective.
-
•
Cannot estimate main effects of exposures.
-
•
Three individuals need to be genotyped for each case.
-
•
Relies on Mendelian proportions in the source population, which may not hold.
-
•
Estimation of maternally mediated effects relies on assumed genetic mating symmetry in the population, an assumption that can fail.
-
•
Assessment of gene-by-exposure interaction relies on within-family independence of genotype and exposure.
-
•
Needed assumptions cannot be tested with case-parents data alone.
One would like an approach that can bring the strengths of these two designs together into a single design/analytic framework. One choice would be a two-component study, where one identifies loci related to risk based on a population-based, case-control comparison and then performs a family-based confirmatory analysis of apparent genetic effects by genotyping the parents of the cases and analyzing the case-parents triads. The confirmatory second-stage, family-based tests are, however, not statistically independent of the first-stage, case-control analysis. Martin and Kaplan (2000) addressed this issue by devising a Monte-Carlo procedure to correct the type I error rate for the two-stage procedure, but they did not allow for possible maternally mediated effects.
Refining an approach introduced by Nagelkerke et al. (2004), Epstein et al. (2005) recently proposed a likelihood-based, combined analysis of multicomponent studies, where genotype data are available for cases and their parents and also for unrelated cases and/or unrelated controls. They show that statistical tests based on the combined data offer improved power over analyses based on either the case-control substudy or the case-parents substudy alone. While it generalizes an earlier approach (Nagelkerke et al. 2004) by not requiring random mating or Hardy-Weinberg equilibrium, the proposed analysis is not inherently protected against bias due to population stratification, although they describe some indirect approaches for assessing the combinability of the affected-family–based and population-based data components. Epstein et al. do not allow for maternally mediated genetic effects. However, our proposal is related to those of Nagelkerke et al. and Epstein et al., in that both these approaches and ours gain efficiency by using an auxiliary sample to increase the precision of inference on mating frequencies in the population. The main difference is that a sample of unrelated controls gives only partial information on these frequencies, whereas a sample of parents of unaffected children, as called for in our proposal, gives direct information on mating frequencies. As we show, this difference not only allows for greater efficiency but also allows more options for detecting and accounting for population stratification.
Material and Methods
A natural way to combine the advantages of population-based and affected-family–based designs is to randomly sample cases and controls and also enroll their parents. The cases and their parents are genotyped, as are the parents of the controls, but the controls themselves are not. Both cases and controls provide exposure information. The unit of analysis is the family.
We require a few assumptions. We assume that the condition under study is rare for the offspring of each parental genotype combination for the diallelic locus under study and that Mendelian proportions are the rule for the locus in the population at large. We assume neither Hardy-Weinberg equilibrium nor random mating.
The information provided by this design comes from two separate components. The parents of cases and of controls provide for population-based inferences related to both maternally mediated genetic effects that act during gestation and genetic effects that act directly on the offspring through the inherited genotype. This component allows the analyst to exploit the fact that an association with a particular allele serves to enrich the relative frequency of that allele in parents of affected (compared with unaffected) offspring in ways that are simple to characterize mathematically. This enrichment is captured under this hybrid design but is neglected in affected-family–based analyses that condition on the parental genotypes and test only for distortions in transmissions to affected offspring. The second component in the hybrid is the affected-family–based component, which provides statistically independent additional evidence related to genetic effects and offers protection against population stratification.
Reflecting the two components of the design, one possible analytic approach employs two stages. One can perform population-based comparisons at the level of the parental generation. Findings based on apparent offspring genetic effects can then be confirmed in a second, statistically independent analysis that, by use of the case-parents triads, now conditions on the parental genotypes and analyzes transmissions to affected offspring.
If, through a test that we will describe, the data support the validity of inference based on the population-based component, then a single log-linear model can pull the two components into a unified and powerful analytic framework. But first, one needs to test for bias due to population stratification. If the no-bias null hypothesis is rejected, one cannot safely include the population-based parent-parent component of the study, but one can still fall back to the case-parents-triad analysis, which remains valid even with population stratification.
