

Abstract
Bacterial ribonuclease P (RNase P) belongs to a class of enzymes that utilize both RNAs and proteins to perform essential cellular functions. The bacterial RNase P protein is required to activate bacterial RNase P RNA in vivo, but previous studies have yielded contradictory conclusions regarding its specific functions. Here, we use biochemical and biophysical techniques to examine all of the proposed functions of the protein in both Escherichia coli and Bacillus subtilis RNase P. We demonstrate that the E. coli protein, but not the B. subtilis protein, stabilizes the global structure of RNase P RNA, although both proteins influence holoenzyme dimer formation and precursor tRNA recognition to different extents. By comparing each protein in complex with its cognate and noncognate RNA, we show that differences between the two types of holoenzymes reside primarily in the RNA and not the protein components of each. Our results reconcile previous contradictory conclusions regarding the role of the protein and support a model where the protein activates local RNA structures that manifest multiple holoenzyme properties.
Keywords: RNA, structural biology
Introduction
Ribonucleoprotein complexes (RNPs) are ubiquitous and essential components of all cells. The compositions of RNPs vary from one RNA and one protein (e.g. the bacterial ribonuclease P (RNase P) and the bacterial signal recognition particle) to multiple RNAs and many (>50) proteins (e.g. the ribosome and the splicesome). Since the discovery of RNA-based catalysis (Kruger et al, 1982; Guerrier-Takada et al, 1983), RNA function in RNPs has been the subject of substantial research. In the case of the ribosome, for example, biochemical and structural studies have yielded sophisticated insight into the mechanism of protein synthesis catalyzed by RNA (Noller and Chaires, 1972; Nissen et al, 2000). However, the same wealth of structural information has revealed little about the functions of most ribosomal proteins, which are considered at a general level to stabilize the global architecture of RNA (Brodersen et al, 2002; Klein et al, 2004).
Like the ribosome, bacterial RNase P is an RNA-based enzyme (ribozyme) that requires a protein component in vivo and performs an essential multiple-turnover reaction as part of the RNA processing machinery in all three domains of life. RNase P catalyzes a phosphodiester cleavage that results in the 5′ maturation of tRNA, one of several post-transcriptional modifications necessary for the functional synthesis of tRNA. Bacterial RNase P RNA (∼130 kDa) can catalyze this reaction without the RNase P protein (∼14 kDa) under conditions of elevated ionic strength in vitro, but requires the protein under physiological conditions (Guerrier-Takada et al, 1983; Reich et al, 1988). Bacterial RNase P, therefore, provides a simple model system in which to elucidate the function of the protein in this RNP. Towards this aim, considerable study has focused on RNase P proteins from Escherichia coli and Bacillus subtilis, two phylogenetically distinct model bacteria. Although specific functions have been proposed for the bacterial RNase P protein, a general understanding of its role has been muddled by inconsistencies in the literature and a void of high-resolution structural information.
Previous studies with E. coli and B. subtilis RNase P support three different functions of the protein. First, based on studies with the E. coli holoenzyme the protein has been suggested to stabilize the active tertiary structure of the RNA (Guerrier-Takada et al, 1983; Westhof et al, 1996; Kim et al, 1997). Second, experiments with the B. subtilis holoenzyme have shown that the protein mediates holoenzyme dimer formation (Fang et al, 2001). Third, studies with the B. subtilis protein have indicated that the protein enhances the specificity of RNase P for precursor tRNA (pre-tRNA) compared to 5′ matured tRNA (mat-tRNA) (Crary et al, 1998; Kurz et al, 1998; Niranjanakumari et al, 1998). In order to build a global model for the role of the protein in E. coli and B. subtilis RNase P, we use a systematic comparative analysis to examine all three previously proposed functions in these two enzymes.
One rationale for a side-by-side comparison of the protein function in these particular RNPs stems from the occurrence of substantial differences in the secondary structures of the E. coli and B. subtilis RNase P RNAs. These RNAs are classified into ‘A' (Ancestral) and ‘B' (Bacillus) types, respectively, due to the presence or absence of certain structural elements in an otherwise conserved core (Brown and Pace, 1992; Haas et al, 1996). On the other hand, the RNase P proteins from organisms with A-type RNA (Thermotoga maritima) and B-type RNAs (B. subtilis and Staphylococcus aureus) show remarkable structural conservation (Stams et al, 1998; Spitzfaden et al, 2000; Kazantsev et al, 2003). Any functional implication of variable RNA structure and conserved protein structure in the holoenzyme is not known.
In this comparison of E. coli and B. subtilis RNase P, we show that the protein activates intrinsic RNA properties (presumably local structures) that dictate holoenzyme functions. A key finding is that E. coli and B. subtilis holoenzymes have somewhat different functional properties in vitro, which have contributed to conflicting results regarding the role of the protein in these holoenzymes (referenced throughout text). By examination of heterologous holoenzymes, E. coli RNA with B. subtilis protein and visa versa, we distinguish between the contributions of the RNA and protein components in the holoenzymes. The comparison illustrates the dominant role and versatile nature of RNA in this enzyme and provides important groundwork for future studies of more complex RNPs, such as eucaryal RNase P.
Results
Folding of RNase P RNA in the absence and presence of protein
The global tertiary structure of RNase P RNA is required for its function and possibly is influenced by the protein. Previous work by Tao Pan and colleagues identified three thermodynamic species in the folding pathway of B. subtilis RNase P RNA: unfolded (U), intermediate (I) and native (N) (Pan and Sosnick, 1997; Fang et al, 1999). The I state was shown to be uniform under a variety of conditions and was therefore postulated to represent a well-defined thermodynamic state rather than an ensemble of conformations (Pan and Sosnick, 1997). According to circular dichroism (CD) and small-angle X-ray scattering (SAXS), the transition from the U state to the I state involves substantial secondary structure formation and results in a significant compaction of the RNA (Fang et al, 2000). The transition from the I state to the N state involves tertiary structure formation and requires the cooperative binding of at least three Mg2+ ions; this results in a subtle compaction of the structure (Fang et al, 1999, 2000). In the current study, we examine the effect of the protein on the stability of the tertiary structure of RNase P RNA, which is related to the free energy difference between the native state, N, and the thermodynamic reference state, I.
Gradient-denaturing polyacrylamide gel electrophoresis (PAGE) was used to probe the tertiary folding transition of E. coli and B. subtilis RNAs in the absence or presence of protein. The transition that we observe is labeled I↔N since the folding parameters are identical to those previously reported for the B. subtilis catalytic domain RNA (see below) (Fang et al, 1999) and we confirm that the transition does not involve substantial secondary structure formation (CD data not shown). In these assays, the fractions of I and N can be quantified because the two states partition according to differences in their shapes (LeCuyer and Crothers, 1993; Pan and Woodson, 1998). Stabilization of the RNA structure by the protein is expected to reduce the magnesium dependence of the native state, to increase its resistance to denaturation and to increase the melting temperature of the N → I transition.
