

Abstract
Dimethylglycine dehydrogenase (DMGDH) (E.C. number 1.5.99.2) is a mitochondrial matrix enzyme involved in the metabolism of choline, converting dimethylglycine to sarcosine. Sarcosine is then transformed to glycine by sarcosine dehydrogenase (E.C. number 1.5.99.1). Both enzymes use flavin adenine dinucleotide and folate in their reaction mechanisms. We have identified a 38-year-old man who has a lifelong condition of fishlike body odor and chronic muscle fatigue, accompanied by elevated levels of the muscle form of creatine kinase in serum. Biochemical analysis of the patient’s serum and urine, using 1H-nuclear magnetic resonance NMR spectroscopy, revealed that his levels of dimethylglycine were much higher than control values. The cDNA and the genomic DNA for human DMGDH (hDMGDH) were then cloned, and a homozygous A→G substitution (326 A→G) was identified in both the cDNA and genomic DNA of the patient. This mutation changes a His to an Arg (H109R). Expression analysis of the mutant cDNA indicates that this mutation inactivates the enzyme. We therefore confirm that the patient described here represents the first reported case of a new inborn error of metabolism, DMGDH deficiency.
Introduction
The catabolism of choline in mitochondria is a cyclical process, involving the formation of betaine, dimethylglycine, sarcosine, glycine, and serine, with the ultimate regeneration of choline (fig. 1). The interconversion of these species involves the transfer of “1-carbon” units at the level of active formaldehyde (Mackenzie and Abeles 1956; Mackenzie and Frisell 1958; Wittwer and Wagner 1981a). One methyl group is removed from dimethylglycine by dimethylglycine dehydrogenase (DMGDH) to form the modified amino acid sarcosine. A second methyl group is removed from sarcosine by sarcosine dehydrogenase (SDH) to form glycine (Frisell and Mackenzie 1962; Lang et al. 1994; Wittwer and Wagner 1981a, 1981b). Both enzymes are associated with a folate derivative as a coenzyme (Wittwer and Wagner 1980, 1981a, 1981b; Steenkamp and Husain 1982; Wagner et al. 1984). In each reaction, one methyl group is donated to one tetrahydrofolate (H4PteGlu) molecule, forming 5,10-methylenetetrahydrofolate (5,10-CH2-H4PteGlu), which is termed “active formaldehyde.” This activated form of formaldehyde, in turn, is able to donate a methyl group in the formation of serine from glycine via serine hydroxymethyltransferase. In addition, 5,10-CH2-H4PteGlu can be used in the conversion of deoxyuridine monophosphate to deoxythimidine monophosphate; it can be reduced to 5-methyltetrahydrofolate (5-CH3-H4PteGlu) for synthesis of methionine from homocysteine via homocysteine methyltransferase (methionine synthase); and it may be oxidized to 10-formyltetrahydrofolate (10-CHO-H4PteGlu) for purine synthesis. In the case of DMGDH and SDH, the preferred folate derivative in the reaction is H4PteGlu5, although other derivatives may be used (Strong et al. 1989; Wittwer and Wagner 1981b). The DMGDH and SDH reactions may proceed in the absence of folate if folate becomes disassociated from the enzymes or if the person has a folate deficiency. However, the end products of the reactions when the folate cofactor is missing are formaldehyde and formate (Steenkamp and Husain 1982; Wittwer and Wagner 1981a). Both SDH and DMGDH have been shown to be major folate-binding proteins (Wittwer and Wagner 1980, 1981a, and 1981b).
Figure 1.

Metabolic pathways involving choline. The key relationships between glycine, serine, dimethylglycine, and sarcosine are indicated, as are the various fates of “active formaldehyde” (adapted from Wittwer and Wagner 1981a).
In addition to their folate cofactor, both DMGDH and SDH have a covalently bound flavin adenine dinucleotide (FAD) (Wittwer and Wagner 1981a; Cook et al. 1984). Flavin peptides with a FAD covalently attached have been isolated from both rat DMGDH (rDMGDH) and rat SDH (rSDH) (Cook et al. 1984 and 1985). The covalent attachment occurs via a His(N3)/flavin (8α carbon) linkage at H84 in rDMGDH, and the recent cloning of rSDH cDNA shows that covalent FAD attachment occurs at H109 in rSDH (Cook et al. 1984; Lang et al. 1991; Bergeron et al. 1998). Although the exact reaction mechanism for these two enzymes is unknown, electrons are subsequently transferred from this FAD to electron-transferring flavoprotein (ETF), then to ETF dehydrogenase, and finally to the mitochondrial electron-transport system (Hoskins and Mackenzie 1961).