Whereas the assessment of offspring-mediated genetic effects uses information both from the parent-based comparison and from the apparent distortions in transmission to affected individuals conditional on their parents, the assessment of maternally mediated genetic effects relies necessarily on the parental generation. Mating symmetry refers to the frequent assumption (Schaid and Sommer 1993; Wilcox et al. 1998) that, for any possible pair of parental genotypes, the frequency in the population for one assignment to mother and father is the same as that for the reverse assignment. The choice of analytic approach for use with our hybrid design depends on mating symmetry. If one suspects mating asymmetry, then the inference related to maternal effects can and should rely on a comparison of case parents and control parents, now stratified into the nine categories based on the ordered parental genotypes. If, on the other hand, the data support mating symmetry, then one can exploit that assumption to gain a more powerful analysis by stratifying on only the six unordered mating-type categories to be defined.
In short, within our log-linear framework, one can test for population structure and for mating asymmetry, as well as tailor analyses to eliminate the potential biases induced by those problems. Although the best power is achieved when population stratification is absent and mating is symmetric, failure of either assumption can be readily accommodated.
First, we require some notation. Let M, F, and C denote the number of copies (0, 1, or 2) of the variant allele carried by the mother, the father, and the affected child, respectively. Either allele may be designated as the “variant,” since all the estimation and testing results are predictably interchangeable under the complementary designation. Let D be an indicator variable that equals 1 for families with an affected offspring and 0 for control families.
As originally proposed elsewhere (Schaid and Sommer 1993), we define six parental mating-type categories based on the unordered values of M and F. The genetic outcomes fall into 24 possible cells, forming the multinomial distribution shown in table 1, under a multiplicative model. In this table, the parameters μ1, μ2, … μ6 are proportional to the relative frequencies in the population for the mating-type categories. Those proportions are important nuisance parameters because they allow stratification on parental mating types, which confers robustness for the case-parents analysis against bias due to population structure. R1, R2, S1, and S2 are the relative risk parameters for offspring-mediated effects, corresponding to offspring with C of 1 or 2 (relative to C=0), and maternally mediated effects, corresponding to mothers with M of 1 or 2 (relative to M=0), respectively (Weinberg et al. 1998). B is a stratification and normalization parameter corresponding to disease status for the offspring. Inclusion of B ensures that the total across the fitted expected counts of triads and the total across the fitted expected number of control couples each equal the corresponding observed total. The logarithm of each expected count is linear in parameters, which implies that standard software for Poisson regression (e.g., SAS) can be used to estimate parameters and perform likelihood-ratio tests for hypotheses of interest.
Table 1.
Structure of Data from Proposed Hybrid Design, Using Parents of Unrelated Controls
|
Parameters |
||||
| MFa | Cb | Mating Type | Affected | Expected Count |
| 00 | 0 | 1 | Yes | Bμ1 |
| 01 | 0 | 2 | Yes | Bμ2/4 |
| 01 | 1 | 2 | Yes | Bμ2R1/4 |
| 10 | 0 | 2 | Yes | Bμ2S1/4 |
| 10 | 1 | 2 | Yes | Bμ2R1 S1/4 |
| 02 | 1 | 3 | Yes | Bμ3R1/2 |
| 20 | 1 | 3 | Yes | Bμ3R1S2/2 |
| 11 | 0 | 4 | Yes | Bμ4S1/4 |
| 11 | 1 | 4 | Yes | Bμ4R1S1/2 |
| 11 | 2 | 4 | Yes | Bμ4R2S1/4 |
| 12 | 1 | 5 | Yes | Bμ5R1S1/4 |
| 12 | 2 | 5 | Yes | Bμ5R2S1/4 |
| 21 | 1 | 5 | Yes | Bμ5R1S2/4 |
| 21 | 2 | 5 | Yes | Bμ5R2S2/4 |
| 22 | 2 | 6 | Yes | Bμ6R2S2 |
| 00 | … | 1 | No | μ1 |
| 01 | … | 2 | No | μ2/2 |
| 10 | … | 2 | No | μ2/2 |
| 02 | … | 3 | No | μ3/2 |
| 20 | … | 3 | No | μ3/2 |
| 11 | … | 4 | No | μ4 |
| 12 | … | 5 | No | μ5/2 |
| 21 | … | 5 | No | μ5/2 |
| 22 | … | 6 | No | μ6 |
No. of copies of variant allele carried by the mother and father.