Based on a cooperative binding model, a plot of the fraction of folded (N) RNA as a function of Mg2+ yields the Hill coefficient (n), and the [Mg2+] at which the concentration of I=N, defined here as [Mg2+]1/2. Notably, the folding parameters are quite similar for the B. subtilis and E. coli RNAs (n=3.0 and 2.8; [Mg2+]1/2=0.22 and 0.25 mM, respectively; Figure 1A and B). The [Mg2+]1/2 is sensitive to both the monovalent ion concentration and temperature of the assay, but the values that we obtain are nearly identical to those reported previously for the B. subtilis catalytic domain with hydroxyl radical protection, CD and catalytic activity under similar buffer conditions at the same temperature (Fang et al, 1999). From the Hill coefficient and [Mg2+]1/2, the Mg2+-dependent free energy of the N state relative to the I state (ΔGIN) is calculated to be −1.4 kcal/mol for B. subtilis RNA and −1.1 kcal/mol for E. coli RNA at 0.5 mM Mg2+ and 10°C (Materials and methods).
Figure 1.
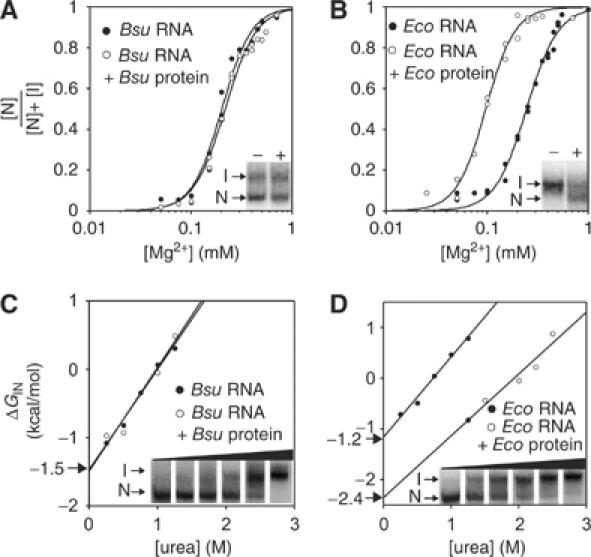
Protein stabilizes the global tertiary structure of the RNA in E. coli RNase P but not B. subtilis RNase P. (A) Fraction of folded B. subtilis (Bsu) RNA in the absence or presence of B. subtilis protein, determined with PAGE (1 × THE, pH 7.4, 4.5% acrylamide and 10°C) at varying concentrations of Mg2+. The data are fit to a cooperative binding model. (B) Same as (A), but with E. coli (Eco) RNA and protein. Insets show I and N folding states of the RNAs in the absence (−) and presence (+) of proteins in representative gels that contained 0.25 mM Mg2+ (A) and 0.10 mM Mg2+ (B). (C) Free energy difference between folding states of B. subtilis RNA in the absence and presence of B. subtilis protein, determined with PAGE (1 × THE, pH 7.4, 0.5 mM Mg2+, 4.5% acrylamide and 10°C) at varying concentrations of urea. The free energy difference between the I and N states is determined by ΔGIN=−RTln([N]/[I]). ΔGIN at zero urea is determined by linear extrapolation and is noted with an arrow. (D) Same as (C), but with E. coli RNA and protein. Insets show the partitioning of I and N folding states of the RNAs in the absence of proteins at urea concentrations between 0 and 1.5 M.
The Mg2+ dependence of the B. subtilis RNA folding transition does not change in the presence of B. subtilis protein (Figure 1A), consistent with a previous report (Loria et al, 1998). However, the E. coli protein decreases the Mg2+ dependence of the E. coli RNA folding transition and thereby increases the stability of the RNA structure in the holoenzyme (ΔGIN=−2.7 kcal/mol compared to −1.1 kcal/mol in the absence of protein at 0.5 mM Mg2+). Thus, in contrast to the B. subtilis protein, the stabilization effect of the E. coli protein appears substantial, as it more than doubles the free energy difference between the N and I states of E. coli RNA at physiological concentrations of Mg2+ (∼1.0 mM).
To validate the ΔGIN calculations obtained by the Mg2+ titration, we monitored stability of the folded RNA by urea denaturation, at constant 0.5 mM Mg2+. The ΔGIN values at zero urea were obtained from the plots shown in Figure 1C and D and are nearly identical to those obtained from the Mg2+ titrations for B. subtilis and E. coli RNAs in the absence of proteins (−1.5 and −1.2 kcal/mol, respectively). The surface area exposed in the N to I transition is related to the ‘m' value, which is calculated according to the urea dependence of unfolding (the slope of the lines in Figure 1C and D). The m values that we obtain are 1.5 and 1.6 kcal/mol M for the folding transitions of B. subtilis and E. coli RNAs, respectively. The m value for the transition of B. subtilis RNA is similar to the previously reported values obtained with CD and SAXS and is consistent with a minor portion of the RNA (<20%) being buried in the folding transition (Pan and Sosnick, 1997; Fang et al, 1999; Shelton et al, 1999; Fang et al, 2000).
The presence of the B. subtilis protein does not affect the stability of the B. subtilis RNA tertiary structure in this assay, consistent with the results of the Mg2+ titration (Figure 1A and C). In contrast, the E. coli protein stabilizes the native E. coli RNA against urea denaturation (Figure 1D); however, its contribution to RNA stability is slightly smaller when calculated by the titration with urea compared to Mg2+ (ΔΔGIN=−1.2 kcal, compared to −1.6 kcal/mol, respectively). These differences are in the range of the calculated errors of the assays (<20%), but they could also reflect inaccurate assumptions employed in the ΔGIN calculations from either the urea or Mg2+ titrations. For instance, the structure of the protein and thereby the RNA–protein interactions could be influenced by urea, or the protein could differentially affect nonspecific associations of Mg2+ in the I and the N states that are not taken into consideration in the ΔGIN calculations based on the Mg2+ titration. Nonetheless, according to either assay, the E. coli protein appears to stabilize the global tertiary structure of the RNA, but the B. subtilis protein does not do so.
Additional evidence for the stabilization of E. coli RNA by E. coli protein is provided by temperature-gradient gel electrophoresis, as shown in Figure 2. The temperature at which [N]=[I] is defined as the melting temperature (TM(IN)) in this assay; this parameter is distinct from the TM of RNA that typically refers to the global melting of secondary structure, which occurs at higher temperatures (>60°C; data not shown). As shown in Figure 2, the TM(IN) values of B. subtilis, Bacillus stearothermophilus, and E. coli RNAs (35°C, 50°C and 33°C, respectively) were determined from the change in RNA mobility as a function of temperature (Szewczak et al, 1998).
Figure 2.

The E. coli protein increases the melting temperature of E. coli RNA. (A) Temperature-gradient gel electrophoresis (1 × THE, pH 7.4, 4.5% acrylamide and either 0.65 or 1.0 M Mg2+) with RNAs in the absence of proteins. The melting temperatures (TM(IN)) of E. coli (Eco), B. subtilis (Bsu) and B. stearothermophilus (Bst) RNase P RNAs are indicated. (B) E. coli RNA in the absence or presence of either E. coli or B. subtilis protein, at 0.65 mM Mg2+. The difference in the melting temperature (ΔTM(IN)) of E. coli RNA upon addition of the E. coli protein is indicated. (C) Sypro ruby stain of the same gel shown in (B) indicates that both E. coli and B. subtilis proteins are bound to the E. coli RNA in this assay.
