

Abstract
Computational constraints currently limit exact multipoint linkage analysis to pedigrees of moderate size. We introduce new algorithms that allow analysis of larger pedigrees by reducing the time and memory requirements of the computation. We use the observed pedigree genotypes to reduce the number of inheritance patterns that need to be considered. The algorithms are implemented in a new version (version 2.1) of the software package GENEHUNTER. Performance gains depend on marker heterozygosity and on the number of pedigree members available for genotyping, but typically are 10–1,000-fold, compared with the performance of the previous release (version 2.0). As a result, families with up to 30 bits of inheritance information have been analyzed, and further increases in family size are feasible. In addition to computation of linkage statistics and haplotype determination, GENEHUNTER can also perform single-locus and multilocus transmission/disequilibrium tests. We describe and implement a set of permutation tests that allow determination of empirical significance levels in the presence of linkage disequilibrium among marker loci.
Introduction
All inheritance information available in large pedigrees genotyped with hundreds of polymorphic markers cannot be extracted by current methods. The computational complexity of the problem grows rapidly as the number of markers and pedigree members considered simultaneously increases. Available algorithms scale exponentially either with the number of markers (Elston and Stewart 1971) or with the number of pedigree members (Lander and Green 1987). Recent advancements in both types of algorithms have made it practical to perform simultaneous analysis of several markers by the Elston-Stewart algorithm (Cottingham et al. 1993; O’Connell and Weeks 1995), as well as analysis of general pedigrees of moderate size by the Lander-Green hidden-Markov-model (HMM) approach (Kruglyak et al. 1996; Kruglyak and Lander 1998; Gudbjartsson et al. 2000). In addition to methods that offer exact likelihood estimates, there have been some promising developments utilizing sampling (Monte Carlo)-based techniques (Sobel and Lange 1993, 1996; Heath 1997). This progress in multipoint linkage analysis has aided the genetic dissection of human diseases. Nonetheless, multipoint analysis is still limited by the computational burden involved in the analysis of large families. Such limitations hamper computation of linkage statistics, as well as reconstruction of haplotypes.
The ability of the HMM approach to simultaneously consider a large number of genetic markers has made it a widely used method for analysis of data generated with today’s dense genetic maps. In this article, we present a set of improvements to this approach that allow rapid multipoint analysis of larger pedigrees, provided that the majority of the pedigree members are available for genotyping. We have implemented the new algorithms in the genetic analysis program GENEHUNTER version 2.1. Compared with the previous release (version 2.0), version 2.1 has efficiency gains in central-processing-unit (CPU) and memory requirements that are 10–1,000-fold in typical families. GENEHUNTER provides an integrated approach to parametric and nonparametric linkage analysis. Multipoint analysis of a given pedigree is separated into two steps (Kruglyak et al. 1996): (1) computation of probability distribution of inheritance patterns (which depends only on the marker genotypes) and (2) computation of a statistic that quantifies linkage (which, conditional on an inheritance pattern, depends only on the phenotypes). The first step is computationally intensive, whereas the second step is, in most cases, computationally trivial. Thus, most of the new methods described below are aimed at significantly reducing the time and memory needed to perform the first step. The improvements are based on reducing the number of inheritance patterns (inheritance vectors) that must be considered in the computation. The exponential growth in time and memory is due to the growth of the inheritance-vector space (state space). Observation of marker genotypes typically restricts the vector space, because inheritance patterns incompatible with the observed marker genotypes have probability exactly equal to zero. For example, a single meiosis with a known outcome reduces by half the number of inheritance vectors with nonzero inheritance probability at that marker location. Therefore, we can reduce both time and memory requirements by considering only the subspace of the vector space whose members are inheritance vectors with nonzero probability. Because state-space reduction through use of genotyping information is marker specific, we face the challenge of computing the HMM transitions, which are necessary for combining the information from multiple markers, without resorting to the full state-space representation. Our innovation here is a method (formally a change of the coordinate system) that permits us to execute the transitions by using only meioses that are not fixed by observation of marker genotypes. In addition, we present an algorithm that speeds computation of the NPL_all (Whittemore and Halpern 1994) statistic for nonparametric linkage analysis. Although performance of the new algorithms is highly dependent on genotype information content, we do not impose any restrictions on the pedigree structures that can be analyzed by GENEHUNTER. No approximations are used, and, in the limit of low information content, the new version has the same time and memory requirements as the does the original code (GENEHUNTER version 2.0).
In addition to computation of linkage statistics and haplotype determination, GENEHUNTER also can perform the transmission/disequilibrium test (TDT). Single- and multilocus TDTs have been available since the release of GENEHUNTER version 2.0 (Daly et al. 1998). Here we present a procedure to compute empirical significance levels when several closely spaced markers are genotyped. In such cases, significance of results must be corrected for multiple testing; however, the traditional Bonferroni correction is likely to be extremely conservative, because of the nonindependence of the multiple tests (i.e., alleles at the same marker and alleles at nearby markers in linkage disequilibrium [LD]). Here we implement a permutation approach that accurately estimates significance in the presence of these factors.
Throughout this article, we use the conventions employed in a previous publication by Kruglyak et al. (1996). Definitions of inheritance vectors and the HMM methodology can be found in that publication. The present article is organized as follows. In the next section, we describe the algorithms used to speed multipoint analysis; users of GENEHUNTER who are not interested in mathematical details may skip this section. In the Computer Implementation section, we introduce a set of commands that allow efficient use of the software. We next describe in detail the permutation method that allows definition of TDT significance levels in the presence of LD. We then apply the new software to simulated and actual data, present performance results, and comment on the influence that pedigree structure and marker polymorphism have on efficiency gains. We conclude with a brief discussion.
Algorithmic Improvements
In the HMM approach, time and memory requirements for multipoint linkage analysis scale exponentially with the number of meioses considered. Currently, the number of inheritance patterns that are evaluated and stored per marker is N=22n-f, where n is the number of individuals with ancestors in the pedigree and f is the number of pedigree founders. We present three methods that effectively reduce N.
For a pedigree genotyped with m markers, storage requirements are easily shown to be (m×3+4.5)×8×N bytes. Unlike the situation with memory requirements, the time that it takes to compute multipoint scores is not a simple function of markers and meioses considered. Although computation time scales with N, it also depends on details such as the presence of inbreeding loops and the fraction of pedigree members available for genotyping, We can divide the computation into three parts:
-
1.
Calculation of inheritance probabilities at each marker, based only on the data for that marker (single-point probabilities);
-
2.
Calculation of multipoint-inheritance probabilities, through use of HMM marker-to-marker transitions;
-
3.
Calculation of linkage statistics (of these, the most CPU intensive is the calculation of the Halpern-Whittemore statistic for nonparametric linkage, NPL_all).
Historically, the most significant time requirement for multipoint analysis has been the vector-matrix multiplication necessary to perform the second step. After speedups resulting from the application of fast Fourier transform (FFT) methods (Kruglyak and Lander 1998), approximately equal time is spent on each one of the three steps in GENEHUNTER version 2.0. Thus, in order to achieve practical speed gains, all three steps must be improved. We introduce the following:
-
1.
Efficient identification of classes of inheritance vectors that are not compatible with the observed genotypes. We use this information to effectively reduce the inheritance-vector space. For classes of inheritance vectors that are not compatible with the observed phenotypes, we do not compute or store single-point–inheritance probabilities or multipoint-inheritance probabilities.
-
2.
Formalism that allows us to compute multipoint-inheritance probabilities (the vector-matrix multiplication necessary for the transition step of the HMM) without resorting to the full state-space representation.
-
3.
Efficient computation and storage of the NPL_all statistic.
In this presentation, we make a distinction between transition-probability distributions and cumulative-probability distributions. The transition-probability distribution is the inheritance probability anywhere on the genome, based on marker data to the left or right of that position (but not at the position itself); the cumulative-probability distribution at a genotyped marker is the product of transition probabilities and single-point probabilities at that marker. From this definition, it follows that state-space restrictions to the single-point distribution at a marker also apply to the cumulative-probability distribution at that marker.