No. of copies of variant allele carried by the child.
The log-linear model corresponding to the multinomial distribution of table 1 is
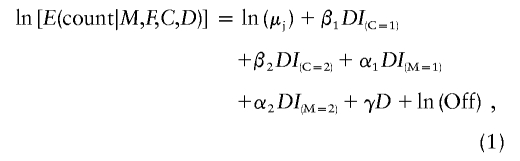 |
where “Off” is the probability multiplier (1, 1/2, or 1/4) shown in table 1 for the particular cell. The relative risk R1 corresponds to exp(β1), R2 to exp(β2), S1 to exp(α1), S2 to exp(α2), and γ to ln(B). Likelihood-ratio tests for any subset of the four relative risk parameters can be performed by computing twice the change in the maximized log likelihood for models with, compared to without, the selected term(s), and comparing that difference with the critical value for a χ2 distribution with df equal to the difference in the number of parameters being fitted. Although difficult to justify when studying a complex condition, the model can also be simplified in the usual ways to allow for dominant, recessive, or log-additive effects, the latter reduction being advisable if the data are sparse for a particular locus.
An important feature of model (1) is that it implicitly imposes the assumption that the baseline risk of disease does not vary across subpopulations and, hence, does not vary across parental mating types. Thus, it specifies that there is no bias due to population structure. This assumption is crucial for the case-control component of the data to provide unbiased information related to the relative risk parameters. It is also testable within the log-linear framework.
A more general model, which allows for bias due to population structure, augments model (1) with an interaction term between disease status D and the mating-type stratification parameters. The improvement in fit that is thereby achieved can be tested by a 5-df likelihood-ratio test. In this way, one can test whether the combined-data inference regarding the relative risk parameters is vulnerable to bias due to population structure. A relatively large P value provides reassurance that the family-based case-parents-triad component and the population-based parent-parent component are safe to combine through the simplified model (1).
Note that if disease-by-mating-type parameters are required, on the basis of the above likelihood-ratio test, then their inclusion in the model indicates that the mating-type stratum parameters are different for case parents and control parents. A direct consequence of preferring the enlarged model is that the control portion of the data will not contribute to inference related to the risk parameters, and the population-based component, in effect, becomes statistically irrelevant. This drastic consequence is the appropriate penalty, given that the need for that set of interaction parameters implies that the parent-level population-based estimates are not to be trusted because the data show bias due to population stratification. Thus, in such a scenario, we can and must confine ourselves to the case-parents-triad substudy.
Because the test for bias due to population stratification yields a χ2 statistic with 5 df, it may have limited power against realistic alternatives. One possible approach for increasing sensitivity is to replace the 5-df likelihood-ratio test with a 1-df test for a trend across mating-type parameters in their departure from equality for case and control parents. To do this, we fit the full model (1) and then include the predictor, (M+F)D, testing for improvement in fit. Under the no-bias null hypothesis, the coefficient of this added predictor is 0, and the corresponding likelihood-ratio statistic is χ2 with 1 df.