The melting temperatures of the RNAs depend on the concentration of Mg2+ in the gels, evidenced by the ∼5°C decrease in the TM(IN) of E. coli RNA at 0.65 mM Mg2+ compared to 1.0 mM Mg2+ (Figure 2A and B). Consistent with the previous assays, the TM(IN) of the E. coli RNA increases in the presence of the E. coli protein (ΔTM(IN)∼14–17°C at <1.0 mM Mg2+), whereas the B. subtilis protein does not affect the melting temperature of either RNA (Figure 2B and data not shown). Specific staining of the proteins with Sypro ruby (Molecular Probes) confirms that E. coli and B. subtilis proteins are bound to the I and N states in this assay (Figure 2C) and the other assays shown above.
Holoenzyme dimer formation
The B. subtilis protein has been reported to induce dimerization of B. subtilis monomers (Fang et al, 2001), but the oligomerization state of the E. coli holoenzyme is not known. Here, we used native PAGE and light scattering to characterize oligomer formation of E. coli and B. subtilis RNase P holoenzymes. Under the native PAGE conditions employed in the folding assays (<1.0 mM Mg2+), both B. subtilis and E. coli holoenzymes migrate at one constant mobility over the concentration range of 1–500 nM holoenzyme. However, under conditions that are typically employed for the substrate binding assays (10 mM Mg2+ and 100 mM NH4+, with either Cl− or OAc− as counterions), we identified at least two forms of E. coli and B. subtilis holoenzymes with native PAGE (Figure 3A). The relative fractions of each form depend on the concentration of the holoenzyme and are consistent with a monomer–dimer equilibrium (Figure 3A). One previous study reported a dimerization constant (KD) of ∼50 nM for the B. subtilis holoenzyme, according to SAXS measurements under similar conditions (Barrera et al, 2002). Based on our native PAGE assay, the KD of the B. subtilis dimer is ∼50–100 nM, and the KD of the E. coli dimer, which previously has not been detected, is ∼500–1000 nM (Figure 3A). An additional higher-order aggregate of the E. coli holoenzyme is sometimes detected (Figure 3A), but this represents <5% of the total concentration of the holoenzyme and could be due to a slight excess of protein in the assay. The binding affinities of the dimers are 100- to 1000-fold weaker than the binding affinities of the proteins for the RNAs within the monomers (Talbot and Altman, 1994).
Figure 3.
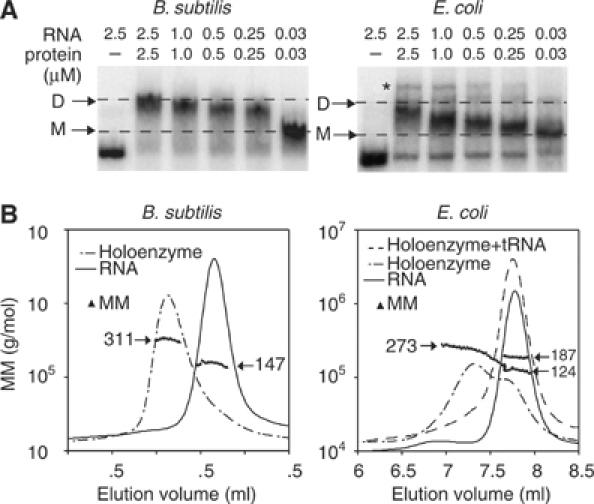
Native gel and light scattering analysis reveals dimerization of both B. subtilis and E. coli holoenzymes. (A) The gel mobility of the RNase P holoenzymes at concentrations between 2.5 and 0.03 μM; dashed lines indicate the migration of dimers (D) and monomers (M) in gels that contain 1 × THE, pH 7.4, 4.5% acrylamide, 100 mM NH4OAc and 10 mM MgCl2. Migration of RNA unbound by protein is shown in the first lane. Higher-order aggregation of the E. coli holoenzyme is noted by ‘*'. (B) Molecular mass (MM) distribution of RNase P RNA, holoenzyme, or holoenzyme+mat-tRNAGlu determined by light scattering under the same buffer conditions as the native gels (the concentration of RNase P in the elution peaks is between 200 and 500 nM). The MM values are plotted at each elution volume with an overlay of the chromatographic peaks from the refractive index values at these elution volumes. Masses are plotted in grams, but indicated in terms of kDa.
In order to substantiate the native PAGE results, we measured the molecular masses of E. coli and B. subtilis holoenzymes by light scattering as the molecules eluted from a size exclusion column, at concentrations between 200 and 500 nM. In the absence of protein, the measured mass of the B. subtilis RNA was ∼147 kDa (Figure 3B), within 10% of its calculated mass. With the addition of equimolar B. subtilis protein, the mass of the B. subtilis holoenzyme was ∼311 kDa (Figure 3B), within 5% of the calculated mass of a dimer containing two RNA and two protein subunits. The precision of the light scattering assay does not enable us to deduce the exact number of proteins bound in the complex. However, considering the equimolar input of RNA and protein in these assays and the large difference in the masses of RNA (∼140 kDa) and protein (∼14 kDa), our results are consistent with a 2 RNA:2 protein B. subtilis dimer, as reported previously (Fang et al, 2001). The E. coli holoenzyme, in contrast, elutes from the size exclusion column as a heterogeneous population (Figure 3B). However, the first third of the molecules that elute from the column have a measured mass of ∼273 kDa, within ∼5% of the calculated mass of a 2 RNA:2 protein E. coli dimer. Indeed, the elution profile and molecular mass distribution suggest an exchange between dimers and monomers or RNA unbound to protein on the time scale (∼15 min) of this assay (Figure 3B). These results are consistent with the native PAGE results, where the B. subtilis holoenzyme occurs predominantly as a dimer, but the E. coli holoenzyme exists as a heterologous mixture at concentrations between 200 and 500 nM (Figure 3A). Although the E. coli holoenzyme has a weaker dimerization constant than the B. subtilis holoenzyme (Figure 3A), the molecular mass distribution suggests that both E. coli and B. subtilis dimers represent distinct forms rather than a combination of aggregates since there is no evidence for additional high-molecular-weight species (Figure 3B).
Rough estimates indicate that the RNase P concentration in cells is ∼20–50 nM (Reich et al, 1986), which is comparable to the KD of the B. subtilis dimer. However, these in vitro assays may lack cofactors or conditions that affect the monomer–dimer equilibrium in vivo (Barrera et al, 2002). To determine if the dimers bind mat-tRNA, we monitored the mobility of the holoenzymes in the absence and presence of radiolabeled mat-tRNA under the native PAGE conditions where both E. coli and B. subtilis dimers were detected (1 μM holoenzyme). We used both B. subtilis mat-tRNAAsp and T. maritima mat-tRNAGlu to confirm that our results did not depend on the identity of the tRNA (both tRNAs are cleaved with comparable efficiency by E. coli and B. subtilis RNase P; data not shown). The B. subtilis holoenzyme dimer does not bind to either version of mat-tRNA under these conditions (Figure 4; data shown is for mat-tRNAGlu). In contrast, in the presence of mat-tRNA, the mobility of the E. coli holoenzyme shifts to a faster-migrating form in which radiolabeled mat-tRNA is present (Figure 4). We therefore presume that the E. coli dimer reduces to a monomer in the presence of mat-tRNA; this was corroborated with light scattering, where the measured mass of the E. coli holoenzyme upon addition of mat-tRNA is 187 kDa, within 10% of the calculated mass of the holoenzyme monomer bound to mat-tRNA (Figure 3B).
