

Abstract
Gentamicin is a widely used ototoxic agent. In this study, we shed light on the mechanisms underlying gentamicin-induced hearing loss. More importantly, we demonstrate in vivo and in vitro the effectiveness of a strategy for preventing drug-induced hearing loss using l-carnitine (LCAR), a safe micronutrient that plays a key role in energy metabolism and detoxification [Rebouche, C. J. & Seim, H. (1998) Annu. Rev. Nutr. 18, 39-61]. We show that LCAR prevents changes in hearing threshold and cochlear damage in newborn guinea pigs exposed to gentamicin in utero. Mechanistically, gentamicin-induced apoptosis of auditory cells is mediated by the extracellular signal-regulated kinase (ERK) 1/2 mitogen-activated protein kinase (MAPK) pathway through up-regulation of the proapoptotic factor Harakiri (Hrk). Most important, small interfering RNA (siRNA) experiments demonstrate that Hrk up-regulation is crucial for gentamicin-induced apoptosis. LCAR, in contrast, prevents both gentamicin-induced Hrk up-regulation and apoptosis acting by means of c-Jun N-terminal kinase (JNK). Together, these results outline pathways for gentamicin-induced hearing loss and its prevention and assign a key role to Hrk in these processes. Thus, our data offer a conceptual framework for designing clinical trials using a safe micronutrient, LCAR, as a simple preventive strategy for iatrogenically induced ototoxicity.
Keywords: apoptosis, ototoxicity, l-carnitine, extracellular signal-regulated kinase 1/2, HEI-OC1 cells
Low cost and high efficacy make gentamicin a common choice for treatment of infections due to Gram-negative bacteria (1). Unfortunately, gentamicin is both nephrotoxic and ototoxic. Although renal impairment is generally mild and reversible, ototoxicity results from the drug-induced apoptosis of auditory and vestibular sensory cells, and it is irreversible. The benefits provided by gentamicin, however, often outweigh the risks. For instance, infection during pregnancy has been causally linked to premature birth, and, as a result, an increasing number of pregnant women are being exposed to gentamicin during the perinatal period (2). Thus, their offspring are also being exposed to this ototoxic agent during a period of development when they are more vulnerable (3). Protective strategies aimed at decreasing drug-induced sensorineural hearing loss in unborn children or neonates, however, have not been reported.
Gentamicin has the ability to catalyze the formation of free radicals by a mechanism that may involve the formation of a complex with iron, which is vital for normal mitochondrial function (4). Free-radical formation, as an underlying cause of ototoxicity, has received strong support because antioxidants attenuate aminoglycoside-induced hearing loss (5). Altogether, these studies point to a key role of the mitochondria, an organelle involved in both iron metabolism and oxidation, as a target of ototoxic drugs. Unfortunately, the defined molecular effectors mediating the toxic effects of these agents remain poorly understood. This gap in the existent knowledge has limited the research for anti-ototoxic drugs rather to an empirical matter of trial and error. Therefore, there is an urgent need of mechanistic studies that, by revealing specific molecular pathways involved in ototoxicity, provide the conceptual basis for the rational design of therapeutic approaches to prevent this phenomenon.
Because a growing body of evidence places mitochondrial homeostasis at the core of aminoglycoside ototoxicity (6, 7), we focused our attention on defining novel apoptotic pathways associated with this common iatrogenic effect and testing the potential preventive action of l-carnitine (LCAR), a naturally occurring neuroprotective agent that plays a crucial role in mitochondrial functioning. LCAR is required for the transport of long-chain fatty acids across the mitochondrial membrane before they can undergo β-oxidation, resulting in ATP formation. In addition, LCAR modulates the intramitochondrial acyl-CoA/CoA ratio and is responsible for scavenging toxic compounds before they have a chance to accumulate in the mitochondria. Interestingly, LCAR and one of its esters, acetyl-l-carnitine (ALCAR), have been shown to reverse many age-associated deficits in cellular function linked to oxidative damage and mitochondrial decay, including age-related hearing loss (presbycusis) (8-10).
We now provide evidence that supplementation of pregnant mothers with LCAR prevents neonatal mortality and sensorineural hearing loss induced by gentamicin in newborn guinea pigs. In addition, we demonstrate that gentamicin induces, and LCAR prevents, the expression of Harakiri (Hrk), a proapoptotic member of the Bcl2 family of proteins, and that Hrk expression is crucial for gentamicin cell toxicity and its prevention.