In addition to the assumption of no bias due to population stratification, an assumption implicitly embodied by table 1—and one that can also be tested—is that parental mating is symmetric. This symmetry is implied, for example, in the splitting of the frequency for the second mating type for control parents into simply μ2/2 for M=0, F=1; and μ2/2 for M=1, F=0. To relax this stricture, define an indicator variable I(M>F) to be 1 when M > F and 0 otherwise. Then add to model (1) an interaction between I(M>F) and mating type, in effect producing nine parental strata to take the place of the original six. Again, a likelihood-ratio test (3 df) can be used to test the improvement in fit achieved by allowing for mating asymmetry. When this goodness-of-fit test suggests that we cannot trust the symmetry assumption (e.g., if P<.10), then we need to base inference on models with nine ordered parental genotype categories instead of the usual six. This means that the inference regarding maternal genetic effects must be based on the comparison of case parents and control parents. Again, this consequence is appropriate, given that the analysis based on case-parents triads relies on symmetry and should not be trusted for assessment of maternal effects in the absence of parental symmetry in the population. As would be expected, some loss of power is the price for the now more trustworthy analysis that abandons the assumption of mating symmetry. But, again, the hybrid design has saved the study here by providing a still-valid analytic framework for inference.
To assess the relative efficiency of various design/analytic options, we used expected cell counts to calculate χ2 noncentrality parameters for likelihood-ratio tests of the four relative risk parameters (4 df) (Agresti 1990). We compared analyses based on our hybrid design with those for the case-parents-triad design and the case-mother/control-mother design, under selected scenarios with offspring-mediated and maternally mediated genetic effects. We also considered a design in which a case-parents-triad study with a particular number of case families is augmented by an equal number of unrelated population-based controls, following the design considered by Nagelkerke et al. (2004) and Epstein et al. (2005).
We considered two forms of our hybrid design’s likelihood-ratio test: one designated “Hybrid MS,” which imposes mating symmetry by including six dummy variables for mating type, and one designated “Hybrid MA,” which, instead, permits valid inference under asymmetry of genotypes for M and F by including nine dummy variables, as discussed above. For simplicity, our calculations assumed Hardy-Weinberg equilibrium, although, again, the method does not require that assumption.
Although Epstein et al. did not consider maternally mediated genetic effects, if one is willing to assume mating symmetry in the population at large, then a study that genotypes controls rather than their parents can also be used for inference related to all four relative risk parameters, including the two maternal parameters. We need three key assumptions: that unaffected offspring genotypes follow Mendelian proportions in relation to their parents, that there is no population stratification, and that there is mating symmetry. Under those assumptions, the control children serve as useful genetic surrogates for their parents, and missing-data methods can be used to maximize the likelihood, even allowing for maternal genetic effects. The expected cell counts for unaffected offspring with 0, 1, or 2 copies of the variant are μ1+μ2/2+μ4/4, μ2/2+μ3+μ4/2+μ5/2, and μ4/4+μ5/2+μ6, respectively. The expectation-maximization (EM) algorithm (Dempster et al. 1977) can be used to estimate the risk parameters, including possible maternal effects. If there is mating asymmetry or population stratification, then combined analysis using the unrelated controls yields neither valid tests nor unbiased estimation of the relative risk parameters. Moreover, without genotype data for control parents, evaluation of mating symmetry is impossible.
For all of our calculations, we used 150 affected individuals. When population-based controls were included, we used equal numbers of cases and unrelated controls. The case-parents-triad design requires genotyping three people for each case. Our case-mother/control-mother scenario required the genotyping of four people for each case. Our hybrid design scenario genotyped five for each case. To explore our ability to detect bias due to population stratification, we considered a null relative-risk scenario and calculated noncentrality parameters for both the 5-df test and the 1-df trend test, both based on a population with two nonintermarrying subpopulations of equal size. The two subpopulations had allele prevalences of 0.1 and 0.5, with corresponding baseline disease risks of 0.001 and 0.003. We also calculated the relative risks under that scenario to get a sense for how strong the bias for the relative risk parameters could be.
Results
Noncentrality parameters are shown as a function of the allele prevalence for selected scenarios in figures 1–4. Cutoffs corresponding to specific values of statistical power appear as horizontal lines in the figures. The results for a scenario with only maternally mediated effects (not shown) are similar to those shown in figure 1. To derive noncentrality parameters for other sample sizes, multiply the curve shown by N/150, where N is the contemplated number of cases. Also, if one is comfortable in assuming that there are no maternally mediated effects, then the noncentrality parameters given in figures 1 and 2 apply, but with 2 df, which confers much higher power.