Efficient Reduction of Inheritance-State Space
We identify classes of inheritance vectors incompatible with the observed genotypes, using an iterative approach. We apply the algorithm used by GENEHUNTER for the computation of single-point–inheritance probabilities in subsets of the pedigree. In the previous implementation, single-point–inheritance probabilities were computed for the entire pedigree, once per inheritance vector. If no allele configuration was compatible with the inheritance vector, the single-point probability was set to zero. Although this approach provides the complete set of incompatible inheritance vectors, CPU and memory requirements grow exponentially with the size of the pedigree. We can dramatically reduce the computational cost if we apply the algorithm in subsets of the pedigree and accumulate the restrictions found. We can then compute and store single-point–inheritance probabilities for the full pedigree, but only for the subset of inheritance vectors compatible with the observed genotypes. This iterative approach is profitable because most restrictions are local; they are defined by the siblings, children, parents, and grandparents of a pedigree member. Once a restriction is found by means of such local relationships, consideration of additional pedigree subsets may provide further restrictions to allele assignment, but it cannot remove already-existing restrictions.
It is not memory efficient to store lists of incompatible inheritance vectors; instead, a few simple rules can capture the restrictions. As an example, consider the pedigree of figure 1; we represent the inheritance pattern at each point on the chromosome by a binary inheritance vector, 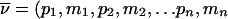 ), whose coordinates describe the outcomes of paternal and maternal meioses. Founder-phase symmetry (Kruglyak et al. 1996) allows us to set the bits of individual 21 to (p21,m21)=(0,0). At the marker, the maternal bits of individuals 22 and 23 are then obligated to be 0 and 1, respectively. After dropping the bits of individual 21 because they are fixed by founder-phase symmetry, we can write (p22,m22,p23,m23)=(x,0,x,1). Here x denotes a bit that is not restricted and that can take a value of either 0 or 1. In this case, observation of genotypes reduces the number of compatible inheritance vectors from 16 to 4. We can store this restriction, using just two integers. One integer is used as a mask that specifies the bits that are fixed by the observed genotypes, whereas the other integer contains the obligatory values of the fixed bits. In this example, the two integers in binary format are (0101) and (0100). Given an inheritance vector, a mask-and-compare operation is sufficient to check for compatibility with the observed genotypes. This also holds if the example given above is a subset of a larger pedigree; as more meioses are constrained, we can keep accumulating any restrictions, using the same pair of integers.
), whose coordinates describe the outcomes of paternal and maternal meioses. Founder-phase symmetry (Kruglyak et al. 1996) allows us to set the bits of individual 21 to (p21,m21)=(0,0). At the marker, the maternal bits of individuals 22 and 23 are then obligated to be 0 and 1, respectively. After dropping the bits of individual 21 because they are fixed by founder-phase symmetry, we can write (p22,m22,p23,m23)=(x,0,x,1). Here x denotes a bit that is not restricted and that can take a value of either 0 or 1. In this case, observation of genotypes reduces the number of compatible inheritance vectors from 16 to 4. We can store this restriction, using just two integers. One integer is used as a mask that specifies the bits that are fixed by the observed genotypes, whereas the other integer contains the obligatory values of the fixed bits. In this example, the two integers in binary format are (0101) and (0100). Given an inheritance vector, a mask-and-compare operation is sufficient to check for compatibility with the observed genotypes. This also holds if the example given above is a subset of a larger pedigree; as more meioses are constrained, we can keep accumulating any restrictions, using the same pair of integers.
Figure 1.

Example of 1-bit restrictions
We can define additional constraints. For the pedigree in figure 2 there are no meioses that are exactly determined, but there are constraints on the relative values of bits that belong to the same individual. Depending on the observed genotype, bits that belong to the same individual obey one of two rules: either they are equal or they are opposite. For individual 22 we can write pi=mi, and for individuals 23 and 24 we can write 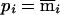 . Here we note as
. Here we note as 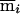 the opposite of meiosis mi. Again, storage of the constraints requires just a few integers (in this case, four), and we can quickly check for compatibility with the observed genotypes, using a series of mask, shift, exclusive-or, and compare operations. In the same example, we can illustrate an additional constraint. For individuals 23 and 24, the choice of the configuration (p23,m23)=(0,1) makes the configuration (p24,m24)=(1,0) the only choice compatible with the observed genotypes. Among the subset of inheritance vectors that obey the first constraint, we can define the relationship
the opposite of meiosis mi. Again, storage of the constraints requires just a few integers (in this case, four), and we can quickly check for compatibility with the observed genotypes, using a series of mask, shift, exclusive-or, and compare operations. In the same example, we can illustrate an additional constraint. For individuals 23 and 24, the choice of the configuration (p23,m23)=(0,1) makes the configuration (p24,m24)=(1,0) the only choice compatible with the observed genotypes. Among the subset of inheritance vectors that obey the first constraint, we can define the relationship 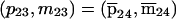 .
.
Figure 2.

Example of 2-bit restrictions. For each individual, all possible meioses are shown, and, next to each pair of meioses, compatibility or noncompatibility with the observed genotype is indicated, by a plus sign (+) or a minus sign (−), respectively.
The examples illustrate 1-bit, 2-bit, and 4-bit constraints. We can continue to define more-complicated constraints that span an ever-larger number of bits, but the gains in state-space reduction must be weighed against the number of operations necessary to check each inheritance vector for compatibility. In the present implementation, we use only the simplest, 1-bit restrictions, The specific algorithm, executed once per marker, is as follows:
-
1.
Define all sibships in the pedigree.
-
2.
For each sibship, find all restricted meioses, using as many as four grandparents, two parents, and sibs. The remaining steps are executed once per sibship.
-
3.
Full siblings who share the same genotype must share the same inheritance-vector restrictions. Because there is a maximum of four unique genotypes per sibship, we can set an upper limit to the computation. Define as “representative sibs” the first n sibs (n⩽4) with a unique genotype, plus a sib used to set founder phase. The use of representative sibs defines an upper limit to the number of inheritance vectors that we examine per nuclear family. The worst-case scenario is a consanguineous pedigree, in which 214=16,384 inheritance vectors must be examined. A more typical situation is a nonconsanguineous pedigree in which at least one of the parents is a founder. In such a situation, we need to examine 211=2,048 inheritance vectors. Because there is a well-defined upper limit, identification of state-space restrictions grows linearly with the number of nuclear families in the pedigree.
-
4.
Given the grandparents, parents, and representative sibs, apply the method used for the computation of single-point probabilities and temporarily store the result.
-
5.
For each sibship, use the list of incompatible inheritance vectors to update the set of constrained meioses.
The method is general and does not impose any restrictions on either the pedigree structure or the number of missing genotypes. Relationships between meioses are deduced by simple inspection of the temporary list of incompatible inheritance vectors. To increase the efficiency of the algorithm, the sibships are ordered so that ancestors always precede descendants. Therefore, some of the parental meioses are often fixed by a previous iteration, further reducing the number of bits per nuclear family. Not only is the calculation faster, but information on parental meioses from the rest of the pedigree is passed to the “current” sibship.
In addition to exact multipoint analysis, this algorithm can be applied to sampling (i.e., Monte Carlo) approaches. The set of integer-bit maps defines a convenient transformation to a contiguous state space where all bits that are not fixed by genotyping are adjacent and allowed to vary freely. Then the sampling can be restricted to a smaller state space with a much higher percentage of inheritance vectors compatible with the observed marker genotypes. In general, the procedure does not produce the complete set of inheritance vectors that are not compatible with the observed genotypes. The question of completeness can be separated into two: (1) What is the fraction of incompatible inheritance patterns found after consideration of subsets of the original pedigree? and (2) Even if all incompatible inheritance patterns are identified, what is the fraction that can be described by use of sets of integers for mask and compare operations? Answers to both questions are highly dependent on pedigree structure, number of missing genotypes, and number of alleles per marker. This makes it difficult to provide a general, concise answer to the question of completeness. Instead, we offer practical considerations that can guide alternative implementations of the algorithm. Our choice of subpedigree structure is practical because, for a set of full sibs, there is an information cutoff after two generations of genotyped ancestors. Consideration of additional ancestors does not produce additional constraints. We can consider sibships with genotyped ancestors separated by several ungenotyped generations, but this is an unlikely scenario in human and animal pedigrees. A promising alternative structure is simultaneous consideration either of full sibs and half-sibs or of first cousins. In the examples given above, we have shown how, by using integer-bit maps that allow simultaneous consideration of an ever larger number of bits, we can increase the number of incompatible inheritance patterns that we can describe. An alternative approach is to keep one full list of compatible inheritance patterns per sibship. Then we can generate compatible inheritance patterns for the whole pedigree, by combining lists of valid vectors. This is practical only when we consider pedigrees with a limited number of sibs per family. In the current implementation, we chose to use only 1-bit restrictions. It is the only class of restrictions that we currently use for computation of multipoint-inheritance probabilities, without resorting to the full state-space representation. Incorporation of 2-bit and 4-bit restrictions would accelerate the computation of single-point–inheritance probabilities but, without additional algorithmic improvements, would not have a significant impact on overall performance.