Materials and Methods
Animal Experiments. Procedures involving laboratory animals were approved by the House Ear Institute Institutional Animal Care and Use Committee (IACUC). Pregnant guinea pigs at 28 ± 4 days of gestation (dg) were purchased from Simonsen Laboratories (Gilroy, CA) and randomly distributed in four groups of four animals each. Animals from group 1 (control) received i.p. injections of normal saline once a day for 7 consecutive days at 51 ± 2 to 57 ± 2 dg. Guinea pigs in group 2 (gentamicin) were injected with gentamicin (100 mg/kg per day, once a day for 7 days at 51 ± 2 to 57 ± 2 dg). Groups 3 and 4 were injected with gentamicin as described for group 2 but received LCAR supplementation in their water supply (1 mg/ml ad libitum, ≈100 mg/kg per day) starting either 2 weeks before (LCAR plus gentamicin) or simultaneously with gentamicin (gentamicin plus LCAR). A total of 71 puppies were born, and newborn mortality was defined as either stillborn or death within the first 48 h of life. Two weeks after delivery, mothers and newborns were tested by auditory brainstem response (ABR) and immediately euthanized with CO2. Cochleae were then harvested for light microscopy (frozen sections), confocal microscopy, and scanning electron microscopy (SEM) studies. In a particular series of experiments, 3-week-old guinea pigs (n = 4) received a single i.p. injection of either saline solution or gentamicin (400 mg/kg of body weight) and were euthanized 6 h or 24 h later. Cochleas were then harvested and processed for light microscopy.
ABR. All experiments were performed inside an acoustically insulated booth (Industrial Acoustics, Bronx, NY). Guinea pigs were anesthetized by i.p. injections of 60 mg/kg ketamine and 5 mg/kg xylazine. The subject's core body temperature was maintained with a thermostatically controlled heating blanket (Baxter K-MOD 100). Broadband clicks (10-ms duration, 50-μs sample rate) were generated by a computer-controlled BioSig Amplifier (siggen Computer Software, Tucker Davis Technologies, Alachua, FL), calibrated with a digital sound level meter (RS 33-2055), and presented alternately to the left and right auditory meatus through an ear bar connected to a Beyer DT-48 transducer. Platinum subdermal needle electrodes (F-E2, Grass Instruments, West Warwick, RI), fed by an Opti-Amp 3000 bioamplifier (Intelligent Hearing Systems, Miami) with a gain of 100,000 and a 300-Hz to 5-kHz filter setting, were placed under each auricle and at the vertex. Averaged responses from 512 stimuli were obtained at 5-dB intervals in the range from 80 dB to 0 dB. Auditory thresholds, defined as the lowest intensity to yield a reproducible deflection in the evoked response trace, were estimated for each ear, and threshold shifts were evaluated by comparison of the means calculated for each experimental group with respect to the control by using ANOVA techniques with Bonferroni correction factor for multiple comparisons (statview 4.1 and superanova, Abacus Concepts, Berkeley, CA).
Frozen Sections. Otic bullae were fixed with 4% paraformaldehyde overnight at 4°C, washed out with PBS for 30 min, and decalcified with 120 mM EDTA for 4 weeks. Samples were washed with PBS twice for 30 min and then placed in 15% sucrose solution for 20 min and in 30% sucrose solution at 4°C overnight. Next, samples were molded with OCT compound embedding medium (Sakura Finetek, Torrance, CA) in proper orientation, sectioned in a cryostat at 10-μm thickness, and stored at -20°C. Sections were labeled with anti-Hrk (Q-17, Santa Cruz Biotechnology), anti-phosphorylated (p)-ERK1/2, and anti-p-JNK (Cell Signaling Technology, Beverly, MA) at 1:100 dilution following standard protocols, and observed with a Zeiss Axiovert 135 TV inverted microscope with Neo-fluar ×10, ×20, and ×40 objectives.