Figure 1.

χ2 noncentrality parameters for a likelihood-ratio test of the four genetic risk parameters based on a scenario with an offspring-mediated effect (R1=2; R2=3) and no maternally mediated effects (S1=1=S2). The calculations assume Hardy-Weinberg equilibrium and use 150 cases for each design. To calculate corresponding noncentrality parameters for a different number, N, of cases, multiply the value shown by N/150. Solid dark curve, hybrid design (Hybrid MS and MA analyses coincide when there are no maternal effects), five individuals genotyped per case. Solid light curve, Epstein et al. (2005) design, four individuals genotyped per case. Dotted curve, case-parents design, three individuals genotyped per case. Dashed curve, case-mother/control-mother design, four individuals genotyped per case. Horizontal reference lines indicate power at α=0.05.
Figure 2.

χ2 noncentrality parameters for a likelihood-ratio test of the four genetic risk parameters based on a scenario with an offspring-mediated recessive effect (R1=1, R2=3) and no maternally mediated effects (S1=1=S2). The calculations assume Hardy-Weinberg equilibrium and use 150 cases for each design. To calculate corresponding noncentrality parameters for a different number, N, of cases, multiply the value shown by N/150. Solid dark curve, hybrid design (Hybrid MS and MA analyses coincide when there are no maternal effects), five individuals genotyped per case. Solid light curve, Epstein et al. (2005) design, four individuals genotyped per case. Dotted curve, case-parents design, three individuals genotyped per case. Dashed curve, case-mother/control-mother design, four individuals genotyped per case. Horizontal reference lines indicate power at α=0.05.
Figure 3.

χ2 noncentrality parameters for a likelihood-ratio test of the four genetic risk parameters based on a scenario with an offspring-mediated recessive effect (R1=1; R2=2) and a maternally mediated recessive effect (S1=1; S2=3). The calculations assume Hardy-Weinberg equilibrium and use 150 cases for each design. To calculate corresponding noncentrality parameters for a different number, N, of cases, multiply the value shown by N/150. Solid dark curve, Hybrid MS, five individuals genotyped per case. Long dash curve, Hybrid MA. Solid light curve, Epstein et al. (2005) design, four individuals genotyped per case. Dotted curve, case-parents design, three individuals genotyped per case. Dashed curve, case-mother/control-mother design, four individuals genotyped per case. Horizontal reference lines indicate power at α=0.05.
Figure 4.

χ2 noncentrality parameters for a likelihood-ratio test of the four genetic risk parameters based on a scenario with an offspring-mediated recessive effect (R1=1; R2=3) and a maternally mediated dominant effect (S1=2; S2=2). The calculations assume Hardy-Weinberg equilibrium and use 150 cases for each design. To calculate corresponding noncentrality parameters for a different number, N, of cases, multiply the value shown by N/150. Solid dark curve, Hybrid MS, five individuals genotyped per case. Long dash curve, Hybrid MA. Solid light curve, Epstein et al. (2005) design, four individuals genotyped per case. Dotted curve, case-parents design, three individuals genotyped per case. Dashed curve, case-mother/control-mother design, four individuals genotyped per case. Horizontal reference lines indicate power at α=0.05.
For detecting genetic effects on risk, including offspring-mediated effects, maternally mediated effects, or both, the Hybrid MS approach for all scenarios considered is more powerful than the case-parents design, the triads-plus-controls design, and the case-mother/control-mother design. Some power is lost if one needs to revert to the more finely stratified Hybrid MA approach and maternal effects are present. Although the triads-plus-controls approach provides good power, it offers no way to verify mating symmetry, and, consequently, its use implies uncertain validity in the face of possible maternally mediated genetic effects.