It is easy to quantify the computational gains. The computational cost per sibship has a well-defined upper bound, and in practice it is negligible. Only a fraction of a second is needed to accumulate all inheritance-pattern restrictions for any pedigree, independent of size or pedigree structure. We store single-point–inheritance and multipoint-inheritance probabilities only for inheritance patterns that satisfy the 1-bit constraints; For all other inheritance patterns, the implied value is zero. Because each 1-bit constraint cuts the vector-space size by a factor of two, the computational gains are exponential as a function of the number of bits constrained by genotyping. At a specific marker, if k of 2n-f meioses are constrained, then we need 2k times less time and memory to complete the calculation. Obviously, maximum gains in speed and memory are attained for pedigrees that are fully genotyped with highly polymorphic markers.
In addition to state-space restrictions, we use a simple rule to further reduce the computation of single-point–inheritance probabilities. Meioses of a genotyped, homozygous parent can be either paternal or maternal, with equal probability. Therefore, the bits representing these meioses define an equivalence class. We calculate single-point–inheritance probabilities for only one member of the class and assign the result to all class members.
Vector-Matrix Multiplication
We limit the discussion to 1-bit restrictions. Consider two markers, m1 and m2, with recombination fraction θ. Assume that k1⩽2n-f and k2⩽2n-f are the number of meioses with known outcome at m1 and m2, respectively. We have shown that we can reduce storage requirements per marker for single-point probabilities, from N=22n-f to N1=N/2k1 and N2=N/2k2, for m1 and m2, respectively. Although there are N nonzero transition probabilities from m1 to m2, we need not compute them all. At marker m2, we are interested in computation and storage of cumulative left/right probabilities (the product of transition probabilities × single-point probabilities), and we know in advance that only N2=N/2k2 of them can be different than zero. In Appendix A we show how to replace the (2n-f )-bit problem with a (2n-f-k1)-bit problem and the N2 operations necessary to map the result to the reduced state space at m2.
For marker-to-marker transitions, we have exponential gains in time and memory requirements, because we can take advantage of state-space restrictions at both markers. This is not the case when we compute linkage statistics between markers, because all inheritance vectors have a finite, nonzero probability of occurrence. Although we are forced to calculate cumulative probabilities for N states, we can still perform the calculation without allocating additional memory. For statistics between markers, we simply accumulate the product of inheritance probability and the scoring function for each inheritance vector. Therefore, we do not need to save the N inheritance probabilities to memory. We first execute two reduced FFTs (two HMM transitions, one with 2n-f-k1 bits and one with 2n-f-k2 bits) and use these results to compute each inheritance probability, as needed.
Computation of the Whittemore-Halpern Statistic (NPL_all)
This statistic quantifies the degree of allele sharing among affected individuals. Let a denote the number of affected individuals in a pedigree. Given an inheritance vector 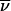 , there are 2a ways in which we can choose one of the two founder alleles for each affected individual. We denote by h such a sampling configuration and by bi(h) the multiplicity of founder allele i in configuration h. The statistic is defined as
, there are 2a ways in which we can choose one of the two founder alleles for each affected individual. We denote by h such a sampling configuration and by bi(h) the multiplicity of founder allele i in configuration h. The statistic is defined as
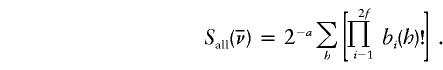 |
Computation of the statistic requires 22n-f×2a×S operations, where 22n−f is the number of inheritance vectors, 2a is the number of sampling configurations, and S is the number of operations needed per configuration.
We modify the original algorithm as follows.
-
1.
We reduce the number of inheritance vectors that we need to evaluate. We do not repeat the calculation for sequential inheritance vectors that produce the same founder-allele configuration among affected individuals and that therefore produce the same value for the statistic. This is the case for two inheritance vectors that are identical except for bits that correspond to meioses of individuals who are not affected and who have no descendents in the pedigree. In addition to reducing the number of operations necessary, we use the modification to reduce the memory necessary to store the result of the computation.
-
2.
We decrease the number of operations per configuration. Instead of counting the number of shared founder alleles for each one of the 2a configurations, we execute S operations once and then find the value of the statistic relative to the previous configuration. A detailed description can be found in Appendix B.
After these changes, the computation of nonparametric statistics becomes several times faster than the computation of parametric LOD scores.
Computer Implementation
In this section we present new commands that add flexibility to the use of the software package, and we discuss some limitations of the current implementation. We introduce three new commands:
-
1.
Compute sharing off: While scanning a pedigree, the program by default accumulates the identity-by-descent (IBD)–sharing probabilities (i.e., the IBD matrix) for all pairs of relatives in the pedigree. The IBD matrix is used for the computation of TDT scores, sib-pair statistics, and variance-components analysis. If such analyses are not required, the user should turn off storage of the IBD matrix, using the command compute sharing off. Time requirements for the accumulation of the IBD matrix are negligible, but there are cases in which storage requirements are significant; for example, for a large collection of highly informative pedigrees, storage of the IBD matrix exceeds the memory required for storage of multipoint-inheritance probabilities. Note that to accumulate the IBD matrix we first have to compute multipoint inheritance. Therefore, all analyses methods benefit from the speedups introduced by state-space reduction.
-
2.
Dump requirements: It would be useful to provide a set of rules for the estimation of computational requirements, given pedigree structure and marker heterozygosity. Unfortunately, human pedigree structures are irregular, and it is difficult to provide a concise answer. For rough estimates, we can use the following argument. On the basis of marker genotypes, our method separates meioses into two classes: those for which the outcome is exactly known and those for which both outcomes are possible. Meioses for which the outcome is known do not enter the calculation; therefore, the problem scales as the exponent of the meioses that do not have fixed outcomes, rather than as the exponent of the total number of meioses. An important issue in the estimation of computational requirements is the presence of markers that offer very little inheritance information; for example, consider a pedigree with 80% of its members available for genotyping but only 20% genotyped for a particular marker. In this case, we will have dramatic improvements for all but the marginal marker. At that position, the scale of the problem reverts to (or close to) the original size, and state-space reduction has little impact on overall performance. For a more accurate estimation, we would need a complex list of rules. We choose instead to let the software quickly estimate computational requirements, given a specific, genotyped pedigree. This simple calculation takes advantage of rapid identification of state-space restrictions and requires just a fraction of a second for any pedigree structure or size. The command dump requirements on/off sets a flag that modifies the behavior of the program. When the flag is set, scanning a pedigree simply reports overall memory requirements and memory requirements per marker. The researcher can add/subtract pedigree members or modify the marker map and get immediate feedback before proceeding with the full analysis. Time requirements are not reported, but, in the absence of inbreeding loops, it is safe to assume that time and memory requirements scale proportionally. We report only memory allocation that scales exponentially with pedigree size and do not report requirements for storage of the IBD matrix.
-
3.
Haplotype method Viterbi and haplotype method MaxProb: There are two possible solutions to haplotype reconstruction (Rabiner 1989; Kruglyak et al. 1996). One selects the most likely inheritance vector at each locus, whereas the other (based on the Viterbi algorithm) selects the most likely set of vectors, considering all loci simultaneously. Although the second solution has theoretical appeal because it finds a global maximum, in practice both methods yield similar results, especially when used for the analysis of pedigrees with high information content. The first method is mentioned in an article by Kruglyak et al. (1996) but is available, for the first time, with this release of GENEHUNTER. State-space reduction can be used to reduce the time and memory requirements of both algorithms. Currently, only the first approach is improved, and it is used by default (or through the command haplo method MaxProb). The Viterbi algorithm is still available (command haplo method Viterbi), but it requires the same amount of computer resources as was required in previous releases. With the first approach, haplotype reconstruction adds only a few percent to the overall computational time and does not add to the memory requirements.