Figure 4.
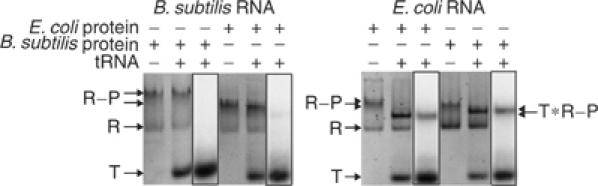
The E. coli holoenzyme, but not the B. subtilis holoenzyme, reduces to a monomer in the presence of mat-tRNA. Native gel analysis of homologous and heterologous holoenzymes (1 μM) in the absence and presence of 32P-labeled mat-tRNAGlu (2 μM) at 1 × THE, pH 7.4, 4.5% acrylamide, 100 mM NH4OAc and 10 mM MgCl2. Holoenzymes were stained with ethidium bromide and visualized with a trans-UV illuminator (Materials and methods); 32P-labeled mat-tRNAGlu was detected in the same gel with a phosphorimager (boxed lane). ‘T'=unbound tRNA, ‘R'=RNA, ‘R–P'=holoenzyme, ‘T * R − P '=holoenzyme bound to tRNA. Results are the same as observed for mat-tRNAAsp (data not shown).
Thus, the E. coli holoenzyme has a weaker dimerization constant than the B. subtilis holoenzyme and readily reduces to a monomer in the presence of mat-tRNA. To determine if these dimerization differences are attributable to differences in the RNAs or proteins between E. coli and B. subtilis, we examined the heterologous holoenzymes. The E. coli RNA, with either protein, reduces to a monomer and binds mat-tRNA (Figure 4). On the other hand, the mobility of B. subtilis RNA, with either protein, does not significantly change in the presence of mat-tRNA; only a faint fraction of the holoenzyme with the B. subtilis RNA and E. coli protein appears to bind mat-tRNA (Figure 4). Clearly, therefore, the differences in the dimerization properties of E. coli and B. subtilis holoenzymes are attributable primarily to the RNAs rather than the proteins, although both components could be a part of the dimerization interface (see Discussion).
Substrate recognition
RNase P RNA is a ribozyme but efficient binding of substrate (pre-tRNA) requires the presence of the protein under low ionic strength conditions (Reich et al, 1988; Kurz et al, 1998). The mechanism of this protein effect is not clear. It was reported that the B. subtilis protein increases the affinity of RNase P for pre-tRNA compared to mat-tRNA by making direct contacts with the 5′ precursor sequence (Crary et al, 1998; Kurz et al, 1998; Niranjanakumari et al, 1998). It was proposed, therefore, that the protein functions in vivo to prevent product inhibition in the cell (Hsieh et al, 2004). However, a different study, with the E. coli holoenzyme, showed that product (mat-tRNA) in fact is an efficient competitive inhibitor of catalysis (Tallsjö and Kirsebom, 1993).
Both E. coli and B. subtilis proteins activate their cognate and noncognate RNAs under low ionic strength conditions (Figure 5A), as demonstrated previously (Guerrier-Takada et al, 1983). Consistent with previous conflicting reports (referenced above), mat-tRNA appears to inhibit catalysis by the E. coli holoenzyme more so than it does the B. subtilis holoenzyme (Figure 5A). This difference in the inhibition of E. coli and B. subtilis holoenzymes by mat-tRNA appears to be inherent to the RNA rather than the protein component of the holoenzyme (Figure 5A). To examine this difference, we directly measured the binding affinities of pre-tRNA and mat-tRNA for both E. coli and B. subtilis RNase P RNAs in the absence and presence of proteins. Mg2+ was replaced by Ca2+ to ensure that the pre-tRNA was not cleaved during the binding assay (Smith et al, 1992). The results are shown in Figure 5B and are summarized in Figure 5C.
Figure 5.
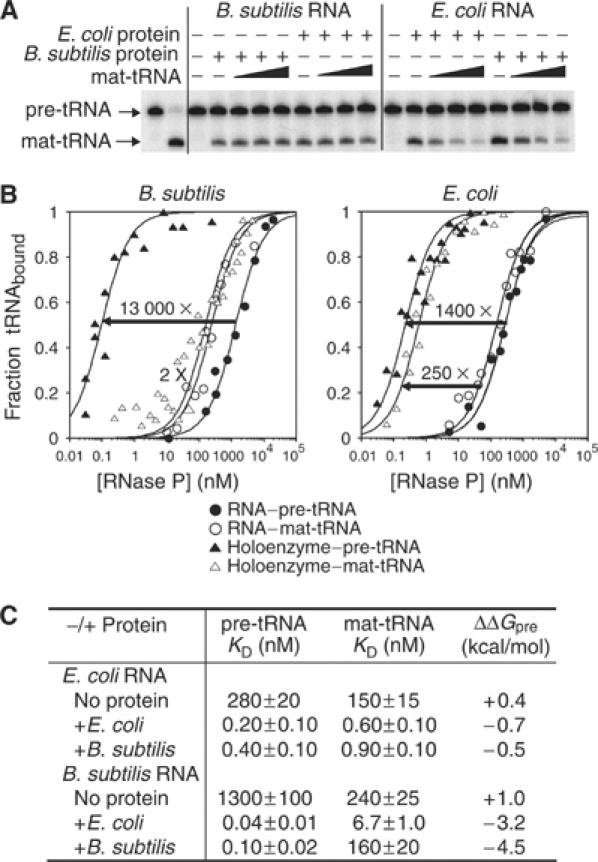
B. subtilis RNase P displays enhanced binding affinity for pre-tRNA compared to mat-tRNA that is not matched by E. coli RNase P. (A) Cleavage of 32P-labeled pre-tRNAAsp (100 nM) by RNase P RNA (100 nM) in the presence of equimolar cognate or noncognate protein for 8 s at 37°C in 50 mM MES, pH 6.0, 100 mM NH4Cl, 10 mM MgCl2 and 0.05% NP40. The first lane shows pre-tRNA at time zero and the second lane shows the end point of the reaction. Increasing concentrations of unlabeled mat-tRNAAsp (100, 500 nM or 2.5 μM) were included in the reactions as indicated. (B) Fractions of pre-tRNAAsp and mat-tRNAAsp bound to RNAs in the absence or presence of proteins, based on the gel filtration assay under the same buffer conditions as (A), but MgCl2 was replaced with CaCl2. The arrows indicate the increase in binding affinity for pre-tRNAAsp and mat-tRNAAsp when the protein is present. (C) Summary of the binding affinities of pre-tRNAAsp and mat-tRNAAsp for RNase P RNAs and holoenzymes as obtained in (B). ΔΔGpre=−RTln(KD(mat-tRNA)/KD(pre-tRNA)).