A deficiency of SDH was first identified in a 10-mo-old male infant, who died soon after reaching the age of 1 year. He had elevated levels of sarcosine in his urine and serum (Gerritsen and Waisman 1966). A total of 20 patients who are thought to have SDH deficiency have been described in the literature (Scott 1995). Clinical symptoms have been difficult to correlate with sarcosinemia/sarcosinuria, and some researchers believe that the diagnosis of sarcosinemia/sarcosinuria is unrelated to any clinical symptoms (Gerritsen 1972; Scott 1995). All patients, however, have been characterized by elevated levels of sarcosine in urine and/or serum.
Dimethylglycine is normally found in urine and serum at very low levels, and elevated levels of dimethylglycine are sometimes found to be secondary to another disorder, such as folate or cobalamin deficiency (Porter et al. 1985; Allen et al. 1993; Dudman et al. 1993). We recently identified a patient who had elevated levels of dimethylglycine in serum and urine. The patient was identified through 1H-nuclear magnetic resonance (NMR) studies of serum and urine that revealed dimethylglycine levels much higher than normal values (reported in detail by Moolenaar et al. [1999]). rDMGDH cDNA has been used to clone the cDNA for human DMGDH (hDMGDH) from expressed-sequence tag (EST) databases. A homozygous mutation was identified in DMGDH cDNA and genomic DNA from this patient (GenBank accession number AF111858). Studies of recombinant mutant protein show that the mutation inactivates the enzyme.
Patient and Methods
hDMGDH cDNA Cloning
Databases were searched using the rDMGDH cDNA sequence (Genbank accession number X55995, E.C. 1.5.99.2). The Human Genome Sciences Database was searched using facilities at SmithKline Beecham. All sequencing was performed by the Molecular Biology Core Sequencing Facility at the Mayo Clinic, using an ABI Prism model 377 sequencer (Applied Biosystems). PCR reactions to amplify gaps between ESTs were performed using PCR Supermix (Gibco BRL). Cycling conditions were as follows: denaturing at 95°C for 3 min; 35 cycles of denaturing at 94°C for 1 min; annealing at 55°C for 1 min; extension at 72°C for 1 min; extension at 72°C for 7 min; and soaking at 4°C. Human fibroblast cDNA served as a template. Primers used in the reaction were: 5′-AAGAGATGTTCCCTTTACTC-3′ (forward) and 5′-TTTAATCTGTTTCAGTGCTTG-3′ (reverse). A PAC clone containing hDMGDH sequence was obtained from Genome Systems. PAC DNA was induced to high copy number and was prepared according to the supplier's protocol. For direct sequencing of the PAC clone, DNA was prepared using kb-100s DNA purification columns (Genome Systems). The rDMGDH primer designed to the sequence of the flavin binding site and used for indirect PAC clone sequencing was 5′-GTGCCAGGTAGAGCCAGCCGTGAGTTCCGA-3′. A second genomic clone (P1) was also obtained from Genome Systems, since the PAC clone was found to be missing at least one exon at the 5′ end of the gene.
Patient-cDNA/Genomic-DNA Analysis
cDNA was prepared from patient and control fibroblast cultures, as described elsewhere (Mohsen et al. 1998). After the H109R mutation was identified, control alleles were sequenced around the mutated region. PCR reactions to amplify the region from cDNA were performed with the following primers: 5′-CAACTTACTTTCATCCTGGA-3′ (forward) and 5′-GAGTAAAGGGAACATCTCTT-3′ (reverse). PCR was performed using the genomic DNA as a template to amplify a region around the H109R mutation with the following primers: 5′-TGGCCTCCCCCATAGTTTTA-3′ (forward) and 5′-GCCTATGGATACTCTTTTGC-3′ (reverse).