Confocal Microscopy and SEM. For confocal microscopy, otic bullae were opened and fixed in 3% paraformaldehyde (PF) in PBS (pH 7.4) for 1 h. Next, cochlear turns were separated with small scissors and individually processed. Samples were permeabilized with 0.5% Triton X-100 in PBS for 30 min, followed by another 30-min incubation in blocking solution (10% goat serum plus 1% BSA in PBS). Finally, samples were incubated at 37°C for 30 min with 33 nM rhodamine/phalloidin (Molecular Probes) diluted in PBS from a stock solution of 3.3 μM in methanol (100 units/ml), mounted, and observed. HEI-OC1 cells, in turn, were fixed in 4% PF for 1 h at 4°C and then washed with PBS three times for 5 min each. Primary antibodies were used at 1:100 dilution in PBS plus 1% Tween 20 (PBST) in overnight incubations at 4°C. Gentamicin uptake was monitored with a monoclonal antibody against gentamicin (Fitzgerald Industries International, Chelmsford, MA). Anti-mouse and anti-rabbit FITC-, CY2-, and CY3-bound secondary antibodies (Jackson ImmunoResearch) were used at 1:1,000 dilutions in PBST in 1-h incubations at room temperature. Annexin V/propidium iodide labeling was performed by using the Vybrant Apoptosis Assay Kit No. 2 (Molecular Probes) following the manufacturer's protocol. Samples were observed with a Zeiss LSM-410 laser confocal microscope with objectives C-Apo ×40 and ×63 (N.A. = 1.2). For SEM, otic bullae were opened and fixed by immersion in 2.5% glutaraldehyde buffered in 100 mM sodium cacodylate (pH 7.2) for at least 4 h. Next, they were decalcified for 3-5 days in 120 mM EDTA (Sigma), washed with PBS, dissected out to expose the organ of Corti, reimmersed in 2.5% glutaraldehyde, and sequentially exposed to tannic acid, osmium tetroxide, and thiocarbohydrazide as described (11). Finally, samples were dried by using a critical-point dryer, and examined in an FE-SEM (XL30 S-FEG, FEI-Philips, Hillboro, OR).
Evaluation of Drug-Induced Cochlear Damage. SEM and confocal samples of guinea pig cochleae were examined thoroughly, and the percentage of missing hair cells was obtained by dividing the number present by the total number counted plus the scars showing missing hair cells × 100. Results were evaluated with ANOVA techniques by using arcsin transformation of the data.
Caspase-3 Activation. HEI-OC1 cells were cultured at 33°C, 10% CO2 in DMEM (GIBCO/BRL) supplemented with 10% FBS (GIBCO/BRL) without antibiotics, in uncoated dishes 100 mm in diameter (12). Untreated cells and cells exposed to 50 μM gentamicin (Sigma) for 24 h, with and without a 48-h preincubation with 2 μg/ml LCAR (Sigma), and with and without PD98059 [extracellular signal-regulated kinase (ERK) inhibitor (ERK-I) 100 μM] and SP600125 [c-Jun N-terminal kinase (JNK) inhibitor (JNK-I) 5 μM] (both from Calbiochem), were used in caspase-3 activation assays (CaspACE Assay System, Promega), following the manufacturer's protocols. Absorbance at 405 nm was measured in 96-well plates (flat-bottom) by using the computer-controlled microplate reader GENios (Tecan, Research Triangle Park, NC) with magellan 5.0 software.
Western Blotting. Cells were lysed at 4°C in a 50 mM Tris buffer solution (pH 7.4) containing 1% Nonidet P-40, 2 mM EDTA, 100 mM NaCl, 1 mM vanadate, 10 μl/ml 0.1 M PMSF, 2 μl/ml 10 mg/ml leupeptin, and 2 μl/ml 10 mg/ml aprotinin. Samples were mixed with loading buffer (2 g of SDS/0.002 g of bromophenol blue/1.54 g of DTT/8 ml of 1 M Tris (pH 6.8)/10 ml of glycerol), heated at 95°C for 5 min, analyzed by SDS/PAGE gels (30 μg of protein per lane), transferred to poly(vinylidene difluoride) (PVDF) membranes, and incubated with primary antibodies. The reaction was detected by ECL (Amersham Pharmacia) by using peroxidase-labeled secondary antibodies.