In the absence of protein, pre-tRNA binds to B. subtilis RNA marginally better (∼7-fold) than mat-tRNA. Addition of the B. subtilis protein dramatically increases the binding affinity of B. subtilis RNA for pre-tRNA (∼13 000-fold), with relatively little effect on the binding affinity for mat-tRNA (Figure 5B), consistent with a previous report (Kurz et al, 1998). In contrast, in the presence of the E. coli protein, the binding affinities of pre-tRNA and mat-tRNA for E. coli RNA are both substantially increased (1400- and 250-fold, respectively; Figure 5B). Therefore, presence of the precursor sequence confers substantial binding energy to the substrate interaction with the B. subtilis holoenzyme (ΔΔGpre=−4.5 kcal/mol), but makes only a subtle energetic contribution to the substrate interaction with the E. coli holoenzyme (ΔΔGpre=−0.7 kcal/mol). The difference in precursor discrimination between the B. subtilis and E. coli holoenzymes is not idiosyncratic to the precursor sequence or tRNAAsp used in these assays; the same trend is observed with T. maritima tRNAGlu (data not shown). The holoenzymes exist in the monomer form in these assays, according to their concentrations, with the possible exception of the B. subtilis holoenzyme in the presence of mat-tRNA. The presence of the B. subtilis dimer in the binding assay could decrease the apparent affinity of the holoenzyme for mat-tRNA since only the monomer is expected to bind to mat-tRNA (Figure 4). It is possible, therefore, that holoenzyme dimerization influences the observed substrate specificity of B. subtilis RNase P (discussed below).
A previous crosslinking study suggested that the B. subtilis protein may make direct contacts with the precursor sequence of pre-tRNA (Niranjanakumari et al, 1998). One interpretation of our data would be that the E. coli protein lacks these proposed contact sites. In order to address this, we measured the binding affinities of pre-tRNA and mat-tRNA to heterologous holoenzymes. The holoenzyme composed of B. subtilis RNA and E. coli protein displays enhanced binding affinity for pre-tRNA compared to mat-tRNA (ΔΔGpre=−3.2 kcal/mol, Figure 5C). In contrast, the complement holoenzyme, E. coli RNA with B. subtilis protein, exhibits little difference in the binding affinity for pre-tRNA compared to mat-tRNA (ΔΔGpre=−0.5 kcal/mol, Table I). Thus, variability in precursor recognition between B. subtilis and E. coli holoenzymes is primarily attributable to differences between the RNAs. This could involve RNA–precursor interactions and/or RNA-induced protein–precursor interactions. We do observe a slight difference in the protein properties since the B. subtilis RNA binds with 20-fold higher affinity to mat-tRNA when it is paired with the E. coli protein compared to the B. subtilis protein (Figure 5C, consistent with Figure 4), although this could be related to dimerization of the B. subtilis holoenzyme in the binding assay. Nonetheless, the differential influences of the proteins are minor compared to the differential influences of the RNAs in the E. coli and B. subtilis holoenzymes.
Discussion
The primary role of the E. coli and B. subtilis RNase P proteins is activation of RNase P RNA for catalysis under conditions of low ionic strength, as occurs in vivo. However, the generalities of this role have been unclear and a variety of protein functions have been suggested, depending on the version of RNase P examined. We address this problem by examining the multiple steps in holoenzyme and enzyme–substrate assembly for both E. coli and B. subtilis RNase P. Our results indicate that the protein functions at a local level to influence intrinsic RNA properties that dictate substrate binding and dimerization, depicted as a cartoon in Figure 6. We speculate that these intrinsic RNA properties are specific local RNA structures that are stabilized by protein binding.
Figure 6.
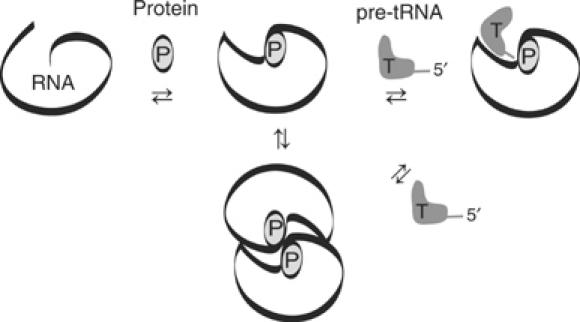
Cartoon depicting the manifestations of local RNA structure activation by RNase P protein.
A key finding in this study is that the E. coli and B. subtilis RNAs do not have identical functional properties, and their differences are manifest upon activation by the proteins. For example, the B. subtilis holoenzyme displays a substantial preference for pre-tRNA compared to mat-tRNA (−4.5 kcal/mol), which is not matched by the E. coli holoenzyme (−0.7 kcal/mol) (Figure 5B and C). Our results with the B. subtilis holoenzyme are nearly identical to those reported previously (Kurz et al, 1998). However, analysis of the heterologous holoenzymes shows that the preference for pre-tRNA over mat-tRNA is a property dictated primarily by the RNA (Figure 5C). These results are consistent with data that suggest RNA–precursor interactions, particularly with the first nucleotide 5′ to the cleavage site (LaGrandeur et al, 1994; Zuleeg et al, 2001; Brannvall et al, 2002; Zahler et al, 2003). The difference in pre-tRNA recognition that we observe between E. coli and B. subtilis RNase P RNAs is also supported by previous crosslinking and phosphorothioate-modification studies on the two RNAs (LaGrandeur et al, 1994; Warnecke et al, 1999).
We do not disregard the possibility that protein–precursor interactions may contribute to pre-tRNA recognition in the holoenzyme, as suggested previously (Kurz et al, 1998; Niranjanakumari et al, 1998). However, it is unlikely that protein–precursor interactions are a main mechanism by which the protein activates RNase P RNA, particularly since the effects of the protein do not appear to be conserved between the E. coli and B. subtilis holoenzymes (Figure 5C). On the other hand, it is possible that B. subtilis and E. coli RNAs coordinate the protein slightly differently, such that protein–precursor interactions are more favorable when the protein is bound to B. subtilis RNA compared to E. coli RNA. Further work is required to distinguish between RNA–precursor and protein–precursor contacts in the enzyme–substrate complex. Nonetheless, either directly or indirectly, the main responsibility for precursor recognition by the holoenzyme is assumed by the RNA (Figure 5C).
The domination of RNA is also evident in the dimerization properties of RNase P. Although both the E. coli and B. subtilis holoenzymes form dimers, only those composed of B. subtilis RNA are present at physiologically relevant concentrations. Indeed, those composed of the E. coli RNA with either protein have a ∼10-fold lower dimerization constant (Figure 3), and readily dissociate in the presence of mat-tRNA (Figure 4). In contrast, the B. subtilis holoenzyme remains as a dimer and does not bind mat-tRNA (Figure 4), even when mat-tRNA is in 50-fold excess (data not shown). However, at similar holoenzyme concentrations (2.4 μM compared to 1.0 μM in this study), a previous report showed that equimolar pre-tRNA caused dissociation of the B. subtilis dimer, under conditions (Ca2+) where the precursor sequence was not cleaved (Barrera et al, 2002). It is possible that some of the residues that confer precursor specificity in B. subtilis RNase P also influence dimerization, such that pre-tRNA competes more strongly than mat-tRNA with dimerization. However, the monomer is presumed to be the active species in the cell and the functional relevance of the dimer structure is not known. Like the substrate recognition properties of the holoenzymes, the dimerization properties are primarily dictated by the RNAs (Figure 4), but this does not rule out the possibility that RNA–protein or protein–protein contacts occur at the dimer interface.