The powers given are based on large sample approximations. We verified that the approximations are reliable via simulations (not shown) and also verified that the empirical type I error rate is compatible with the nominal rate for level-0.05 testing, based on studying 150 cases. The powers of the 5-df and 1-df tests for bias due to population stratification (at a level of 0.10) were 0.41 and 0.53, respectively, for the admixture scenario considered. These correspond to noncentrality parameters of 3.93 for the 5-df test and 3.0 for the 1-df test. If the number of affected individuals studied is doubled to 300, these powers become 0.68 and 0.79. This population stratification scenario produces biased relative risks of R1=1.30, R2=1.40, S1=1.32, and S2=1.39, based on the expected counts. The noncentrality parameter for the corresponding relative-risk 4-df test is 6.24, yielding a type I error rate of 0.49 (for a 0.05-level test). Under the same scenario, the case-control test statistic has a noncentrality parameter of 7.67 when the design uses 150 affected individuals and their mothers and 150 controls and their mothers, for a type I error rate of 0.58. The corresponding biased parameter estimates for heterozygous and homozygous carriers of the variant allele, based on case-control analysis, are 1.41 and 1.49 for both maternal and offspring effects.
Discussion
The proposed design seems, at first, counterintuitive, in that it calls for genotyping the parents of controls, but not the controls themselves. However, the parental genotype data enables one to assess the distribution of parental genotypes in the population, which, in turn, enables tests for violations of mating symmetry and for bias due to population stratification. Moreover, once we have the parental genotypes, the control offspring genotypes are not informative, given that we assume Mendelian proportions for offspring in the population at large.
The hybrid design proposed by Nagelkerke et al. (2004) and considered further by Epstein et al. (2005) differs from ours in several respects. Both designs provide for a full-data analysis that is valid if there is no bias due to population stratification, if there is mating symmetry, and if there are no maternal effects. Neither Hardy-Weinberg equilibrium nor random mating is required. One could extend the likelihood-based analysis of Epstein et al. to include maternal effects, as we have done in our power calculations, and one could also allow for imprinting. Without control parent genotypes, however, one has no way to verify the required mating-symmetry assumption or to abandon it if symmetry fails. Our proposed modification, in which parents of controls are genotyped rather than the controls themselves, improves power for detecting genetic effects (see figs. 1–4) and also allows one to verify mating symmetry and increase power whenever that assumption is warranted. Because control parents are genotyped, one can also directly test for bias due to population stratification and revert to the case-parents data when necessary.
One practical problem that inevitably arises concerns missing genotypes: How can we make the best analytic use of partial families? Under the assumption that missingness has nothing to do with the missing genotype, the EM algorithm (Dempster et al. 1977) can be usefully applied to the structure of table 1, so that all data can contribute to the analysis. If, for a given control family, one of the parents is not available for genotyping, one would do well to genotype an available unaffected offspring, since that offspring’s genotype can inform the E stage of the EM algorithm to make better use of the family’s data. For other families, the genotype might be available for both parents but not for the affected offspring. This kind of missing data frequently arises when studying a major birth defect, such as anencephaly, because some affected pregnancies are electively terminated. Such partial families offer useful information and should be included to avoid bias. In principle, unrelated cases could also be included by treating their parental genotypes as missing data.
The proposed hybrid design provides a kind of Swiss army knife, a tool with components that can be taken out and used separately as needed. With this multicomponent tool, one can exploit the best features of the case-parents-triad design, such as the ability to investigate parent-of-origin effects and, at the same time, make use of the advantageous features of the case-control design, such as the ability to assess effects of exposures. When the data permit combined inference and support a mating-symmetry assumption, a simple log-linear analysis provides a powerful test of the role of genetic variants, allowing estimation of relative risks and allowing maternally mediated effects to be distinguished from offspring-mediated effects. The analyses can be performed through use of the LEM software (loglinear and event history analysis with missing data using the EM algorithm), which was developed by Vermunt (van den Oord and Vermunt 2000). Script files for running the various models in LEM, under differing assumptions, together with instructions for use, are provided in downloadable zipped formats, suitable for running on a PC with Windows.