Expression of hDMGDH in Mammalian Cells
The precursor sequence for hDMGDH was cloned into the KpnI site of the pcDNA6/V5-His plasmid (Invitrogen). Two hundred ninety-three cells (American Type Culture Collection) were grown in Dulbecco's minimal essential medium supplemented with 10% fetal calf serum. Cells grown in 6-cm dishes were transfected with 3 μg of either the wild-type or mutant vectors, along with a pcDNA3.1 His/LacZ control plasmid (Invitrogen), using the Calcium Phosphate Transfection kit from the same company. Forty hours after transfection, the cells were harvested for western blot analysis or RNA extraction. Control plates were stained with the β-Gal staining kit (Invitrogen). For western blots, the isolated cell pellets were resuspended in PBS, pH 7.4, and were sonicated briefly; 30 μg of supernatant was loaded onto a 12% SDS polyacrylamide gel. Western blot analysis was performed, as described elsewhere, using a 1:1000 dilution of rabbit antiserum, which was raised against native DMGDH that had been purified from pig liver, as the primary antibody (Mohsen et al. 1998). To quantitate the level of expression of the wild-type and mutant vectors, mRNA was extracted from transfected cells with the QuickPrep Micro mRNA Purification kit (Amersham Pharmacia Biotech). Then 0.5 μg of RNA was used, with the First-Strand cDNA synthesis kit (Amersham Pharmacia Biotech), to generate first-strand cDNA. One microliter of the reverse-transcription product was subjected to PCR with primers specific to DMGDH (5′-ATGGATTGAAGAAGAAGCAGTC-3′ and 5′-GATGTCGTGTTGCCAACCAC-3′) or β-ETF (5′-CACCCTGTAAGTGGCTGC-3′ and 5′-ATGTTGAGAGTCAGTTTTATTGCC-3′), with cycling conditions as described above. Amplification cycle number was varied to identify conditions that avoided saturation of the PCR reaction. PCR products were examined on 1% agarose gels.
DMGDH Expression Studies
Mature wild-type rat and human DMGDH sequences (with amino termini beginning at Ser22 and Val29, respectively) were subcloned into the pKK223-3 expression vector (Amersham Pharmacia Biotech) in the EcoRI/HindIII sites. The H109R mutant, which represents residue 81 in the mature protein, was created using PCR-based mutagenesis and was inserted into compatible restriction sites in the wild-type vectors. Cultures (100 ml) of XL1-Blue cells containing wild-type, mutant DMGDH, and pKK223-3 with no insert were grown in 2xYT media plus ampicillin at 37°C, were induced in log-phase growth with 0.05 mM isopropyl-β-D-thiogalactoside, and were allowed to grow overnight at 30°C. Cell extracts were prepared as described elsewhere (Binzak et al. 1998). Extracts were tested for DMGDH activity using the ETF reduction assay, as described elsewhere, with 0.5 M dimethylglycine as substrate (Binzak et al. 1998) (N,N-dimethylglycine [hydrochloride] was purchased from Sigma). Protein assays were performed using the Bio-Rad DC Protein Assay (Bio-Rad). Western blot analysis was performed as described above.
1H NMR Spectroscopy
1H NMR spectroscopy of serum and urine specimens was performed essentially as described elsewhere (Wevers et al. 1994).
Clinical History
The patient is a 38-year-old man of African ancestry who presented with a fishlike body odor that was first noticed when he was 5 years old. The intensity of the odor increases with physiological stress, such as illness, as well as during times of increased physical activity. The odor has led to severe psychological problems and professional difficulties. The patient had normal growth and development and is of normal intelligence with no unusual CNS symptoms, but he has experienced chronic muscular fatigue since adolescence. He has had no known hematologic problems or liver dysfunction. Routine clinical chemistry determinations yielded unremarkable results, with normal levels of cobalamin, folate, and homocysteine in serum. Creatine kinase levels were consistently found to be four- to fivefold higher than normal levels. Metabolic investigations—including testing of amino acids, organic acids, carnitine ester profile, and serum levels of very-long-chain fatty acids—were normal. Trimethylaminuria, the only known specific defect leading to “fish-odor syndrome” was excluded using NMR spectroscopy of urine (Moolenaar et al. 1999). The patient does not use any dietary supplements containing either carnitine or dimethylglycine. There is no known consanguinity in the pedigree, but the patient’s parents were unavailable for interview. The brothers and sisters (a precise pedigree was unavailable) and the two sons of the patient are asymptomatic.