We conclude that the mechanism of protein activation of RNase P RNA is stabilization of intrinsic RNA properties, which we presume to be local RNA structures (depicted in Figure 6). This activation can have multiple manifestations, to increase the affinity for pre-tRNA and mat-tRNA, to induce holoenzyme dimer formation and, in some cases, to provide additional stabilization to the global tertiary structure of the RNA. The influence of RNase P protein on the local RNA properties is distinct from the functions of chaperone or cofactor proteins, which are required to facilitate the global tertiary folding of RNA (Weeks, 1997). Although we provide direct evidence that the E. coli protein stabilizes the tertiary structure of E. coli RNA, this protein function does not appear to be conserved in the B. subtilis RNase P protein (Figures 1 and 2) and is not sufficient to explain the RNA-activation properties of the protein. Although the global structure and the activation properties of the RNase P protein are conserved in bacteria, there is only ∼30% sequence identity between the proteins from each bacterial kingdom, which leaves room for some diversity in protein function. Indeed, our folding results suggest that at least some properties differ between the E. coli and B. subtilis proteins (Figures 1 and 2). However, these differences could either be a reflection of in vivo differences between the proteins or they could be related to the in vitro conditions that we employ. Although both E. coli and B. subtilis holoenzymes display optimal activity under similar ionic conditions (data not shown), there could be subtle differences in their responses to the buffer conditions of each assay, which is one caveat of our in vitro results. It is possible, for example, that both the E. coli and B. subtilis holoenzymes form dimers in the cell, but different cofactors might stabilize the dimers in each organism. It is also possible that both proteins stabilize the global structures of the RNAs in vivo, even though we only detect this function with the E. coli protein in vitro. On this note, our report demonstrates that a global, inclusive understanding of RNase P is not possible without a comparison of multiple versions of the enzyme.
This study illustrates the complexity and idiosyncrasy of separating RNA and protein functions in bacterial RNase P, one of the simplest of RNPs. High-resolution structure models of the holoenzyme will be needed for a specific structural interpretation of the RNase P protein properties described here. However, these results provide a critical functional basis with which to interpret future models of this universally conserved enzyme.
Materials and methods
Purification of RNAs and proteins
B. subtilis RNase P RNA, E. coli RNase P RNA, B. subtilis pre-tRNAAsp and T. maritima pre-tRNAGlu were transcribed in vitro from linearized plasmids pDW152, pDW98, pDW128 and pTGlu as described previously (Milligan and Uhlenbeck, 1989; Chen et al, 1998). pTGlu was constructed by inserting the T. maritima tRNAGlu gene (Thermotoga_maritima.trna30 from the genomic tRNA database) into a pGEM vector, with a 24 nucleotide precursor sequence (5′-gggcgaauucggugcacgcuucau-3′) introduced by the 5′ primer. RNAs were internally labeled by the addition of [α-32P]GTP (Perkin-Elmer) to the transcription reactions or 5′-labeled with T4 polynucleotide kinase (Fisher) and [γ-32P]ATP (MP Biomedicals) following transcription. A fraction of transcribed pre-tRNA was incubated with excess B. subtilis RNase P to produce mat-tRNA. RNAs were purified as previously described, except that SDS was omitted from all buffers (Chen et al, 1998).
The E. coli and B. subtilis RNase P proteins were expressed from plasmids pECPE and pWT1, respectively. pECPE was constructed by insertion of the PCR-amplified rnpA gene from genomic E. coli DNA into vector pET-11a; pWT1 was a generous gift from Dr Carol Fierke. Both proteins were expressed and purified essentially as described for the T. maritima protein, except neither required a GST fusion (Krivenko et al, 2002). For the E. coli protein purification, 100 mM DTT was included in the lysis buffer and 1 mM DTT in all other buffers.
Gradient-denaturing gel electrophoresis assays
RNase P RNAs were folded prior to all experiments in 66 mM HEPES, 33 mM Tris, pH 7.4, 0.1 mM EDTA (1 × THE), 100 mM NH4OAc, 10 mM MgCl2 and 0.05% NP40 for 10 min at 50°C, followed by 20 min at 37°C. In the folding, dimerization and substrate binding assays, NH4Cl can be substituted for NH4OAc and the results are the same. For holoenzyme reconstitutions, the RNAs were incubated with equimolar protein for an additional 20 min at 37°C, followed by slow cooling to room temperature (RT). All samples were tested for homogeneity on native gels prior to the experiments. For the Mg2+ titration, samples were diluted to final concentrations of 25 mM NH4OAc, 2.5 mM MgCl2, 1 × THE, 0.01% NP40, 5% glycerol and 50 nM RNA or holoenzyme before they were loaded onto a series of gels (>20) that contained 4.5% acrylamide, 1 × THE and between 0.0 and 2.0 mM MgCl2. It was not necessary to dialyze the samples to the exact gel conditions because they equilibrated rapidly once in the gel. The concentration of Mg2+ noted in the figures represents the free [MgCl2], which assumes chelation of the Mg2+ by the EDTA present. At concentrations below 0.30 mM MgCl2, experiments were repeated in the absence of EDTA without this assumption and results were the same. Fractions of folded RNAs were plotted based on the cooperative binding model:
![]()
The magnesium-dependent free energy of the N state relative to the I state was calculated by
![]()
as described previously (Fang et al, 1999).
For the urea titrations, RNAs and holoenzymes were reconstituted as described above, but samples were incubated with the appropriate concentrations of urea for 20 min at RT prior to being loaded onto a series of gels (>15) that contained 1 × THE, 0.6 mM MgCl2 and 0–4 M urea. In order to ensure that the folding states were at equilibrium, the samples were also incubated for >2 days at the different urea concentrations prior to analysis and results were the same. The reversibility of the I↔N transition was confirmed by examining both the folding and unfolding pathways of the RNA (in the folding experiments, RNAs were prefolded at elevated temperatures as described above and then unfolded in urea and refolded by dialyzing out the urea). The ΔGIN values at each urea concentration were calculated by ΔGIN=−RTln([N]/[I]) and the ΔGIN at zero urea was obtained by linear extrapolation (Santoro and Bolen, 1992). All gels were run for 3.5 h at constant voltage (350 V) with an internal temperature of 10°C. The temperature-gradient gels were performed as described previously (Szewczak et al, 1998), using water baths at 4°C and 65°C to create the 11–59°C gradient in the gels. Samples were loaded 20 min apart onto gels that contained 4.5% acrylamide, 1 × THE and either 0.75 or 1.1 mM MgCl2. At least three replicate gels were used to calculate the melting temperatures, and the error between replicates was <2°C. Gels were stained with Sypro ruby (Molecular probes) and visualized with a trans UV-illuminator (Gel Logic) prior to drying. In all of the folding assays, the relative intensities of 32P-labeled RNA bands were visualized with a Phosphorimager (Molecular Dynamics), quantified with ImageQuant and plotted with KaleidaGraph software.