With this release of GENEHUNTER, we have made an effort to avoid storage of inheritance probabilities at uninformative positions. An example in which uninformative markers enter the calculation is joint analysis of pedigrees collected by different research groups that use different marker sets for gonotyping. Performing a joint analysis requires a unified marker map that includes all markers in the data set. Therefore, each pedigree is genotyped only for a subset of the unified marker map. GENEHUNTER version 2.0 computed and stored inheritance probabilities for each pedigree, at every position on the map, even at positions where the pedigree was not genotyped. With the new implementation (version 2.1), a unique map of informative markers is automatically created for each family, and uninformative markers are treated as “positions between markers.” To identify uninformative markers, we use a simple rule: if, at most, one individual is genotyped at a marker, the marker is flagged as uninformative. The rule is useful not only when data sets are merged, but also when genotyping fails. In addition to the automatic removal of this trivial case, the program issues a warning if a marker is found to be uninformative after computation of single-point–inheritance probabilities. In cases in which only a few pedigrees are being analyzed, we encourage investigators to first use the command dump requirements (see above) and then inspect and, if it is appropriate, remove markers that have significant memory requirements. We have used this procedure for one of the pedigrees analyzed in the Simulated 30-Bit Pedigree subsection (below).
There are two limitations in this release of GENEHUNTER, related to the specific computer implementation. First, the maximum number of meioses that the program can process is 2n=32, where n is the number of nonfounders. The upper limit is not related to lack of computational resources but to the representation of inheritance vectors by 32-bit integers. Second, computation of parametric LOD scores requires a disproportionate amount of computational resources. We take advantage of pedigree symmetries to reduce time and storage requirements for inheritance probabilities and nonparametric statistics (i.e., “NPL” score). This is not yet the case for parametric statistics. If parametric LOD scores are not required, the user should turn LOD computation off (command analysis NPL). In a future release, we plan to remove both limitations.
TDT Significance Levels in the Presence of LD
The TDT was implemented in GENEHUNTER version 2.0 (Daly et al. 1998). Standard single-marker TDT is available; in the case of markers with more than two alleles, the transmission of each allele is tested against the collection of all other alleles. Multilocus TDT is similarly performed by counting the cases of transmission and nontransmission of each haplotype whenever it is observed in a parent who carries two different haplotypes.
One of the major challenges in the interpretation of any individual TDT result is the estimation of significance. In a typical LD study designed to follow up linkage to a region, tens to hundreds of markers may be genotyped, and the actual number of tests performed after examination of each marker (along with multiple-marker haplotypes of various lengths) can be considerable. Clearly, in such a case, the χ2 P value for a single result is not a useful indicator of significance, without some correction for the multiple testing performed. The simplicity of a Bonferroni correction for the number of tests performed (e.g., markers × alleles) is appealing, but the nature of the data renders such a correction prohibitively conservative. Trivially, the nonindependence of multiple alleles (microsatellites) and the presence of very rare alleles (microsatellites and single-nucleotide polymorphism [SNPs]) prevent a clean application of the Bonferroni correction. Much more important, however, is the issue of nonindependence of alleles at different markers (i.e., LD). In densely genotyped regions where strong LD exists in the overall population under examination (and not specifically in the disease population), many markers can provide nonindependent (and, in the case of closely spaced SNPs, frequently identical) TDT results. To apply the Bonferroni correction in this case could be dangerously conservative. Although this may be considered an extreme example now, available SNP maps (Mullikin et al. 2000) are quickly approaching the density at which LD is to be expected in general populations. As genotyping technologies follow, it will be quite reasonable and desirable for researchers to genotype hundreds to thousands of SNPs in very small genomic regions, in attempts to follow up a convincing linkage result or to clarify the involvement of a candidate gene by exhaustively examination of the variation in and around that gene.
To better address this problem, we have developed a permutation approach for the estimation of significance, an approach that is similar to the method that we used for case-control haplotype comparisons (Laitinen et al. 1997). The method involves generation of artificial data sets, perfectly matched to the observed data, by randomly permuting whichever chromosome from each parent is transmitted. Because this process leaves haplotypes intact, any LD present in the population is maintained fully in the permuted data sets. Each such data set is analyzed in the same fashion as are the original data, and the number of permutations in which a marker (or haplotype) exceeds the maximum value seen in the observed data is reported. Also reported is the number of permutations in which the number of observations of P<.01, .001, and .0001 equals or exceeds the number of such observations in the real data set. Depending on the degree of background LD, the presence of several nearby markers with strong TDT results may be an indicator of association that should be considered in addition to the magnitude of the highest result. In certain cases in which haplotyping is made difficult (e.g., incomplete genotyping, small sample size, inability to resolve transmission from doubly-heterozygous parents, etc.), attention to a cluster of high results may be a valid surrogate for complete multilocus haplotype analysis. Along these lines, in an analysis of SNPs surrounding the APO-E4 variant (Martin et al. 2000), it was noted that no nearby SNP provided evidence of association nearly as strong as the evidence provided by the E4 variant itself—but that the association might be best recognized by way of the cluster of nominally significant results.
So far, we have made no distinction between single-marker–based TDT and haplotype-based TDT. As noted by Dudbridge et al. (2000), when the haplotype test is used, failure to reconstruct multilocus haplotypes can introduce a modest inflation of type I error; for example, it is not possible to reconstruct haplotypes when two parents share the same heterozygous genotype and there is no additional information from nearby markers to help resolve the phase of the transmission. Not counting these cases leads to an overestimate of the strength of gene effect as estimated by transmission ratio. For this reason, an option exists to eliminate such data from all TDT counts and to present transmission counts that allow a robust estimate of gene effect (command dhskip). The correction recommended by Dudbridge et al. (2000) can be trivially recovered from these data. Dudbridge et al. recommend that each case in which unreconstructed heterozygotes are recovered by a homozygous offspring be counted as one transmission rather than as two. With dhskip off these cases are counted twice, and with dhskip on they are not counted at all; therefore, addition of half the difference to the result produces the recommended test.
Performance
We demonstrate the performance of the new algorithms, using the following sample data sets:
A simulated (2n-f)=30-bit two-generation family (fig. 3): This data set shows how the overall scale of the problem is reduced and illustrates some practical limitations of the current software implementation.
An actual (2n-f)=26bit pedigree with five of the six founders missing (fig. 4): This data set is more typical of human pedigrees and illustrates the ability of the new code to cope with missing genotypes.
A single pedigree structure simulated 10 times (fig. 5): The pedigree structure remains the same, but meioses and marker genotypes are generated independently, to produce 10 replicas of the pedigree. These pedigrees demonstrate how changes in information content influence performance, independent of pedigree structure.
A collection of published pedigrees from Genetic Analysis Workshop 10 (GAW10) (Goldin et al. 1997). Here we stress improvements specific to the handling of large collections of pedigrees genotyped by different research groups.
Unless otherwise noted, we report time requirements for computation of linkage statistics at marker locations. We used two computer systems to test the code: A SUN enterprise 450 workstation with 4 GB of memory and a commodity PC with a 350-MHz Pentium II processor and 256 MB of memory running Linux. All time benchmarks given below are for the SUN workstation. In terms of speed, the two computers required roughly the same amount of time to complete calculations that needed <256 MB of memory.
Simulated 30-Bit Pedigree
This is a (2n-f=2×16-2)=30-bit pedigree (fig. 3 and table 1). After removal of 3 of the 20 markers from consideration (see below), nonparametric scores at marker positions can be computed by use of just 12 MB of memory. In comparison, the memory required by the previous version of the software is almost 0.5 terabytes (500,000 MB), a factor of 38,000 higher. The time required for the computation was ∼2.5 h (9,392 s). For this problem, we cannot run the previous version of GENEHUNTER. If a computer with sufficient storage capacity were available, we extrapolate that the previous version would need 5 mo to complete the same calculation (a factor of 1,400).