Results
1H NMR
1H NMR spectroscopic analysis of blood samples from the patient showed a dimethylglycine concentration of 449 mM creatinine (∼20-fold higher than normal values) in urine and a concentration of 221 μM/liter (∼100-fold higher than normal values) in serum. No other unusual metabolites were present. Figure 2 shows part of the 1H NMR spectrum of serum from the patient. A singlet at 2.93 parts per million (ppm) is caused by dimethylglycine. Dimethylglycine was observed in serum spectra from healthy individuals only in low concentration (3.80-ppm singlet; data not shown). The identity of the dimethylglycine was confirmed using 13C-NMR spectroscopy and gas chromatography–mass spectroscopy. Similar findings had been observed in the patient’s urine (complete details of biochemical analyses of the urine have been reported elsewhere [Moolenaar et al. 1999]).
Figure 2.

1H NMR spectrum of serum from the DMGDHdeficient patient. Peaks of relevant metabolites are labeled. The singlet peak at 2.93 ppm is caused by an accumulation of dimethylglycine in the patient’s serum.
cDNA Cloning
Because of the biochemical findings in this patient, we suspected a deficiency of DMGDH. To confirm this, we initiated the cloning of hDMGDH cDNA. The cDNA sequence for rDMGDH (Lang et al. 1991) was used to screen several databases to identify a human homologue. An EST was identified from the Human Genome Sciences database that aligns to the 3′ end of the rat cDNA and is 92% identical over ∼400 nucleotides. This clone also contains 1.1 kb of 3′ UTR, including the poly A tail. A second human EST that aligns near the 5′ end of the rat cDNA sequence was identified from Genbank. This EST is 81% identical to the rat sequence over ∼170 nucleotides. PCR was then used to amplify the region between these two short sequences from human fibroblast cDNA made from control fibroblast mRNA. Various techniques to clone the extreme 5′ end of the cDNA were unsuccessful; therefore, a human PAC clone was isolated using ∼1.8 kb of the hDMGDH cDNA as a probe. Both Southern blot analysis and direct sequencing of this PAC clone confirm that it contains the hDMGDH genomic sequence (Binzak et al. 2000). Since the extreme 5′ end of the gene was still missing, a second genomic clone (P1) was obtained from Genome Systems, and it proved to contain the final 5′-most exon of coding sequence of the gene.
The hDMGDH cDNA is 2,598 nucleotides long, which suggests the presence of a protein with 866 amino acids and a 3′ UTR of ∼1,100 nucleotides (fig. 3). Overall, the precursor cDNA sequences for hDMGDH and rDMGDH share 85.6% nucleotide identity, as calculated using the GAP program from the University of Wisconsin GCG sequence-analysis software, although the mature proteins show 91.4% amino acid identity (93.9% amino acid similarity). The flavin peptide of rDMGDH (Wittwer and Wagner 1980) is 100% conserved, at the amino acid level, with the same region in hDMGDH; however, the area just upstream shows reduced homology. In fact, the cDNA sequence of hDMGDH suggests that seven additional amino acids are present in the mitochondrial targeting peptide, compared with rDMGDH. Val29 is predicted, on the basis of the known pattern of processing of mitochondrial leader peptides, to be the amino terminus of the mature human protein.
Figure 3.

The hDMGDH cDNA sequence. Only the sense strand is shown. Amino acids are represented by their one-letter codes. The italicized amino acid sequence with a single underline is the putative flavin-binding peptide. The Val residue at position 29 (precursor numbering), outlined by the triangle, was taken as the mature 5′-end of the cDNA. The boxed His residue is the putative flavin-binding site (corresponding to His84 in rDMGDH). The circled His residue is mutated in the patient. The underlined nucleotide sequence denotes the probe used to isolate the hDMGDH PAC clone.