Native gel-shift and light scattering assays
In order to determine the concentration dependence of holoenzyme mobility (Figure 3A), unlabeled and internally 32P-labeled RNAs were mixed to achieve constant 32P counts/min at each concentration of RNA. The holoenzymes were then reconstituted as described above in 100 mM NH4OAc, 1 × THE, 10 mM MgCl2 and 0.05% NP-40. Glycerol was added to the samples (at a final concentration of 5%) and they were run on gels that contained 4.5% acrylamide and the same buffer conditions but without NP-40. To examine the tRNA-binding properties of the dimers (Figure 4), unlabeled RNase P RNAs were reconstituted with equimolar proteins to final concentrations of 1 μM, with and without 2 μM internally 32P-labeled mat-tRNAGlu or mat-tRNAAsp (results were the same for both tRNAs; data shown if for mat-tRNAGlu). The tRNAs were prefolded in the same buffer as the holoenzymes at 65°C for 2 min, prior to incubation with the holoenzymes for 20 min at RT. RNAs were stained with ethidium bromide and visualized with a trans UV-illuminator (Gel Logic), after which the gels were dried and 32P-labeled tRNA visualized with a Phosphorimager (Molecular Dynamics).
For the light scattering assays, holoenzymes (7 μM) were reconstituted as described above; the E. coli holoenzyme was also incubated with 14 μM mat-tRNAGlu. Samples were loaded (30 μl) onto a size exclusion column (Shodex KW803) that was coupled to instrumentation measuring light scattering at 690 nM (Wyatt Tech Corp.), absorption at 260 nM (SpectraSystem) and the refractive index (Wyatt Tech Corp.) of the molecules as they eluted. Buffer was the same as for the native gel assays with a flow rate of 1.0 ml/min. Data analysis was performed with the Astra software program using bovine serum albumin (dn/dc=0.185) to test the accuracy of the system. The dn/dc value 0.17 (Rambo and Doudna, 2004) was used for both RNAs and holoenzymes, since RNA made up >90% of the complexes by mass.
tRNA cleavage and binding assays
RNase P RNAs and holoenzymes were reconstituted as described above but in buffer that contained 100 mM NH4Cl, 50 mM MES, pH 6.0, 10 mM MgCl2 and 0.05% NP40 (these buffer conditions are the same as those used in the spin column assay, except for the identity of the divalent cation, see below). Internally 32P-labeled pre-tRNAAsp was prefolded and incubated (at a final concentration of 100 nM) with the RNAs or holoenzymes (at final concentrations of 100 nM) in the absence or presence of mat-tRNAAsp (at a final concentrations of 100 nM, 500 nM or 2.5 μM) for 8 s at 37°C. The reactions were quenched by the addition of 25 mM EDTA; pre-tRNA and mat-tRNA were separated with denaturing (7 M urea) PAGE (7% acrylamide) and visualized with a Phosphorimager (Molecular Dynamics). The binding affinities of pre- and mat versions of tRNAAsp and tRNAGlu for RNase P were determined using a G-75 sephadex spin column assay, as described elsewhere (Beebe and Fierke, 1994). RNAs and holoenzymes were reconstituted in buffer that contained 100 mM NH4Cl, 50 mM MES, pH 6.0, 10 mM CaCl2 and 0.05% NP40; we used these buffer conditions and show the data for tRNAAsp in order to compare our results to those reported previously with the B. subtilis holoenzyme (Kurz et al, 1998). The 5′-labeled pre-tRNA (0.01 nM) and mat-tRNA (0.1 nM) were mixed with the RNAs or holoenzymes (0–10 μM) for 30–180 min at 37°C prior to loading onto the gel matrix. Bound and unbound tRNA fractions at each enzyme concentration were separated as described (Beebe and Fierke, 1994) and quantified with scintillation counting. Dissociation constants were determined according to:
![]()
the end points for the total fraction of bound and unbound tRNA were determined in each experiment according to how much tRNA was eluted or retained in the absence of enzyme or at saturating enzyme concentrations. Aliquots of the binding reactions that contained pre-tRNA were tested on a denaturing gel to ensure that pre-tRNA was not cleaved during the assay.
Acknowledgments
We thank Dr Rob Batey and Dr Arthur Pardi for critical review of the manuscript. We also thank the labs of Dr Thomas Cech and Dr Marcelo Sousa for sharing the temperature-gradient gel electrophoresis and light scattering instruments. This research was supported by NIH Grant GM-34527 to NR Pace and NIH Molecular Biophysics Training Grant T32 GM-65103 to AH Buck through the University of Colorado at Boulder.
References
- Barrera A, Fang X, Jacob J, Casey E, Thiyagarajan P, Pan T (2002) Dimeric and monomeric Bacillus subtilis RNase P holoenzyme in the absence and presence of pre-tRNA substrates. Biochemistry 41: 12986–12994 [DOI] [PubMed] [Google Scholar]
- Beebe JA, Fierke CA (1994) A kinetic mechanism for cleavage of precursor tRNA(Asp) catalyzed by the RNA component of Bacillus subtilis ribonuclease P. Biochemistry 33: 10294–10304 [DOI] [PubMed] [Google Scholar]
- Brannvall M, Fredrik Pettersson BM, Kirsebom LA (2002) The residue immediately upstream of the RNase P cleavage site is a positive determinant. Biochimie 84: 693–703 [DOI] [PubMed] [Google Scholar]
- Brodersen DE, Clemons WM Jr, Carter AP, Wimberly BT, Ramakrishnan V (2002) Crystal structure of the 30 S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J Mol Biol 316: 725–768 [DOI] [PubMed] [Google Scholar]
- Brown JW, Pace NR (1992) Ribonuclease P RNA and protein subunits from bacteria. Nucleic Acids Res 20: 1451–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Nolan JM, Harris ME, Pace NR (1998) Comparative photocross-linking analysis of the tertiary structures of Escherichia coli and Bacillus subtilis RNase P RNAs. EMBO J 17: 1515–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary SM, Niranjanakumari S, Fierke CA (1998) The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry 37: 9409–9416 [DOI] [PubMed] [Google Scholar]
- Fang X, Littrell K, Yang XJ, Henderson SJ, Siefert S, Thiyagarajan P, Pan T, Sosnick TR (2000) Mg2+-dependent compaction and folding of yeast tRNAPhe and the catalytic domain of the B. subtilis RNase P RNA determined by small-angle X-ray scattering. Biochemistry 39: 11107–11113 [DOI] [PubMed] [Google Scholar]
- Fang X, Pan T, Sosnick TR (1999) A thermodynamic framework and cooperativity in the tertiary folding of a Mg2+-dependent ribozyme. Biochemistry 38: 16840–16846 [DOI] [PubMed] [Google Scholar]
- Fang XW, Yang XJ, Littrell K, Niranjanakumari S, Thiyagarajan P, Fierke CA, Sosnick TR, Pan T (2001) The Bacillus subtilis RNase P holoenzyme contains two RNase P RNA and two RNase P protein subunits. RNA 7: 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S (1983) The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857 [DOI] [PubMed] [Google Scholar]