Figure 3.

Simulated (2n-f)=30-bit pedigree
Table 1.
Relative Memory and Time Requirements for GENEHUNTER Versions 2.0 and 2.1
|
Memory(MB) |
|||
| Calculation | Version 2.1 | Version 2.0a | Factora |
| 30-Bit family: | |||
| NPL_all | 12 | 454,656 | 37,888 |
| 26-Bit nimh9002: | |||
| NPL_all | 2,529 | 26,880 | 10.63 |
| NPL_all and LOD | 3,041 | 26,880 | 8.84 |
| 10 18-Bit simulations:b | |||
| Missing 0 grandparents: | |||
| NPL_all | .07–10.53 | 75 | 1,041–7 |
| NPL_all and LOD | 2.07–12.53 | 75 | 36–6 |
| Missing 1 grandparent: | |||
| NPL_all | .43–10.57 | 75 | 173–7 |
| NPL_all and LOD | 2.43–12.57 | 75 | 7–3 |
| Missing 2 grandparents: | |||
| NPL_all | 10.63, 25.32 | 75 | 7–3 |
| NPL_all and LOD | 12.63–27.32 | 75 | 6–3 |
| 180 Pedigrees: | |||
| NPL_all, total | (See fig. 7) |
(See fig. 7) |
(See fig. 7) |
| Time(s) |
|||
| 30-Bit family: | |||
| NPL_all | 9,392 | NA | NA |
| 26-Bit nimh9002: | |||
| NPL_all | 3,382 | NA | NA |
| NPL_all and LOD | 9,402 | NA | NA |
| 10 18-Bit simulations:b | |||
| Missing 0 grandparents: | |||
| NPL_all | 31 | 3,163 | 102 |
| NPL_all and LOD | 184 | 3,317 | 18 |
| Missing 1 grandparent: | |||
| NPL_all | 47 | 3,165 | 67 |
| NPL_all and LOD | 201 | 3,320 | 16 |
| Missing 2 grandparents: | |||
| NPL_all | 141 | 3,132 | 22 |
| NPL_all and LOD | 297 | 3,287 | 11 |
| 180 Pedigrees: | |||
| NPL_all, total | 37,890 | 196,525 | 5 |
NA = not available.
Both the highest and the lowest requirements are shown.
We generated genotypes for 20 markers with an average of 4 alleles each. For 3 of the 20 markers, we could not employ state-space reduction, because none of the individual meioses were constrained by observed marker genotypes. These markers were manually removed by a procedure described above, in the Computer Implementation section. Of the three markers, two were completely uninformative because both parents were homozygous. Therefore, the two markers could be removed from consideration, without any loss of inheritance information. The third marker was informative, but the current implementation of the software does not take into consideration this 2-bit restriction—that is, two heterozygous parents with the same genotype. This marker introduces a huge computational burden (24 GB of storage) for a very limited return in terms of inheritance information. Therefore, we chose to remove it from consideration, in favor of analysis of a larger pedigree. Note that this is the only instance in which markers were removed by hand.
Reconstruction of haplotypes increased the analysis time by 5% and required just a fraction of a percent of additional memory. Of the two haplotype-reconstruction algorithms available, we used the maximum-probability-path option. Details on haplotype reconstruction can be found above, in the Computer Implementation section. As mentioned in the Algorithmic Improvements section, calculation of statistics for positions between markers has minimal impact on memory allocation but increases time requirements significantly. For this example, we repeated the calculation of nonparametric statistics, with the additional requirement that scores be calculated at one or four positions between markers. In both cases, 12 MB of memory was sufficient. The time requirements increased from 2.5 h to 30 h or 108 h (5 d), respectively. Evaluation of parametric LOD scores would require an additional 8 GB of memory storage, preventing us from calculating them for this example. This is a section of the code that has not been modified since the previous release of GENEHUNTER and that therefore does not exhibit the overall scale reduction that we see for other parts of the calculation. This example highlights both the orders-of-magnitude efficiency gains afforded by state-space reduction and the limitations of the current software implementation.
Actual 26-Bit Pedigree
This is an actual (2n-f=32-6)=26-bit pedigree from GAW10 (fig. 4 and table 1). Five of the six founders are not genotyped. In addition, among the available individuals several genotypes are missing. For this pedigree, efficiency gains are a factor of 10 for memory and a factor of 25 for speed (extrapolation). It is not an example most favorable for the new computational techniques, but it is representative of what should be expected for pedigrees with several missing ancestors. The high percentage of missing individuals reduces the number of meioses fixed by genotyping. Because our methods rely on informative markers to reduce the overall scale of the problem, one or two markers that lack local restrictions are sufficient to force the problem size back to its original scale. Here, 2 of the 16 markers have only 1 of 26 meioses exactly determined on the basis of local relationships. As a result, of the 2,145 MB used for storage of single-point–inheritance and multipoint-inheritance probabilities, 1,536 MB are required by these two markers.
Figure 4.

Actual (2n-f)=26-bit pedigree
Simulated 18-Bit Pedigree
We compare the efficiency of the previous and current versions of GENEHUNTER (versions 2.0 and 2.1, respectively), using a simulated pedigree that can be analyzed by both versions of the program. The (2n-f=22-4)=18-bit pedigree structure is shown in figure 5. We analyze 11 markers with an average heterozygosity of 75%. The same pedigree structure is simulated 10 times. For each of the 10 simulations, we use the same disease position, marker map, and allele frequencies, but we sample founder alleles and segregate them through the pedigree independently. This sampling generates pedigrees with different information content, which, in turn, has an influence on the efficiency of the computational methods.
Figure 5.

Simulated (2n-f)=18-bit pedigree. The same pedigree was simulated 10 times.
For nonparametric statistics (e.g., NPL_all), version 2.0 requires the same amount of memory for each of the 10 simulations (i.e., 75 MB; see fig. 6); the new version requires 7–1,000 times less memory, depending on the pedigree. Analysis of all 10 pedigrees by version 2.0 requires 3,160 s—versus 31 s for version 2.1, a factor of ∼100 times faster. As with memory, time requirements vary significantly by replicate, with speed-up factors of 40–135.
Figure 6.

Comparison of memory (a and b) and time (c and d) requirements for 10 instances of the simulated (2n-f)=18-bit pedigree shown in figure 5. Results shown are for GENEHUNTER versions 2.0 and 2.1. The linear plots (a and c) present requirements for the new version only; requirements for the previous version, which are 75 MB and 316 s, do not fit at this scale. The logarithmic plots (b and d) present relative requirements scaled to the number of equivalent bits—that is, the number of founder-symmetry–reduced meioses (i.e., 2n-f) that the previous version can process in the same amount of memory or time.
A common problem in human pedigrees is that founder DNA is not available. We tested the effect of missing founders by using the same 10 pedigrees, by deleting first one and then both grandparental genotypes and then rerunning the analysis. The results are shown in table 1. For version 2.0 the computational requirements remain the same, whereas for the new release of GENEHUNTER the memory allocation increases but remains several times less than what is required by version 2.0. When one grandparent is removed, we need 7–173 times less memory; when both are removed, we need 3–4 times less memory. In terms of speed, the newer version is 102, 67, or 22 times faster when no, one, or two grandparents are missing, respectively. If, in addition to the marker locations, we compute statistics for four positions between markers, the newer version is eight times faster, and the speed is almost independent of the number of missing grandparents.
For this example, pedigree structure and affection status are the same for all 10 pedigrees. Therefore, the computational cost for the evaluation of parametric LOD scores remains constant: 2 MB of memory and 15 s of time, for each pedigree. This is a very small fraction of the overall requirements of the older version (75 MB of memory and ∼300 s, per pedigree) but is the dominant component when the new methods are used. For all 10 pedigrees, calculation of parametric LOD scores requires 150 s, almost five times more than the 31 s needed for calculation of everything else.