Analysis of Patient DNA
Analysis of hDMGDH cDNA and genomic DNA from the patient identified a homozygous A→G point mutation at nucleotide 326 of the precursor coding region (fig. 4). This suggests that the alteration of a His to an Arg at amino acid 109 (H109R or residue 81 of the predicted mature protein) occurs 18 amino acids downstream of the site of the FAD attachment that was identified for rDMGDH (fig. 3) (Wittwer and Wagner 1981a; Cook et al. 1984). Southern blot analysis of genomic DNA from patient and control fibroblast DNA gave an identical pattern, suggesting that no genomic DNA deletions were present (data not shown). This region is 100% conserved between rats and humans, and the alteration was not present in 50 DMGDH alleles from control human fibroblast samples. The remainder of the cDNA sequence from the patient matched the control sequence. No information could be obtained about nucleotides 1–12, which were contained within the 5′-most PCR primer; however, this is within the mitochondrial targeting peptide and thus is not represented in the mature enzyme. Enzymatic assay and western blot analysis with DMGDH-specific antiserum were not sensitive enough to detect DMGDH, even in control fibroblasts. This is in keeping with previous observations with this enzyme (Moolenaar et al. 1999). To evaluate the effect of the H109R substitution on DMGDH enzyme activity, therefore, the nucleotide 326 A→G mutation was introduced into hDMGDH cDNA, and both the normal and mutant sequences were expressed in Escherichia coli, Saccharomyces cerevisiae, and a variety of mammalian tissue culture samples. No active enzyme could be detected in E. coli after expression of the normal hDMGDH, although immunoreactive material was present (data not shown). Similarly, expression studies in yeast yielded no hDMGDH antigen. After expression of the wild-type cDNA in 293 cells, DMGDH antigen could be detected, although the level of expression was not high enough to allow measurement of enzyme activity (fig. 5A). When a cDNA containing the patient mutation was similarly expressed, hDMGDH antigen was barely detectable (fig. 5A), although the transfection efficiency was similar for both vectors, as determined by β-galactosidase staining of colonies on the transfection plates (data not shown). The levels of DMGDH-specific mRNA after transfection with the wild-type and mutant plasmids, as determined by a semiquantitative PCR technique (fig. 5B), were much lower in the mock-transfected cells. In contrast, β-ETF–specific primers used as a control showed a similar level of this message in all samples (fig. 5C).
Figure 4.

Patient mutation in the hDMGDH gene. A, Chromatograms of hDMGDH cDNA sequence from a control fibroblast sample and, B, from a patient fibroblast sample; C, genomic DNA sequence from the patient. mRNA, cDNA, and genomic DNA were prepared as described in the Patient and Methods section. Sequence shown is read in the 5′→3′ direction. The sequencing primer reads the sequence on the antisense strand. The arrow indicates the nucleotide that is mutated in the patient: T→C (A→G on sense strand), changing the residue from CAT (His) to CGT (Arg).
Figure 5.

Expression of wild-type and mutant hDMGDH in 293 cells. A, Western blot analysis of cellular extracts after SDS-PAGE. Lanes 1 and 2, extracts from cells transfected in separate experiments with wild-type hDMGDH vector; lanes 3 and 4, extracts from cells transfected in separate experiments with an hDMGDH vector containing the patient mutation; lane 5, mock transfection; lane 6, partially purified pig liver DMGDH. B, RT-PCR results in transfected cells. Lanes 1 and 2, cells transfected with wild-type DMGDH vector; lanes 3 and 4, cells transfected with an hDMGDH vector containing the patient mutation; lanes 5 and 6, mock transfection. Lanes 1, 3, and 5 are products after 16 cycles of amplification. Lanes 2, 4, and 6 are products after 18 cycles of amplification. C, β-ETF message in samples prepared concurrently with those shown in panel B. Lanes 1–3, cells transfected with wild-type DMGDH vector; lanes 4–6, cells transfected with an hDMGDH vector containing the patient mutation; lanes 7–9 mock transfection. Lanes 1, 4, and 7 are products after 20 cycles of amplification. Lanes 2, 5, and 8 are products after 22 cycles of amplification. Lanes 3, 6, and 9 are products after 24 cycles of amplification.
Because the region containing the patient mutation is identical in rat and human genes, the mutation was introduced into rDMGDH in a prokaryotic expression vector previously made in our laboratory, and the wild-type and mutant proteins produced in E. coli were characterized. After overnight induction of a culture containing the wild-type vector, mean±SD DMGDH activity in cellular extracts was 0.14±0.03 mU/mg protein (n=9) (as measured with the anaerobic electron transferring flavoprotein assay), although no activity was detectable in extracts from cells containing a vector with the mutant rDMGDH cDNA or a vector with no insert (<10% control activity). In contrast, western blot analysis of the cellular extracts shows that levels of wild-type and mutant protein were similar after induction, indicating that the mutant enzyme was stable but inactive (fig. 6).
Figure 6.