Gene Profiling. HEI-OC1 cells growing to confluence in 100-mm plastic culture dishes (six per experimental condition) were incubated with 50 μM gentamicin for 24 h at 33°C. Total RNA was extracted from cells by homogenization in TRIzol LS Reagent (Gibco-Invitrogen) following the manufacturer's protocol. Biotin-dUTP-labeled cDNA probes were generated by PCR, added to prehybridized GEArray membranes (GEArray Q series Mouse Apoptosis and Stress & Toxicity PathwayFinders, SuperArray, Frederick, MD), and incubated in a hybridization oven overnight. Next, membranes were incubated with the streptavidin-AP conjugate and developed with the CDP-Star chemiluminescent substrate provided by the manufacturer following GEArray protocols. Changes in gene expression were validated by Western blotting and RT-PCR. For RT-PCR analysis, RNA was extracted from HEI-OC1 cells with TRIzol, and cDNA was prepared from the RNA by using SuperScript (Invitrogen). The following oligonucleotide pairs were used to amplify Hrk-specific transcripts from the normal cell line cDNA: for Hrk, (+) 5′-ATT CCG TAC CTG TGC ATG CCT G-3′ and (-) 5′-TGT GCT GAA CAG TTG GTC CAC G-3′; for GAPDH (control for RNA integrity), (+) 5-TGA TGA CAT CAA GAA GTG GTG AAG-3′ and (-) 5′-TCC TTG GAG GCC ATG TAG GCC AT-3′. PCR was carried by using the following conditions: 20 cycles of denaturation at 95°C for 30 s, annealing 52°C for 30 s, and extension at 72°C for 1 min. PCR product was analyzed by separation on a 2% agarose-TAE gel.
Small Interfering RNA (siRNA). HEI-OC1 cells were transfected with four SMARTselection-designed siRNA oligonucleotides targeting Hrk, alone and pooled (Dharmacon Research, Lafayette, CO). The oligonucleotide sequences are as follows: 1, GTAAAGAGCTGATGGTGGA; 2, GATGTGAACTCTGAGACTT; 3, AAACTTACATGGACCGGTG; and 4, GAACTCTGAGACTTCGTAA. For ectopic reconstitution of the Hrk expression, we used a Hrk-resistant (rHrk) cDNA, kindly provided by G. Nuñez (University of Michigan, Ann Arbor), that lacks the siRNA-targeting sequences (12). HEI-OC1 cells were grown in six-well plates at 33°C until 90-95% confluent, and then incubated with Lipofectamine 2000 (Invitrogen), with and without the siRNA's oligos, for 4 h following the manufacturer's protocol. Next, the transfection mixture was replaced with complete growth medium, and the cells were cultured for another 48 h. Finally, cells were exposed to gentamicin for 24 h and then collected and processed for Western blotting and caspase-3 activation assays as described above.
Results and Discussion
We used guinea pigs at late stages of pregnancy as an animal model to investigate gentamicin-induced hearing loss and the effect of LCAR supplementation on the newborns. Interestingly, we observed a significant difference in neonatal mortality rates among the groups included in our study (Fig. 1a). Although exposure to gentamicin increased mortality of newborn guinea pigs, LCAR supplementation, both before and simultaneously with gentamicin, significantly decreased it. In addition, although it may be presumed that the animals potentially most affected by gentamicin were those stillborn, ABR experiments showed a significant gentamicin-induced increase in the hearing threshold of the survivors (gentamicin = 30 ± 3 dB vs. control = 21 ± 1 dB, P ≤ 0.01, Fig. 1b). LCAR supplementation, either from 28 days of pregnancy or coincidental with gentamicin injections, completely prevented this change (LCAR plus gentamicin = 23 ± 1 dB and gentamicin plus LCAR = 21 ± 1 dB). A similar protective effect of LCAR was observed in the mothers (Fig. 5, which is published as supporting information on the PNAS web site). Importantly, this response would be indeed an oto-protective effect, because LCAR does not interfere with the antibiotic efficacy of gentamicin (Fig. 6, which is published as supporting information on the PNAS web site).
Fig. 1.

Exposure to gentamicin in utero induces, and LCAR prevents, hearing loss and cochlear damage in newborn guinea pigs. (a) LCAR significantly decreases gentamicin-induced neonatal mortality (gentamicin vs. LCAR plus gentamicin, P ≤ 0.003). (b) Typical ABR traces in response to 10-ms clicks. (c) ABR experiments indicate that LCAR prevents the significant shift in hearing threshold associated with exposure to gentamicin. (d) Percentage of lost OHCs in the first, second, and third cochlear turns in newborn guinea pigs. Fourth turn data were not included because of the different nature of the damage (see Results and Discussion and f). (e) SEM image of a full guinea pig cochlea with the smaller, apical fourth turn on the left, and the bigger, basal first turn to the right. (f) Typical appearance of the OHCs in the apical fourth turn of animals exposed to gentamicin. The pattern of distribution is disrupted, many cells are lost, and others show atypical hair bundles. A single giant stereocilia is also visible (arrowhead). (g-i′) SEM and confocal images of more basal turns show the normal structure of the organ of Corti, with three rows of OHCs and scars replacing dead cells (arrows).