An extension to allow for parent-of-origin effects would work by splitting the triple-heterozygous cell shown in table 1 into a cell where the mother was the origin of the variant allele and a cell where the father was the origin (which is usually not knowable), yielding 25 possible outcomes instead of 24. The log-linear structure is expanded to include a multiplier IM for cells where the offspring inherited a single copy, a copy from the mother. One needs to use missing-data methods to account for the ambiguity in parent of origin when the triad is triply heterozygous: the EM algorithm readily permits estimation and testing of hypotheses related to the genetic relative risk parameters, which now number five. In particular, one can test a no-imprinting null hypothesis (i.e., that IM=1).
An extension to allow for a “main effect” of an exposure would be based on the comparison of case parents with control parents. The unit of analysis is, again, the family, regardless of whether the exposure under consideration was experienced by the mother during gestation or experienced by the offspring directly. If the exposure is dichotomous, one can simply add to model (1) an indicator variable for exposure status, together with the product of that indicator variable and the indicator for disease status. The coefficient of that product then estimates the log of the relative risk, corresponding to the exposure. More complex models can be developed to allow for possible confounding of exposure status with the parental mating-type distribution or to accommodate multiple levels of exposure. Ongoing work will provide additional detail.
The hypothetical existence of bias due to population stratification has concerned geneticists and has led some to prefer family-based studies over population-based studies; this concern may have needlessly restricted research into environmental cofactors in the etiology of complex diseases. Although population stratification can, in principle, cause bias in case-control studies, it will do so only to the extent that the baseline risk of disease covaries with allele prevalences across incompletely mixed subpopulations. The 5-df test we propose for testing for bias due to population stratification offers limited power for detecting such bias. (If the Epstein test statistic were adapted to our setting, it would need to carry 4 df or 8 df to allow for possible maternally mediated effects.) The 1-df trend test for bias evidently improves the power but may still fail to detect moderate bias. The full-data analysis of the hybrid design, even when both tests for bias due to population stratification support the inclusion of all of the data, consequently carries some increased vulnerability to this bias. However, the full-data analysis offers improved precision of estimation of genetic effects, together with the ability to include environmental factors, and those are important compensatory benefits. The investigator may want to report both the full-data estimates for the four genetic relative risk parameters and their estimates based on case-parents triads alone, the latter being less precise but more robust.
We have compared power for detecting genetic effects across the various designs/analytic strategies on the basis of the number of cases to be studied. Our hybrid design power calculations presumed genotyping five individuals for each case, whereas the other approaches required fewer individuals to be genotyped, either three (for the case-parents design) or four (for the case-mother/control-mother design or the Epstein-based hybrid we considered). Nevertheless, when a rare disease is under study, the major part of the expense is, typically, in identifying and recruiting subjects and their family members, and not in genotyping, which will continue to become cheaper as biotechnology improves.
The hybrid design suggested by Nagelkerke et al. offers less power than does ours (see figs. 1–4), but requires less genotyping. With only controls and not their parents, however, one has no way to verify the needed mating-symmetry assumption, and one cannot adjust for biasing effects of possible asymmetry. Moreover, without the parents of controls, one cannot estimate the relative frequencies for mating types in the population, and, consequently, testing for population stratification is necessarily indirect and ad hoc. When control parents are genotyped instead, as in our proposed design, a direct likelihood-ratio test for bias due to population stratification becomes possible.
If one uncovers evidence for such bias, and the bias is related to ethnicity or to some other identifiable factor, it may be possible to correct it through stratification. Thus, for example, the bias may disappear if one stratifies on white versus African American families. Instead of the 24-cell multinomial shown in table 1, one would be analyzing a 48-cell multinomial, and the test for bias would now involve 10 df or 2 df.