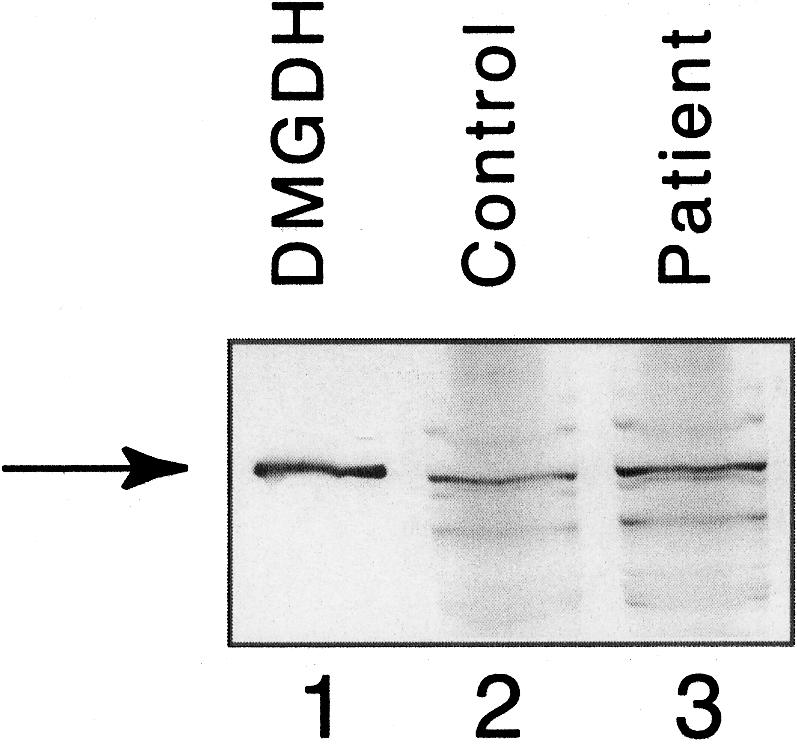
Western blot of wild-type and mutant rDMGDH produced in E. coli. DMGDH purified from pig liver for reference (lane 1). Extracts from induced cells containing the wild-type rDMGDH vector (lane 2) and rDMGDH containing the patient mutation (lane 3) were separated by SDS-PAGE, were transferred to a polyvinyldiene fluoride membrane, and were examined using anti-DMGDH antibodies. No DMGDH-specific band was present in extracts from cells containing no plasmid or plasmid without insert (data not shown).
Discussion
Our findings unequivocally identify a deficiency of DMGDH as the cause of accumulation of dimethylglycine in the urine and serum of this patient. The accumulation appears to be the result of a homozygous point mutation in the DMGDH gene, although the presence of a large deletion on one allele at this locus, leading to hemizygosity for the point mutation cannot be excluded. Although the level of expression of the wild-type and mutant cDNAs in a eukaryotic expression system was too low to allow direct measurement of enzyme activity, the level of mutant enzyme antigen was clearly lower than that of the wild-type enzyme antigen, most likely indicating reduced stability of the mutant protein. This is consistent with the E. coli expression data for the mutant enzyme, because it was unstable in this system. Additional prokaryotic expression studies confirm that the point mutation inactivates enzymes when introduced into the rat coding sequence, which is 100% conserved in this region of the human enzyme. Thus, even if a mutant human enzyme were stably formed in the cell, it would not be active.
In view of the potential consequence of a disruption of metabolism of methyl groups related to DMGDH action, which are necessary for several other important cellular reactions, this patient’s clinical symptoms are surprisingly mild. Clearly, other sources of methyl groups for these reactions must be available when DMGDH is deficient. Because sarcosine in DMGDH deficiency can still be produced directly from glycine via glycine methyltransferase, which uses S-adenosylmethionine as a methyl donor, the remainder of the downstream reactions of choline catabolism can likely still proceed to some extent; however, chronic elevation of serum creatine kinase levels in this patient are indicative of ongoing muscle breakdown, suggesting that DMGDH deficiency does have physiologic repercussions.