GAW10
The final example illustrates the impact that our improvements have on the analysis of a diverse collection of pedigrees, typical of a large genome scan. We reanalyzed 180 families from a published study (Goldin et al. 1997), using both versions of the program. The data set is a collection of studies from different research groups that used different marker sets for genotyping. As mentioned above, in the Computer Implementation section, joint analysis requires computation of linkage statistics at every position on the map, even at positions where a pedigree was not genotyped. With this release of GENEHUNTER, a unique map of informative markers is automatically created for each family, and uninformative markers are treated as “positions between markers.” With this modification and state-space reduction, we have orders-of-magnitude improvements in terms of memory requirements, but the new version is only a few times faster than the previous release (fig. 7 and table 1). Although we take advantage of state-space reduction for storage at informative markers, joint analysis requires computation of linkage statistics at several uninformative positions. The newer version is approximately four to five times faster, depending on the combination of analysis options (e.g., calculation of parametric LOD scores, calculation of scores at five positions between markers, etc.). Unlike what has been seen in the previous examples, the speed improvements are modest; therefore, the choice of analysis options has a minimal (<20%) impact on relative performance.
Figure 7.

Memory requirements for a collection of 180 actual pedigrees (linear and logarithmic scale). Results shown are for GENEHUNTER versions 2.0 and 2.1. The logarithmic plot is scaled to the number of bits that version 2.0 can process with the same amount of memory.
Discussion
We have introduced a new approach that significantly reduces the computational cost of multipoint analysis. We have attacked the exponential growth of the problem by using the observed marker genotypes to restrict the number of inheritance patterns that we need to consider. We have introduced exact formalism that allows us to apply the Markov-chain reconstruction step of the Lander-Green algorithm to only those meioses that have ambiguous outcome. With the new formalism, the scale of the computation per marker drops from O(22n−f) to O(22n−f−k ), where k is the number of meioses with known outcome at that marker. Because the number of meioses with known outcome grows proportionally with pedigree size, the efficiency gains increase exponentially with the size of the pedigree. The formalism is exact—no approximations are applied. As shown in the examples section, we achieve maximum computational efficiency when the markers used are highly polymorphic and the number of missing pedigree members is small. Low marker heterozygosity and missing pedigree members lower the inheritance-information content of the pedigree. In the limit of very low information content, the computational requirements are the same as in the older version of the software. For typical pedigrees, performance gains are 10–1,000-fold. The improvements that we have discussed here do not overlap with those discussed by Gudbjartsson et al. (2000). We achieve exponential efficiency gains in computing not only single-point–inheritance probabilities but also multipoint-inheritance probabilities.
There are classes of local restrictions that we mention but that we do not use in the current software implementation. Most important among them is the case of 2-bit restrictions. This is not an important case when the current generation of highly polymorphic markers is used. For a marker with four equally frequent alleles, <10% of all parental pairs share the same heterozygous genotype; in contrast, if the next generation of more-abundant but less-informative genetic markers (i.e., SNPs) is used, the number of double heterozygote pairs is ∼ 1/4 of all parental pairs. In addition, the number of uninformative meioses due to homozygous parents increases significantly (Kruglyak 1997). Such low-information-content markers, as well as missing pedigree members, reduce the number of meioses that are fixed through local relationships, but they generate classes of equal-probability inheritance vectors. We are working on a complementary set of methods that accelerate the calculation by taking advantage of such equivalence classes. We defer use of 2-bit restrictions to the GENEHUNTER software release that will incorporate the equivalence-class algorithms.
All methods described here have been incorporated into a new release of the computer package GENEHUNTER (i.e., version 2.1). The computer program is written in C and is freely available (source code included) from either the Kruglyak lab website (by anonymous ftp) or via the Index of /ftp/distribution/software/gh2.1 website.
Acknowledgments
We would like to thank Alan Katz, for valuable programming support, and Mike Eberle and Mark Gibbs, for useful discussions. We would also like to thank the research groups that contributed data to GAW10: L. Goldin, E. Gershon, and W. Berrettini, from the National Institute of Mental Health; R. DePaulo and F. McMahon, from John Hopkins University; M. Byron, from Columbia University; H. Gurling and D. Curtis, from the University of London; and P. Propping, from the University of Bonn. This work was supported in part by a DOE/Sloan Foundation Fellowship in Computational Molecular Biology (to K.M.) and by National Institute of Mental Health grant MH-59520 (to L.K.). L.K. is a James S. McDonnell Centennial Fellow.
Appendix A: Vector-Matrix Multiplication
Assume that there are two markers, m1 and m2, with recombination fraction θ, and that there are k1,k2⩽2n-f meioses with known outcome at m1, m2. We refer to such meioses as fixed bits and to all other meioses as variable bits. We have shown that the number of inheritance patterns with nonzero probability of occurrence at m1 and m2 are, at most, N1=N/2k1 and N2=N/2k2, respectively. Here we show how to perform the vector-matrix multiplication necessary for the reconstruction of multipoint-inheritance probabilities, without resorting to the full state-space representation.
The HMM formalism (Lander and Green 1987; Rabiner 1989; Kruglyak et al. 1996) allows us to compute multipoint-inheritance probabilities incorporating one marker at a time. First we compute inheritance probabilities by using single-point information from all markers to the left of the current marker (i.e., the left probabilities). We then repeat the same procedure, incorporating marker information from right to left (i.e., the right probabilities). Computation of left-probability and right-probability distributions is referred to as the “reconstruction” step of the HMM. At each point in the genome the multipoint-inheritance–probability distribution is the product of left-probability and right-probability distributions. To incorporate each additional marker during the reconstruction step of the HMM, we need to execute the vector-matrix multiplication
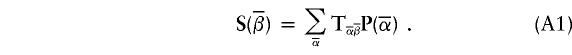 |
Vector P represents the (left/right) cumulative-probability distribution at m1, whereas vector S represents the (left/right) transition-probability distribution at m2. We remind the reader that we have defined the cumulative-probability distribution at a genotyped marker as being the product of transition probabilities and single-point probabilities at that marker. The two probability distributions, P and S, are indexed by inheritance vectors 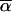 and
and 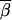 . The matrix
. The matrix 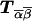 is the transition matrix between the two loci. It is defined as Tα,β(s,θ)=θh(1-θ)s-h, where s is the total number of meioses (bits), and
is the transition matrix between the two loci. It is defined as Tα,β(s,θ)=θh(1-θ)s-h, where s is the total number of meioses (bits), and 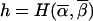 is the Hamming distance (i.e., the number of bits that differ) between the two vectors. The vector-matrix multiplication is an O(24n) operation. It can be reduced to an O[(2n-f)22n-f] operation by use of the FFT formalism described by Kruglyak and Lander (1998). Although we can expand the N1=N/2k1 probabilities at m1 into an N-components inheritance vector and can use the FFT formalism to execute the transition, this procedure would revert the scale of the problem back to O[(2n-f)22n-f]. We show how to execute the transition from m1 to m2 as an O[(2n-f-k1)22n-f-k1+22n-f-k2] operation. First we present an outline of the method, and then we proceed with the formal proof.
is the Hamming distance (i.e., the number of bits that differ) between the two vectors. The vector-matrix multiplication is an O(24n) operation. It can be reduced to an O[(2n-f)22n-f] operation by use of the FFT formalism described by Kruglyak and Lander (1998). Although we can expand the N1=N/2k1 probabilities at m1 into an N-components inheritance vector and can use the FFT formalism to execute the transition, this procedure would revert the scale of the problem back to O[(2n-f)22n-f]. We show how to execute the transition from m1 to m2 as an O[(2n-f-k1)22n-f-k1+22n-f-k2] operation. First we present an outline of the method, and then we proceed with the formal proof.