SEM and confocal techniques confirmed both that gentamicin induces a significant damage of outer hair cells (OHCs) and that this damage can be prevented by LCAR supplementation in newborn guinea pigs (Fig. 1 d-i′). Drug-induced OHC death was similar in the first three turns of the cochlea (Fig. 1 d and g-i′). The apical fourth turn (low frequency region), however, was remarkably affected by gentamicin, with near disappearance of the normal pattern of three parallel rows of OHCs and many OHCs showing single, giant stereocilia or disorganized hair bundles (Fig. 1f). These effects were ameliorated by LCAR supplementation. Because in adult guinea pigs the primary target of ototoxic drugs is the basal (high frequency) region of the cochlea (see Fig. 5b), the apical damage could be associated with the particular period of cochlear development at which the fetuses were exposed to gentamicin.
To further investigate the molecular mechanism activated by gentamicin and LCAR, we used an auditory cell line, HEI-OC1, highly sensitive to ototoxic drugs (13). After 2 h incubation with gentamicin, ≈30% of HEI-OC1 cells incorporated significant amounts of the drug as indicated by labeling with a monoclonal antibody. Gentamicin up-take, in the presence or absence of LCAR, reached a plateau after 6 h of incubation, with ≈50% of the cells showing significant labeling (Fig. 2a). As described in other systems, small vesicles filled with gentamicin were first observed in the perinuclear region of the cell, and later the drug distributed all over the cytoplasm (Fig. 2 b and c). HEI-OC1 cells exposed to gentamicin display several morphological and biochemical features characteristic of apoptosis, including positive immunolabeling with anti-annexin V antibodies and blebbing of the plasma membrane (Fig. 2 d-g).
Fig. 2.

HEI-OC1 cells are sensitive to gentamicin. (a) HEI-OC1 cells quickly incorporate gentamicin, reaching a plateau at 6 h incubation. Gentamicin uptake is not prevented by preincubation with LCAR. (b and c) Gentamicin-filled vesicles (green) accumulate preferentially in the perinuclear region (b), and later distribute in the entire cytoplasm (c). (d and e) Labeling with anti-annexin V antibodies (green) and propidium iodide stain (red) indicates that many gentamicin-exposed cells undergo apoptosis (e), but necrotic (annexin V-negative, propidium iodide-positive) cells were also observed (d). (f and g) SEM studies indicate that plasma membrane blebbing (g) was significantly more frequent in cells exposed to gentamicin than in control cells (f).
Gene profiling showed that gentamicin increases 13-fold the expression of the proapoptotic molecule Hrk in HEI-OC1 cells, whereas LCAR prevented this effect. This result was validated by RT-PCR, Western blotting and immunolabeling (Fig. 3 a-c). Hrk encodes a member of the Bcl-2 family of proteins (14, 15) recently implicated in axotomy-induced neuronal cell death (16) and potassium-induced apoptosis of cerebellar granule neurons (17). Hrk expression in auditory cells was already augmented after a 6-h incubation with gentamicin, and there were further increases with longer incubation times (Fig. 3a). In contrast, preincubation of the cells with LCAR abolished this response. Most importantly, suppression of Hrk expression by siRNA abolished the gentamicin-induced activation of caspase-3, a reliable indicator of apoptosis, in cultured auditory cells. Moreover, reconstitution of Hrk expression using RNA interference (RNAi)-resistant cDNA (rHrk) override this inactivation (Fig. 3d). These results indicate that Hrk up-regulation is necessary for gentamicin-induced apoptosis. Furthermore, gentamicin also increased the expression of Hrk in OHCs and other cell populations of the guinea pig cochlea, and this increase was prevented by LCAR supplementation (Fig. 7 a-c, which is published as supporting information on the PNAS web site). Therefore, LCAR protects auditory cells from apoptosis by preventing the gentamicin-induced up-regulation of Hrk.
Fig. 3.