- Haas ES, Banta AB, Harris JK, Pace NR, Brown JW (1996) Structure and evolution of ribonuclease P RNA in Gram-positive bacteria. Nucleic Acids Res 24: 4775–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Andrews AJ, Fierke CA (2004) Roles of protein subunits in RNA-protein complexes: lessons from ribonuclease P. Biopolymers 73: 79–89 [DOI] [PubMed] [Google Scholar]
- Kazantsev AV, Krivenko AA, Harrington DJ, Carter RJ, Holbrook SR, Adams PD, Pace NR (2003) High-resolution structure of RNase P protein from Thermotoga maritima. Proc Natl Acad Sci USA 100: 7497–7502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Kilani AF, Zhan X, Altman S, Liu F (1997) The protein cofactor allows the sequence of an RNase P ribozyme to diversify by maintaining the catalytically active structure of the enzyme. RNA 3: 613–623 [PMC free article] [PubMed] [Google Scholar]
- Klein DJ, Moore PB, Steitz TA (2004) The roles of ribosomal proteins in the structure assembly, and evolution of the large ribosomal subunit. J Mol Biol 340: 141–177 [DOI] [PubMed] [Google Scholar]
- Krivenko AA, Kazantsev AV, Adamidi C, Harrington DJ, Pace NR (2002) Expression, purification, crystallization and preliminary diffraction analysis of RNase P protein from Thermotoga maritima. Acta Crystallogr D 58: 1234–1236 [DOI] [PubMed] [Google Scholar]
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR (1982) Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 31: 147–157 [DOI] [PubMed] [Google Scholar]
- Kurz JC, Niranjanakumari S, Fierke CA (1998) Protein component of Bacillus subtilis RNase P specifically enhances the affinity for precursor-tRNAAsp. Biochemistry 37: 2393–2400 [DOI] [PubMed] [Google Scholar]
- LaGrandeur TE, Huttenhofer A, Noller HF, Pace NR (1994) Phylogenetic comparative chemical footprint analysis of the interaction between ribonuclease P RNA and tRNA. EMBO J 13: 3945–3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCuyer KA, Crothers DM (1993) The Leptomonas collosoma spliced leader RNA can switch between two alternate structural forms. Biochemistry 32: 5301–5311 [DOI] [PubMed] [Google Scholar]
- Loria A, Niranjanakumari S, Fierke CA, Pan T (1998) Recognition of a pre-tRNA substrate by the Bacillus subtilis RNase P holoenzyme. Biochemistry 37: 15466–15473 [DOI] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC (1989) Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62 [DOI] [PubMed] [Google Scholar]
- Niranjanakumari S, Stams T, Crary SM, Christianson DW, Fierke CA (1998) Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc Natl Acad Sci USA 95: 15212–15217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen P, Hansen J, Ban N, Moore PB, Steitz TA (2000) The structural basis of ribosome activity in peptide bond synthesis. Science 289: 920–930 [DOI] [PubMed] [Google Scholar]
- Noller HF, Chaires JB (1972) Functional modification of 16S ribosomal RNA by kethoxal. Proc Natl Acad Sci USA 69: 3115–3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, Woodson SA (1998) Folding intermediates of a self-splicing RNA: mispairing of the catalytic core. J Mol Biol 280: 597–609 [DOI] [PubMed] [Google Scholar]
- Pan T, Sosnick TR (1997) Intermediates and kinetic traps in the folding of a large ribozyme revealed by circular dichroism and UV absorbance spectroscopies and catalytic activity. Nat Struct Biol 4: 931–938 [DOI] [PubMed] [Google Scholar]
- Rambo RP, Doudna JA (2004) Assembly of an active group II intron–maturase complex by protein dimerization. Biochemistry 43: 6486–6497 [DOI] [PubMed] [Google Scholar]
- Reich C, Gardiner KJ, Olsen GJ, Pace B, Marsh TL, Pace NR (1986) The RNA component of the Bacillus subtilis RNase P. Sequence, activity, and partial secondary structure. J Biol Chem 261: 7888–7893 [PubMed] [Google Scholar]
- Reich C, Olsen GJ, Pace B, Pace NR (1988) Role of the protein moiety of ribonuclease P, a ribonucleoprotein enzyme. Science 239: 178–181 [DOI] [PubMed] [Google Scholar]
- Santoro MM, Bolen DW (1992) A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry 31: 4901–4907 [DOI] [PubMed] [Google Scholar]
- Shelton VM, Sosnick TR, Pan T (1999) Applicability of urea in the thermodynamic analysis of secondary and tertiary RNA folding. Biochemistry 38: 16831–16839 [DOI] [PubMed] [Google Scholar]
- Smith D, Burgin AB, Haas ES, Pace NR (1992) Influence of metal ions on the ribonuclease P reaction. Distinguishing substrate binding from catalysis. J Biol Chem 267: 2429–2436 [PubMed] [Google Scholar]
- Spitzfaden C, Nicholson N, Jones JJ, Guth S, Lehr R, Prescott CD, Hegg LA, Eggleston DS (2000) The structure of ribonuclease P protein from Staphylococcus aureus reveals a unique binding site for single-stranded RNA. J Mol Biol 295: 105–115 [DOI] [PubMed] [Google Scholar]
- Stams T, Niranjanakumari S, Fierke CA, Christianson DW (1998) Ribonuclease P protein structure: evolutionary origins in the translational apparatus. Science 280: 752–755 [DOI] [PubMed] [Google Scholar]
- Szewczak AA, Podell ER, Bevilacqua PC, Cech TR (1998) Thermodynamic stability of the P4–P6 domain RNA tertiary structure measured by temperature gradient gel electrophoresis. Biochemistry 37: 11162–11170 [DOI] [PubMed] [Google Scholar]
- Talbot SJ, Altman S (1994) Gel retardation analysis of the interaction between C5 protein and M1 RNA in the formation of the ribonuclease P holoenzyme from Escherichia coli. Biochemistry 33: 1399–1405 [DOI] [PubMed] [Google Scholar]
- Tallsjö A, Kirsebom LA (1993) Product release is a rate-limiting step during cleavage by the catalytic RNA subunit of Escherichia coli RNase P. Nucleic Acids Res 21: 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke JM, Held R, Busch S, Hartmann RK (1999) Role of metal ions in the hydrolysis reaction catalyzed by RNase P RNA from Bacillus subtilis. J Mol Biol 290: 433–445 [DOI] [PubMed] [Google Scholar]
- Weeks KM (1997) Protein-facilitated RNA folding. Curr Opin Struct Biol 7: 336–342 [DOI] [PubMed] [Google Scholar]
- Westhof E, Wesolowski D, Altman S (1996) Mapping in three dimensions of regions in a catalytic RNA protected from attack by an Fe(II)-EDTA reagent. J Mol Biol 258: 600–613 [DOI] [PubMed] [Google Scholar]
- Zahler NH, Christian EL, Harris ME (2003) Recognition of the 5′ leader of pre-tRNA substrates by the active site of ribonuclease P. RNA 9: 734–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuleeg T, Hansen A, Pfeiffer T, Schubel H, Kreutzer R, Hartmann RK, Limmer S (2001) Correlation between processing efficiency for ribonuclease P minimal substrates and conformation of the nucleotide −1 at the cleavage position. Biochemistry 40: 3363–3369 [DOI] [PubMed] [Google Scholar]