Outline. At each marker, we can define a convenient coordinate system in which all fixed bits are adjacent and in which the ranking among fixed and variable bits remains unchanged; for example, if y denotes fixed bits and x denotes variable bits, a change of base transforms the inheritance vector 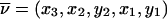 into
into 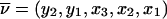 . The advantage is that, in the new coordinate system, all inheritance vectors compatible with 1-bit restrictions are stored sequentially. In general, for the cumulative probability, we can write
. The advantage is that, in the new coordinate system, all inheritance vectors compatible with 1-bit restrictions are stored sequentially. In general, for the cumulative probability, we can write 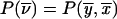 and
and 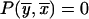 ,
, 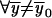 . Here,
. Here, 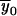 represents the k1-bit vector in which the coordinates are the obligatory values of the meioses dictated by 1-bit restrictions. The Hamming distance H(
represents the k1-bit vector in which the coordinates are the obligatory values of the meioses dictated by 1-bit restrictions. The Hamming distance H(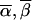 ) between two vectors
) between two vectors 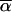 and
and 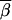 is invariant under such a transformation (if both
is invariant under such a transformation (if both 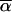 and
and 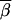 are transformed in the same way). Thus the transition probabilities are also invariant. We execute the vector-matrix multiplication (transition) from m1 to m2 by using the coordinate system defined by 1-bit restrictions at m1. The transition is accomplished in two steps. First we perform a partial transition, using only the 2n-f-k1 variable bits at m1. The partial result is a list of probabilities St(
are transformed in the same way). Thus the transition probabilities are also invariant. We execute the vector-matrix multiplication (transition) from m1 to m2 by using the coordinate system defined by 1-bit restrictions at m1. The transition is accomplished in two steps. First we perform a partial transition, using only the 2n-f-k1 variable bits at m1. The partial result is a list of probabilities St(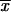 ). Then we use exact formalism that allows us to compute any transition probability S(
). Then we use exact formalism that allows us to compute any transition probability S(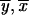 ), as a function of St(
), as a function of St(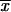 ) and the Hamming distance H(
) and the Hamming distance H(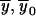 ). For θ≠ 0, all transition probabilities are other than zero. Calculating them all is an O(N) operation. However, there is no need to proceed with the whole calculation. For m2, we use the product of transition probabilities and single-point probabilities. Therefore, we need to compute for only N2=N/2k2 of N states. The rest are, after multiplication, exactly equal to zero. Thus, we first execute a partial FFT and then compute transition probabilities only for inheritance patterns that are compatible with 1-bit restrictions at m2. We transform every one of those vectors to the coordinate system defined at m1, and we use the exact formula to compute the transition probability. Transformations between coordinate systems are performed by a series of precalculated mask-and-shift operations. There is no need either for transformations to the original coordinate system or for additional temporary storage.
). For θ≠ 0, all transition probabilities are other than zero. Calculating them all is an O(N) operation. However, there is no need to proceed with the whole calculation. For m2, we use the product of transition probabilities and single-point probabilities. Therefore, we need to compute for only N2=N/2k2 of N states. The rest are, after multiplication, exactly equal to zero. Thus, we first execute a partial FFT and then compute transition probabilities only for inheritance patterns that are compatible with 1-bit restrictions at m2. We transform every one of those vectors to the coordinate system defined at m1, and we use the exact formula to compute the transition probability. Transformations between coordinate systems are performed by a series of precalculated mask-and-shift operations. There is no need either for transformations to the original coordinate system or for additional temporary storage.
Proof. For a pedigree with n members, there are s≡2n meioses. The vector-matrix multiplication of equation (A1) is an O(22s) operation. When the FFT algorithm (Kruglyak and Lander 1998) is used, the number of operations necessary to execute the matrix multiplication is reduced to O(s2s) additions and O(2s) multiplications. In addition, for f founders in the pedigree, the algorithm can take advantage of founder-phase symmetry, which reduces the FFT from s to s-f bits. We will show how to further reduce the number of operations, by using 1-bit restrictions. First, we derive the formalism, using all meioses (s=2n bits). Later, we extend the method so that it is applicable to the founder-symmetry–reduced state space (s=2n-f bits).
We prove that, after the change in coordinate system, the transition probability S(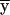 ,
, 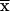 ) at recombination fraction θ from marker m1 is
) at recombination fraction θ from marker m1 is
where St(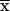 ) is the transition probability that we compute if we consider only the s-k1 variable bits. To prove the relationship, we use the computational method developed by Idury and Elston (1997). We can divide the cumulative-probability vector P at m1 into two vectors—P0 and P1—each half the size of P. The elements of P0 are indexed by inheritance vectors in which the lowest-order bit is equal to zero; in P1, the lowest-order bit is equal to one. Since the sequence of inheritance patterns in P0,P1 is identical up to one bit, we can rewrite the matrix-vector multiplication, as follows:
) is the transition probability that we compute if we consider only the s-k1 variable bits. To prove the relationship, we use the computational method developed by Idury and Elston (1997). We can divide the cumulative-probability vector P at m1 into two vectors—P0 and P1—each half the size of P. The elements of P0 are indexed by inheritance vectors in which the lowest-order bit is equal to zero; in P1, the lowest-order bit is equal to one. Since the sequence of inheritance patterns in P0,P1 is identical up to one bit, we can rewrite the matrix-vector multiplication, as follows:
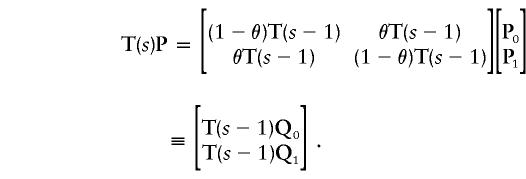 |
We have replaced an s bit problem with two (s-1)-bit problems; the vectors P0 and P1 have been replaced by Q0≡(1-θ)P0+θP1 and Q1≡θP0+(1-θ)P1, respectively. For the second iteration,
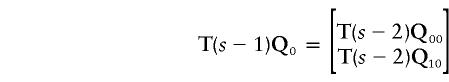 |
and
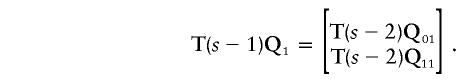 |
After s-k1 iterations, we have 2s-k1 vectors 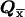 with 2kt components each.
with 2kt components each.
Proposition. 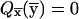 ,
, 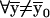 .
.
Proof. After the coordinate-system change, 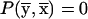 ,
, 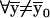 . The components of P0 and P1 are selected by using the lowest-order bit as the selection criterion. Therefore, the components of the new 2s-1-size vectors P0(
. The components of P0 and P1 are selected by using the lowest-order bit as the selection criterion. Therefore, the components of the new 2s-1-size vectors P0(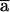 ) and P1(
) and P1(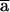 ) obey the same constraint: the first k1 high-order bits of
) obey the same constraint: the first k1 high-order bits of 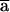 have to be equal to
have to be equal to 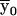 ; otherwise,
; otherwise, 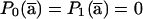 . Since Q0 and Q1 are linear combinations of P0 and P1, the same constraint holds for Q0(
. Since Q0 and Q1 are linear combinations of P0 and P1, the same constraint holds for Q0(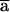 ) and Q1(
) and Q1(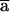 ). We can repeat the same argument up to iteration s-k1, which proves the proposition.
). We can repeat the same argument up to iteration s-k1, which proves the proposition.
After s-k1 iterations, we need to execute 2s-k1 vector-matrix multiplications—that is, T(k1)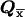 . The result is trivial, since all but one of the components of
. The result is trivial, since all but one of the components of 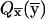 are equal to zero. Thus, the last k1 iterations can be executed in one step:
are equal to zero. Thus, the last k1 iterations can be executed in one step:
This is the desired relationship up to the replacement of 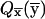 by St(
by St(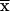 ), in which
), in which 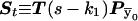 . The substitution replaces s-k1 iterations, each of which requires 2s intermediate results, by the same number of iterations requiring only 2s-k1 intermediate results each. We can compute
. The substitution replaces s-k1 iterations, each of which requires 2s intermediate results, by the same number of iterations requiring only 2s-k1 intermediate results each. We can compute 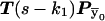 by using the iterative procedure that have we outlined for the computation of T(s)P. In the comparison of intermediate results, it is trivial to show that
by using the iterative procedure that have we outlined for the computation of T(s)P. In the comparison of intermediate results, it is trivial to show that
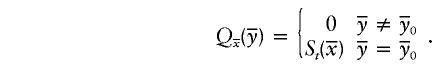 |
This concludes the proof for the case in which all meioses are taken into account.