Although we have not calculated power-for-exposure effects, the power for detecting such effects should be the same for the case-mother/control-mother design as for the hybrid designs, because the analyses involving exposure effects are equivalent, being based on comparing the case families to the control families. Thus, standard power software can be used. For gene-by-environment interactions, the hybrid should then have a marked statistical advantage due to its ability to provide more precise estimates of genotype effects at each exposure level.
Acknowledgments
We thank Drs. Norman Kaplan, Laura Mitchell, Jackie Starr, and Stephanie London, for their helpful comments. This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
Web Resources
The URL for data presented herein is as follows:
References
- Agresti A (1990) Categorical data analysis. John Wiley & Sons, New York [Google Scholar]
- Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B 39:1–38 [Google Scholar]
- Devlin B, Roeder K, Bacanu S (2001) Unbiased methods for population-based association studies. Genet Epidemiol 21:273–284 10.1002/gepi.1034 [DOI] [PubMed] [Google Scholar]
- Epstein M, Veal C, Trembath R, Barker J, Li C, Satten G (2005) Genetic association analysis using data from triads and unrelated subjects. Am J Hum Genet 76:592–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk C, Rubinstein P (1987) Haplotype relative risks: an easy, reliable way to construct a proper control sample for risk calculations. Ann Hum Genet 51:227–233 [DOI] [PubMed] [Google Scholar]
- Lee P, Ridout D, Walter J, Cockburn F (2005) Maternal phenylketonuria: report from the United Kingdom Registry: 1978–1997. Arch Dis Child 90:143–146 10.1136/adc.2003.037762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Kaplan N (2000) A Monte-Carlo procedure for two-stage tests with correlated data. Genet Epidemiol 18:48–62 [DOI] [PubMed] [Google Scholar]
- Mitchell L (2003) Maternal and embryonic genetic effects: case-control studies. Paper presented at the Neural Tube Defects Conference, Seabrook Island, SC, September 27-30 [Google Scholar]
- Nagelkerke N, Hoebee B, Teunis P, Kimman T (2004) Combining the transmission disequilibrium test and case-control methodology using generalized logistic regression. Eur J Hum Genet 12:964–970 10.1038/sj.ejhg.5201255 [DOI] [PubMed] [Google Scholar]
- Pritchard J, Rosenberg N (1999) Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 65:220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman K, Greenland S (1998) Modern epidemiology. Lippincott-Raven, Philadelphia [Google Scholar]
- Schaid D, Sommer S (1993) Genotype relative risks: methods for design and analysis of candidate-gene association studies. Am J Hum Genet 53:1114–1126 [PMC free article] [PubMed] [Google Scholar]
- Self S, Longton G, Kopecky K, Liang K (1991) On estimating HLA-disease association with application to a study of aplastic anemia. Biometrics 47:53–61 [PubMed] [Google Scholar]
- Spielman R, Ewens W (1996) Invited editorial: the TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet 59:983–989 [PMC free article] [PubMed] [Google Scholar]
- Spielman R, McGinnis R, Ewens W (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516 [PMC free article] [PubMed] [Google Scholar]
- Umbach DM, Weinberg CR (2000) The use of case-parent triads to study joint effects of genotype and exposure. Am J Hum Genet 66:251–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Oord E, Vermunt J (2000) Testing for linkage disequilibrium, maternal effects, and imprinting with (in)complete case-parent triads, by use of the computer program LEM. Am J Hum Genet 66:335–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacholder S, Rothman N, Caporaso N (2002) Counterpoint: bias from population stratification is not a major threat to the validity of epidemiological studies of common polymorphisms and cancer. Cancer Epidemiol Biomarkers Prev 11:513–520 [PubMed] [Google Scholar]
- Weinberg CR, Wilcox AJ, Lie RT (1998) A log-linear approach to case-parent triad data: assessing effects of disease genes that act directly or through maternal effects and may be subject to parental imprinting. Am J Hum Genet 62:969–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox AJ, Weinberg CR, Lie RT (1998) Distinguishing the effects of maternal and offspring genes through studies of “case-parent triads.” Am J Epidemiol 148:893–901 [DOI] [PubMed] [Google Scholar]