Gentamicin induces, and LCAR prevents, transcriptional up-regulation of Hrk. (a and b) Microarray results were validated by RT-PCR (a) and Western blot (b). (c) Confocal images of HEI-OC1 cells triple-labeled with anti-gentamicin (green), anti-Hrk (red), and the nuclear stain DAPI (blue) show more abundant Hrk expression in cells that incorporate than in those that do not incorporate gentamicin (arrowhead). (d) siRNA experiments confirm that Hrk expression is necessary for gentamicin-induced apoptosis. All of the siRNA oligonucleotides inhibit the expression of Hrk and its up-regulation by gentamicin, and prevent caspase-3 activation. These effects were abolished by cotransfection with Hrk-resistant (rHrk) cDNA.
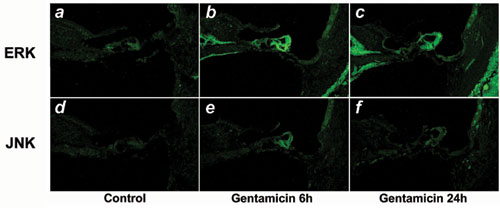
Subsequent studies on signaling cascades that can mediate the gentamicin-induced transcriptional regulation of Hrk revealed a role for mitogen-activated protein kinases (MAPKs). Pharmacological inhibition of ERK1/2 prevented gentamicin-induced activation of caspase-3 in HEI-OC1 cells (control = 100 ± 4% vs. gentamicin plus ERK-I = 160 ± 20%, P = not significant), whereas inhibition of JNK increased the apoptotic effect of gentamicin (gentamicin = 280 ± 20% vs. gentamicin plus JNK-I = 510 ± 30%, P ≤ 0.001) (Fig. 4a). Moreover, inhibition of ERK1/2, but not JNK, significantly reduced gentamicin-induced Hrk expression in auditory cells (Fig. 4b). These results suggest that gentamicin-induced apoptosis is mediated by ERK1/2. Consistently, studies of MAPK activation indicated that gentamicin induces both phosphorylation and nuclear translocation of ERK1/2 (Fig. 4 c-f), a process associated with neuronal apoptosis and neurodegeneration (18). JNK phosphorylation, on the other hand, diminished with 6 h incubation, and it is abolished by 24 h incubation with gentamicin (Fig. 4c). Remarkably, a similar gentamicin-induced ERK activation occurs in OHCs and other cochlear cell populations of guinea pigs exposed to gentamicin (Fig. 8 a-c, which is published as supporting information on the PNAS web site). JNK, in contrast, is weak and transiently activated mainly in supporting cells (Fig. 8 d-f).
Fig. 4.

Gentamicin-induced up-regulation of Hrk is mediated by MAPKs. (a) Caspase-3 activation experiments indicate that gentamicin-induced apoptosis of HEI-OC1 cells is prevented by preincubation with LCAR and ERK1/2 inhibition, but enhanced by inhibition of JNK. JNK inhibition also interferes with the preventive effect of LCAR. Values are normalized (control = 100%). (b) RT-PCR results confirmed that the effects of the inhibitors of MAPK on caspase-3 activation were associated with changes in Hrk expression. (c) Phosphorylation studies indicate that gentamicin activates ERK1/2 and inactivates JNK. LCAR, in turn, is able to reverse the gentamicin-induced inactivation of JNK. (d-f) Confocal microscopy of HEI-OC1 cells triple-labeled with gentamicin (green), anti-p-ERK (red), and DAPI (blue) confirms that gentamicin activates ERK and demonstrates that gentamicin also induces its translocation to the nucleus. A cell with numerous gentamicin-filled vesicles in the cytoplasm and p-ERK concentrated in the nucleus is pointed out with an arrow. An arrowhead indicates a cell that incorporates only a small amount of gentamicin and shows cytoplasmic labeling with anti-p-ERK.
Interestingly, inhibition of JNK, but not ERK, abolished the protective effects of LCAR (gentamicin = 280 ± 20% vs. LCAR plus gentamicin = 88 ± 6%, P ≤ 0.03; vs. LCAR plus gentamicin plus JNK-I = 244 ± 6%, P = not significant; vs. LCAR plus gentamicin plus ERK-I = 112 ± 8, P ≤ 0.05; Fig. 4a). Consistently, inhibition of JNK completely reversed the preventive effect of LCAR on gentamicin-induced up-regulation of Hrk (Fig. 4 a and b), and LCAR ameliorated the gentamicin-induced inactivation of JNK (Fig. 4c). These results suggest that, whereas gentamicin-induced up-regulation of Hrk in auditory cells is mediated by ERK1/2, the preventive effects of LCAR occur via JNK.