The His→Arg mutation demonstrated in the patient’s DMGDH enzyme is in close proximity to the predicted FAD attachment site and could lead to a disruption of flavin binding. Alternatively, the mutation may disrupt interaction with folate. It has not been definitively determined where folate associates with the enzyme, but a compelling case has been made for association near the amino terminus of the protein based on structural information available for another folate-dependent enzyme, thymidylate synthase. The region starting 10 amino acids toward the C-terminus from the His91 (precursor numbering) flavin-binding site in DMGDH contains 3 Lys residues conserved in both the human and rat enzyme (residues 106, 107, and 114 in the precursor) and is homologous to a highly conserved motif present in several thymidylate synthase enzymes from various sources (Maley et al. 1982; Hardy et al. 1987; Lang et al. 1991). A thymidylate synthase has been crystallized from Lactobacillus casei, and the Lys residues in this region are important in binding its folate coenzyme, H4PteGlu7 (Hardy et al. 1987). It is important to note that, in the patient described here, the H109R mutation lies between these Lys residues and may lead to disruption of folate binding in the mutant DMGDH enzyme. However, these Lys residues are not conserved in the evolutionarily related SDH, suggesting that DMGDH and SDH may have a different mechanism of association with their folate coenzyme (Bergeron et al. 1998). Alternatively, some other binding motif may be responsible for folate association. The presence of a Lys/Arg–rich region between Lys740 and Arg764 in rDMGDH has also been implicated in folate binding (Otto et al. 1996). This region is similarly Lys/Arg rich in hSDH and hDMGDH (Bergeron et al. 1998).
Here we present the cloning of the cDNA for hDMGDH and the identification of a homozygous H109R mutation in a patient presenting with a fish odor and having high dimethylglycine levels in his serum and urine. In conjunction with our previous report (Moolenaar et al. 1999), this confirms that this patient represents the first identified case of DMGDH deficiency, a new inborn error of metabolism.
Acknowledgments
We would like to acknowledge Bambi Anderson for technical assistance. J.V. is supported in part by National Institutes of Health grant R01-DK45482.
References
- Allen RH, Stabler SP, Lindenbaum J (1993) Serum betaine, N, N-dimethylglycine and N-methylglycine levels in patients with cobalamin and folate deficiency and related inborn errors of metabolism. Metabolism 42:1448–1460 [DOI] [PubMed] [Google Scholar]
- Bergeron F, Otto A, Blache P, Day R, Denoroy L, Brandsch R, Bataille D (1998) Molecular cloning and tissue distribution of rat sarcosine dehydrogenase. Eur J Biochem 257:556–561 [DOI] [PubMed] [Google Scholar]
- Binzak B, Willard J, Vockley J (1998) Identification of the catalytic residue of human short/branched chain acyl-CoA dehydrogenase by in vitro mutagenesis. Biochim Biophys Acta 1382:137–142 [DOI] [PubMed] [Google Scholar]
- Binzak BA, Vockley JG, Jenkins RB, Vockley J (2000) Structure and analysis of the human dimethylglycine dehydrogenase. Mol Genet Metab 69:181–187 [DOI] [PubMed] [Google Scholar]
- Cook RJ, Misono KS, Wagner C (1984) Identification of the covalently bound flavin of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver mitochondria. J Biol Chem 259:12475–12480 [PubMed] [Google Scholar]
- Cook RJ, Misono KS, Wagner C (1985) The amino acid sequences of the flavin-peptides of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver mitochondria. J Biol Chem 260:12998–13002 [PubMed] [Google Scholar]
- Dudman NP, Wilcken DE, Wang J, Lynch JF, Macey D, Lundberg P (1993) Disordered methionine/homocysteine metabolism in premature vascular disease: its occurrence, cofactor therapy, and enzymology. Arteroscler Thromb 13:1253–1260 [DOI] [PubMed] [Google Scholar]
- Frisell WR, Mackenzie CG (1962) Separation and purification of sarcosine dehydrogenase and dimethylglycine dehydrogenase. J Biol Chem 237:94–98 [PubMed] [Google Scholar]