Now we extend the formalism to the founder-symmetry–reduced space. Details on the application of founder symmetry to multipoint analysis can be found in the work of Kruglyak et al. (1996) and Kruglyak and Lander (1998). Here we summarize the results. For a pedigree with f founders, the inheritance vectors can be organized into equivalence classes with 2f equivalent members each. Working with a representative from each class reduces the number of states in the HMM by a factor of 2f. If a founder has l meioses in the pedigree, one is arbitrarily set to zero (paternal) and the remaining l-1 bits represent the outcome of the founder’s meioses relative to the reference phase. Switching the phase of a founder corresponds to simultaneously switching l bits representing the founder’s meioses. The 2n-bit inheritance vector 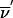 is replaced by the (2n-f)-bit inheritance vector
is replaced by the (2n-f)-bit inheritance vector 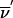 . The transition probability S(
. The transition probability S(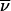 ) is replaced by the transition probability Sclass(
) is replaced by the transition probability Sclass(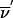 ), the sum of transition probabilities for all members of the class. In principle, to compute Sclass(
), the sum of transition probabilities for all members of the class. In principle, to compute Sclass(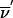 ) by using the formalism outlined above, we have to use equation (A2) 2f times. We will see that the sums factorize. Depending on the effect that 1-bit restrictions have on the l-1 founder bits, each founder phase can be classified into one of the following three categories: α (all bits variable), β (some variable and some bits fixed), and γ (all bits fixed). Here, fα, fβ, and fγ represent the number of phases that belong to each category; therefore, the total number of founder symmetry bits is f=fα+fβ+fγ. We can reduce the sums by a factor of 2fa if we use the FFT algorithm to perform the partial transformation that evaluates St(
) by using the formalism outlined above, we have to use equation (A2) 2f times. We will see that the sums factorize. Depending on the effect that 1-bit restrictions have on the l-1 founder bits, each founder phase can be classified into one of the following three categories: α (all bits variable), β (some variable and some bits fixed), and γ (all bits fixed). Here, fα, fβ, and fγ represent the number of phases that belong to each category; therefore, the total number of founder symmetry bits is f=fα+fβ+fγ. We can reduce the sums by a factor of 2fa if we use the FFT algorithm to perform the partial transformation that evaluates St(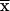 ). Of the remaining 2fβ+fγ sums, 2fγ factorize because changing founder phase does not change the St(
). Of the remaining 2fβ+fγ sums, 2fγ factorize because changing founder phase does not change the St(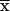 ) used in equation (A2); thus, we have to evaluate equation (A2) only (fγ+2fβ) times.
) used in equation (A2); thus, we have to evaluate equation (A2) only (fγ+2fβ) times.
To summarize, we have replaced an (N=22n-f)-size transition by an (N=22n-f-kt)-size transition plus O[N(fγ+2fβ)] operations necessary, to evaluate Sclass(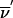 ), by using equation (A2). If there are state-space restrictions at the position where we evaluate Sclass(
), by using equation (A2). If there are state-space restrictions at the position where we evaluate Sclass(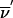 ), then we need to evaluate equation (A2) far fewer times; if k2 of 2n-f bits obey 1-bit restrictions, then the number of operations is reduced to O[N/2k2)(fγ+2fβ)]. In both cases, temporary memory requirements for the transition are reduced by a factor of 2kt.
), then we need to evaluate equation (A2) far fewer times; if k2 of 2n-f bits obey 1-bit restrictions, then the number of operations is reduced to O[N/2k2)(fγ+2fβ)]. In both cases, temporary memory requirements for the transition are reduced by a factor of 2kt.
Appendix B: Whittemore-Halpern Statistic
We represent a configuration of chosen alleles among a affected individuals with an a - bit-integer number C=0,1,2,…,(2a-1). The value of each bit represents the choice of founder allele for a particular individual.
In the previous algorithm, the value of C extends from 0 (all alleles paternal) to 2a−1 (all alleles maternal), and, for each value of C, we execute the following instructions:
-
1.
Count the multiplicity of each one of 2f founder alleles. (This is the most time-consuming step.)
-
2.
Evaluate the (factorial-dependent) statistic.
The new algorithm computes the statistic relative to the previous configuration. Consider that the value of the statistic is known for a configuration and that the next configuration differs only by the founder-allele choice of a single individual. If the individual has founder alleles A and B, then we assume that, in the previous configuration, we had picked A and that, in the current configuration, we pick B. To calculate the current value, we take the following steps:
-
1.
Divide the previous value of the statistic by the multiplicity of A.
-
2.
Decrease the multiplicity of A.
-
3.
Increase the multiplicity of B.
-
4.
Multiply the statistic by the new multiplicity of B.
To implement the method, we need an algorithm that allows us to consider the complete set of configurations while switching one assignment bit at a time; for example, for four affected individuals, instead of the sequence (binary format) C=0000, 0001, 0010, 0011, 0100, …, we use the sequence C=0000, 0001, 0011, 0010, 0110, …, . Such algorithms are used in instrument building (analog-to-digital conversion) and are referred to as “gray code.”
Electronic-Database Information
URLs for data in this article are as follows:
- Kruglyak lab website, http://www.fhcrc.org/labs/kruglyak/Downloads/index.html
- Index of /ftp/distribution/software/gh2.1, http://www-genome.wi.mit.edu/ftp/distribution/software/gh2.1
References
- Cottingham RW Jr, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Kruglyak L, Pratt SC, Houstis N, Reeve-Daly MP, Kirby A, Lander ES (1998) GENEHUNTER 2.0—a complete linkage analysis system. Am J Hum Genet Suppl 63:A286 [Google Scholar]
- Dudbridge F, Koeleman BP, Todd JA, Clayton DG (2000) Unbiased application of the transmission/disequilibrium test to multilocus haplotypes. Am J Hum Genet 66:2009–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston RC, Stewart J (1971) A general model for the genetic analysis of pedigree data. Hum Hered 21:523–542 [DOI] [PubMed] [Google Scholar]
- Goldin LR, Gershon ES, Berrettini WH, Stine OC, DePaulo R, McMahon F, Meyers D, et al (1997) Description of the Genetic Analysis Workshop 10 bipolar disorder linkage data sets. Genet Epidemiol 14:563–568 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Heath SC (1997) Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet 61:748–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idury RM, Elston RC (1997) A faster and more general hidden Markov model algorithm for multipoint likelihood calculations. Hum Hered 47:197–202 [DOI] [PubMed] [Google Scholar]
- Kruglyak L (1997) The use of a genetic map of biallelic markers in linkage studies. Nat Genet 17:21–24 [DOI] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Lander ES (1998) Faster multipoint linkage analysis using Fourier transforms. J Comput Biol 5:1–7 [DOI] [PubMed] [Google Scholar]
- Laitinen T, Kauppi P, Ignatius J, Ruotsalainen T, Daly MJ, Kaariainen H, Kruglyak L, Laitinen H, de la Chapelle A, Lander ES, Laitinen LA, Kere J (1997) Genetic control of serum IgE levels and asthma: linkage and linkage disequilibrium studies in an isolated population. Hum Mol Genet 6:2069–2076 [DOI] [PubMed] [Google Scholar]
- Lander ES, Green P (1987) Construction of multilocus genetic maps in humans. Proc Natl Acad Sci USA 84:2363–2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ER, Lai EH, Gilbert JR, Rogala AR, Afshari AJ, Riley J, Finch KL, Stevens JF, Livak KJ, Slotterbeck BD, Slifer SH, Warren LL, Conneally PM, Schmechel DE, Purvis I, Pericak-Vance MA, Roses AD, Vance JM (2000) SNPing away at complex diseases: analysis of single-nucleotide polymorphisms around APOE in Alzheimer disease. Am J Hum Genet 67:383–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullikin JC, Hunt SE, Cole CG, Mortimore BJ, Rice CM, Burton J, Matthews LH, et al (2000) An SNP map of human chromosome 22. Nature 407:516–520 [DOI] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE (1995) The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set—recoding and fuzzy inheritance. Nat Genet 11:402–408 [DOI] [PubMed] [Google Scholar]
- Rabiner LR (1989) A tutorial on hidden Markov models and selected applications in speech recognition. Proc IEEE 77:257–286 [Google Scholar]
- Sobel E, Lange K (1993) Metropolis sampling in pedigree analysis. Stat Methods Med Res 2:263–282 [DOI] [PubMed] [Google Scholar]
- ——— (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337 [PMC free article] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50:118–127 [PubMed] [Google Scholar]