Altogether, these results demonstrate that different MAPKs play antagonistic roles in gentamicin cell toxicity, setting an important baseline for further defining the effect of LCAR on these important pathways. They are also consistent with reports in the literature identifying MAPKs as important mediators in the apoptotic pathways activated by ototoxic drugs such as neomycin and cisplatin (19-22). However, it is essential to note that, although neomycin, like gentamicin, is an aminoglycoside antibiotic, these drugs induce different cellular and biochemical responses. Noteworthy, for instance, whereas neomycin is mainly cochleotoxic, gentamicin is considered more a vestibulotoxic agent. Ylikoski et al. (23) have previously suggested that JNKs, not ERK, could be mediating in the gentamicin-induced death of inner hair cells of the cochlea and type I hair cells of the vestibular organ in guinea pigs. However, as recognized by the authors, the cochlear damage induced by gentamicin in their study was so extensive as to prevent the actual documentation of JNK activation. Similarly, the proportion of hair cell death associated with necrosis (versus apoptosis) was unknown. Therefore, the conditions reported in the current study have allowed a more defined determination of the role of JNK in gentamicin-induced apoptosis and provide a reliable reference for future studies aimed at evaluating the efficacy of novel compounds that manipulate this pathway.
The findings that Hrk is expressed in auditory cells and the functional characterization of this molecule as a mediator of gentamicin-induced apoptosis, as reported here, are significantly important for better understanding the molecular repertoire that is involved in regulating cell death in the inner ear. The fine details of the mechanisms underlying the proapoptotic effects of Hrk are poorly understood. The dominant theory is that Hrk would inhibit the antiapoptotic function of other Bcl-2 family member by heterodimerizing with them. However, a homotetrameric protein, p32, was recently isolated in a two-hybrid screen by its ability to interact with Hrk (24). Hrk-mediated apoptosis requires tetrameric p32 to form a channel in the mitochondria membrane and destabilize the function of this organelle. Thus, the emerging picture describing the functional mechanisms of Hrk suggests that several protein-protein interactions are necessary for its effects. Thus, it is likely that these types of interactions also underlay the effects of Hrk in gentamicin-induced apoptosis. Future studies focused on characterizing these signaling pathways downstream of Hrk in auditory cells could provide additional therapeutic targets to prevent iatrogenic hearing loss.
In summary, we have used a combination of both animal and cellular models to further investigate the molecular mechanisms underlying gentamicin-induced ototoxicity and to define an effective chemopreventive strategy for this phenomenon. We presented evidence that supplementation of pregnant mothers with LCAR prevents neonatal mortality and sensorineural hearing loss induced by gentamicin in newborn guinea pigs. Our experiments with auditory cells outline a more detailed pathway for gentamicin cell toxicity mediated by activation of the ERK1/2 and inhibition of the JNK pathways, followed by the translocation of ERK to the nucleus, transcriptional up-regulation of Hrk, and initiation of the execution phases of apoptosis. LCAR, on the other hand, would be preventing the gentamicin-induced inhibition of JNK and the consequent up-regulation of Hrk, blocking cell death. Thus, l-carnitine, a natural neuroprotective agent that can be safely used in humans (25), could be central for developing clinical strategies to prevent gentamicin-induced hearing loss.
Supplementary Material
Acknowledgments
We thank Dr. P. Webster and Ms. Siva Wu for helping us with SEM preparations. This publication was made possible by National Institutes of Health Grants DC05335, DC05220, DK52913, and P50-CA102701 and by the support of the House Ear Institute and the S. Mark Taper Foundation.
Author contributions: G.M.K., M.E.F.-Z., S.C., and F.K. performed research; G.M.K., M.E.F.-Z., R.U., and F.K. analyzed data; R.U. contributed new reagents/analytic tools; N.E.-C. and F.K. designed research; and R.U. and F.K. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: LCAR, l-carnitine; Hrk, Harakiri; ABR, auditory brainstem response; SEM, scanning electron microscopy; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; siRNA, small interfering RNA; OHC, outer hair cell; MAPK, mitogen-activated protein kinase; p, phosphorylated; I, inhibitor.
References
- 1.Chambers, H. F. & Sande, M. A. (1996) in Goodman & Gilman's The Pharmacological Basis of Therapeutics, eds. Hardman, J. G., Limbird, L. E., Molinoff, P. B., Ruddon, R. W. & Gilman, A. G. (McGraw-Hill, New York), pp. 1103-1121.