- Gerritsen T (1972) Sarcosine dehydrogenase deficiency, the enzyme defect in hypersarcosinemia. Helv Paediatr Acta 21:33–37 [PubMed] [Google Scholar]
- Gerritsen T, Waisman HA (1966) Hypersarcosinemia: an inborn error of metabolism. N Engl J Med 275:66–69 [DOI] [PubMed] [Google Scholar]
- Hardy LW, Finer-Moore JS, Montfort WR, Jones MO, Santi DV, Stroud RM (1987) Atomic structure of thymidylate synthase: target for rational drug design. Science 235:448–455 [DOI] [PubMed] [Google Scholar]
- Hoskins DD, Mackenzie CG (1961) Solubilization and ETF requirement of mitochondrial sarcosine dehydrogenase and dimethylglycine dehydrogenase. J Biol Chem 236:177–183 [PubMed] [Google Scholar]
- Lang H, Minaian K, Freudenberg N, Hoffmann R, Brandsch R (1994) Tissue specificity of rat mitochondrial dimethylglycine dehydrogenase expression. Biochem J 299:393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang H, Polster M, Brandsch R (1991) Rat liver dimethylglycine dehydrogenase: flavinylation of the enzyme in hepatocytes in primary culture and characterization of a cDNA clone. Eur J Biochem 198:793–799 [DOI] [PubMed] [Google Scholar]
- Mackenzie CG, Abeles RH (1956) Production of active formaldehyde in the mitochondrial oxidation of sarcosine-CD3. J Biol Chem 222:145–150 [PubMed] [Google Scholar]
- Mackenzie CG, Frisell WR (1958) The metabolism of dimethylglycine by liver mitochondria. J Biol Chem 232:417–421 [PubMed] [Google Scholar]
- Maley GF, Maley F, Baugh CM (1982) Studies on identifying the folylpolyglutamate binding sites of Lactobacillus casei thymidylate synthetase. Arch Biochem Biophys 216:551–558 [DOI] [PubMed] [Google Scholar]
- Mohsen A-W, Anderson B, Volchenboum S, Battaile K, Tiffany K, Roberts D, Kim J-J, Vockley J (1998) Characterization of molecular defects in isovaleryl-CoA dehydrogenase in patients with isovaleric acidemia. Biochemistry 37:10325–10335 [DOI] [PubMed] [Google Scholar]
- Moolenaar SH, Poggi-Bach J, Engelke UFH, Corstiaensen JMB, Heerschap A, de Jong JGN, Binzak BA, Vockley J, Wevers RA (1999) Defect in dimethylglycine dehydrogenase, a new inborn error of metabolism: NMR spectroscopy study. Clin Chem 45:459–464 [PubMed] [Google Scholar]
- Otto A, Stoltz M, Sailer H-P, Brandsch R (1996) Biogenesis of the covalently flavinylated mitochondrial enzyme dimethylglycine dehydrogenase. J Biol Chem 271:9823–9829 [DOI] [PubMed] [Google Scholar]
- Porter DH, Cook RJ, Wagner C (1985) Enzymatic properties of dimethylglycine dehydrogenase and sarcosine dehydrogenase from rat liver. Arch Biochem Biophys 243:396–407 [DOI] [PubMed] [Google Scholar]
- Scott CR (1995) Sarcosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 1329–1335 [Google Scholar]
- Steenkamp DJ, Husain M (1982) The Effect of tetrahydrofolate on the reduction of electron transfer flavoprotein by sarcosine and dimethylglycine dehydrogenases. Biochem J 203:707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong WB, Cook R, Schirch V (1989) Interaction of tetrahydropteroyl polyglutamates with two enzymes from mitochondria. Biochemistry 28:106–114 [DOI] [PubMed] [Google Scholar]
- Wagner C, Briggs WT, Cook RJ (1984) Covalent binding of folic acid to dimethylglycine dehydrogenase. Arch Biochem Biophys 233:457–461 [DOI] [PubMed] [Google Scholar]
- Wevers RA, Engelke U, Heerschap A (1994) High-resolution 1H-NMR spectroscopy of blood plasma for metabolic studies. Clin Chem 40:1245–1250 [PubMed] [Google Scholar]
- Wittwer AJ, Wagner C (1980) Identification of folate binding protein of mitochondria as dimethylglycine dehydrogenase. Proc Natl Acad Sci USA 77:4484–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittwer AJ, Wagner C (1981a) Identification of the folate-binding proteins of rat liver mitochondria as dimethylglycine dehydrogenase and sarcosine dehydrogenase: flavoprotein nature and enzymatic properties of the purified proteins. J Biol Chem 256:4109–4115 [PubMed] [Google Scholar]
- Wittwer AJ, Wagner C (1981b) Identification of the folate-binding proteins of rat liver mitochondria as dimethylglycine dehydrogenase and sarcosine dehydrogenase: purification and folate-binding characteristics. J Biol Chem 256:4102–4108 [PubMed] [Google Scholar]