- 2.Romero, R., Espinoza, J., Chaiworapongsa, T. & Kalache, K. (2002) Semin. Neonatol. 7, 259-274. [DOI] [PubMed] [Google Scholar]
- 3.Henley, C. M. & Rybak, L. P. (1995) Brain Res. Brain Res. Rev. 20, 68-90. [DOI] [PubMed] [Google Scholar]
- 4.Priuska, E. M. & Schacht, J. (1995) Biochem. Pharmacol. 50, 1749-1752. [DOI] [PubMed] [Google Scholar]
- 5.Forge, A. & Schacht, J. (2000) Audiol. Neurootol. 5, 3-22. [DOI] [PubMed] [Google Scholar]
- 6.Dehne, N., Rauen, U., de Groot, H. & Lautermann, J. (2002) Hear. Res. 169, 47-55. [DOI] [PubMed] [Google Scholar]
- 7.Fischel-Ghodsian, N. (2003) Ear Hear. 24, 303-313. [DOI] [PubMed] [Google Scholar]
- 8.Shigenaga, M. K., Hagen, T. M. & Ames, B. N. (1994) Proc. Natl. Acad. Sci. USA 91, 10771-10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seidman, M., Khan, M., Bain, U., Shirwany, N. & Quirk, W. (2000) Am. J. Otol. 21, 161-167. [DOI] [PubMed] [Google Scholar]
- 10.Willot, J. F., Chisolm, T. H. & Lister, J. J. (2001) Audiol. Neurootol. 6, 231-249. [DOI] [PubMed] [Google Scholar]
- 11.Jongebloed, W. L., Stokroos, I., Van Der Want, J. J. L. & Kalichara, D. (1999) J. Microsc. (Oxford) 193, 158-170. [DOI] [PubMed] [Google Scholar]
- 12.Inohara, N., Ding, L., Chen, S. & Nunez, G. (1997) EMBO J. 16, 1686-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalinec, G. M., Webster, P., Lim, D. J. & Kalinec, F. (2003) Audiol. Neurootol. 8, 177-189. [DOI] [PubMed] [Google Scholar]
- 14.Cory, S. & Adams, J. M. (2002) Nat. Rev. Cancer 2, 647-656. [DOI] [PubMed] [Google Scholar]
- 15.Borner, C. (2003) Mol. Immunol. 39, 615-647. [DOI] [PubMed] [Google Scholar]
- 16.Imaizumi, K., Benito, A., Kiryu-Seo, S., Gonzalez, V., Inohara, N., Leiberman, A. P., Kiyama, H. & Nunez, G. (2004) J. Neurosci. 24, 3721-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris, C. A. & Johnson, E. M., Jr. (2001) J. Biol. Chem. 276, 37754-37760. [DOI] [PubMed] [Google Scholar]
- 18.Colucci-D'Amato, L., Perrone-Capano, C. & di Porzio, U. (2003) BioEssays 25, 1085-1095. [DOI] [PubMed] [Google Scholar]
- 19.Pirvola, U., Xing-Qun, L., Virkkala, J., Saarma, M., Murakata, C., Camoratto, A. M., Walton, K. M. & Ylikoski, J. (2000) J. Neurosci. 20, 43-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang, J., Van De Water, T. R., Bonny, C., de Ribaupierre, F., Puel, J. L. & Zine, A. (2003) J. Neurosci. 23, 8596-8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsui, J. I., Gale, J. E. & Warchol, M. E. (2004) J. Neurobiol. 61, 250-266. [DOI] [PubMed] [Google Scholar]
- 22.Wang, X., Martindale, J. L. & Holbrook, N. J. (2000) J. Biol. Chem. 275, 39435-39443. [DOI] [PubMed] [Google Scholar]
- 23.Ylikoski, J., Xing-Qun, L., Virkkala, J. & Pirvola, U. (2002) Hear. Res. 166, 33-43. [DOI] [PubMed] [Google Scholar]
- 24.Sunayama, J., Ando, Y., Itoh, N., Tomiyama, A., Sakurada, K., Sugiyama, A., Kang, D., Tashiro, F., Gotoh, Y., Kuchino, Y. & Kitanaka, C. (2004) Cell Death Differ. 11, 771-781. [DOI] [PubMed] [Google Scholar]
- 25.Pons, R. & De Vivo, D. C. (1995) J. Child Neurol. 10, Suppl. 2, S8-S24. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}