

Abstract
The p53 tumor suppressor is activated after DNA damage to maintain genomic stability and prevent transformation. Rapid activation of p53 by ionizing radiation is dependent on signaling by the ATM kinase. MDM2 and MDMX are important p53 regulators and logical targets for stress signals. We found that DNA damage induces ATM-dependent phosphorylation and degradation of MDMX. Phosphorylated MDMX is selectively bound and degraded by MDM2 preceding p53 accumulation and activation. Reduction of MDMX level by RNAi enhances p53 response to DNA damage. Loss of ATM prevents MDMX degradation and p53 stabilization after DNA damage. Phosphorylation of MDMX on S342, S367, and S403 were detected by mass spectrometric analysis, with the first two sites confirmed by phosphopeptide-specific antibodies. Mutation of MDMX on S342, S367, and S403 each confers partial resistance to MDM2-mediated ubiquitination and degradation. Phosphorylation of S342 and S367 in vivo require the Chk2 kinase. Chk2 also stimulates MDMX ubiquitination and degradation by MDM2. Therefore, the E3 ligase activity of MDM2 is redirected to MDMX after DNA damage and contributes to p53 activation.
Keywords: ATM, Chk2, MDM2, MDMX, p53
Introduction
The p53 tumor suppressor is critical for maintenance of genomic stability and protection against malignant transformation. P53 is regulated by multiple signaling pathways and can respond to a wide range of stress conditions, allowing it to act as a tumor suppressor in many cell types (Vousden, 2000). P53 turnover is regulated by MDM2, which binds p53 and functions as an ubiquitin E3 ligase to promote p53 degradation by the proteasomes (Zhang and Xiong, 2001). Stress signals such as DNA damage induce p53 accumulation by phosphorylation (Prives and Hall, 1999). Mitogenic signals activate p53 by induction of the p14ARF tumor suppressor, which inhibits the ability of MDM2 to ubiquitinate p53 (Zhang and Xiong, 2001).
Mammalian cells also express an MDM2 homolog called MDMX (Shvarts et al, 1996). MDMX shares strong homology to MDM2 at the amino-acid sequence level and can bind to p53 and inhibit its transcriptional function. However, MDMX alone does not promote p53 ubiquitination or degradation in vivo (Stad et al, 2001). Knockout of MDM2 in mice results in embryonic lethality due to hyperactivation of p53 (Oca Luna et al, 1995). Several studies showed that MDMX-null mice also die in utero in a p53-dependent fashion, which can be rescued by crossing into the p53-null background (Finch et al, 2002; Migliorini et al, 2002; Parant et al, 2002). Therefore, MDMX is also a critical regulator of p53.
Recent evidence showed that MDMX cooperates with MDM2 to promote p53 degradation (Gu et al, 2002). Human tumor cell lines with wild-type p53 often overexpress MDMX (Ramos et al, 2002), suggesting that MDMX may contribute to p53 inactivation during tumorigenesis. Therefore, regulation of MDMX expression level may be an important mechanism of activating p53 during stress response. Several reports showed that MDMX can be ubiquitinated and degraded by MDM2 (De Graaf et al, 2003; Kawai et al, 2003; Pan and Chen, 2003). MDMX and MDM2 form heterodimers through the C-terminal RING domains and the MDM2 RING domain alone is sufficient to degrade MDMX (Sharp et al, 1999; Tanimura et al, 1999; Pan and Chen, 2003). ARF inhibits the ability of MDM2 to ubiquitinate p53, but stimulates MDM2 ubiquitination of MDMX (Pan and Chen, 2003). This reveals a novel mechanism by which ARF activates p53 function by selective regulation of MDM2 E3 ligase function toward different substrates. DNA damage induced by ionizing irradiation also promotes MDMX nuclear translocation and degradation by the proteasomes (Li et al, 2002; Pan and Chen, 2003). Therefore, regulation of MDMX–MDM2 heterodimer formation may be important for the control of MDMX level.
In cells with functional p53, DNA damage activation of p53 leads to induction of MDM2 expression. This may lead to MDMX ubiquitination and degradation, resulting in a positive feedback effect on p53 activity (Pan and Chen, 2003). However, this mechanism alone is not expected to function in the initial activation of p53, since MDM2 induction by activated p53 is required. A recent study showed that DNA damage also induces MDMX degradation in p53-deficient cells, without induction of MDM2 (Kawai et al, 2003). This observation suggested that downregulation of MDMX may be an early event during DNA damage response and may directly contribute to p53 activation. Therefore, it is important to elucidate the mechanism responsible for MDMX degradation after DNA damage.
Phosphorylation is an important mechanism for activation of p53 in response to DNA damage. Several kinases have been shown to modify p53 and MDM2, including ATM, Chk2, and c-Abl. ATM functions as the primary signal transducer of DNA double-strand breaks (Shiloh, 2003). A major target of ATM that triggers cell cycle checkpoint response is p53. The importance of ATM in rapid p53 activation after ionizing radiation (IR) was revealed by the delay in p53 accumulation in ATM-deficient cells (Kastan et al, 1993). ATM phosphorylates p53 on serine 15, enhancing its transcriptional activity (Banin et al, 1998). ATM also activates Chk2, which in turn phosphorylates p53 on serine 20 and inhibits MDM2 binding (Shieh et al, 1997; Banin et al, 1998; Chehab et al, 2000; Shieh et al, 2000). MDM2 phosphorylation on serine 395 by ATM and tyrosine 394 by c-Abl has also been shown to affect MDM2's ability to regulate p53 (Maya et al, 2001; Goldberg et al, 2003). However, mutations of phosphorylation sites for these kinases on p53 and MDM2 or siRNA knockdown of Chk1 and Chk2 kinases lead to partial or no phenotypes (Wu et al, 2002; Ahn et al, 2003; Chao et al, 2003; Sluss et al, 2004), suggesting that additional targets and mechanisms may also contribute to activation of p53 after DNA damage.
In this report, we present evidence that MDMX is phosphorylated at several C-terminal serine residues after DNA damage. Stimulation of MDMX phosphorylation by DNA damage requires the activity of ATM. MDMX is a direct substrate for Chk1 and Chk2 in vitro. Phosphorylation of MDMX leads to increased binding to MDM2 and more efficient ubiquitination, providing an explanation for the enhanced degradation of MDMX after DNA damage. Downregulation of MDMX by RNAi increases p53 response to ionizing irradiation, suggesting that MDMX degradation by MDM2 is important for efficient activation of p53 by DNA damage.
Results
DNA damage induces degradation of MDMX
Previous experiments in our laboratory revealed that MDMX is an ubiquitination substrate for MDM2 (Pan and Chen, 2003). DNA damage induces proteasome-mediated degradation of MDMX, which correlates with increased MDM2 expression in cells with wild-type p53, suggesting that MDM2 induction plays a role in MDMX degradation (Figure 1A) (Pan and Chen, 2003). However, further tests of a panel of cell lines showed that MDMX downregulation also occur in p53-deficient HCT116 cells and p53-mutant DLD1 and C33A cells without induction of MDM2 (Figure 1B). This result corroborated recent observations made by other investigators (Kawai et al, 2003), suggesting that additional mechanisms cause MDMX degradation after DNA damage. To further characterize the change in MDMX stability, MCF7 treated with 10 Gy irradiation was analyzed for MDMX degradation by cyclohexamide block. MDMX was stable before irradiation but was rapidly degraded after DNA damage (Figure 1C). As expected, MDM2 was unstable before and after DNA damage, and p53 was stabilized after DNA damage. These results confirmed that the decrease in MDMX level was due to reduced stability.
Figure 1.
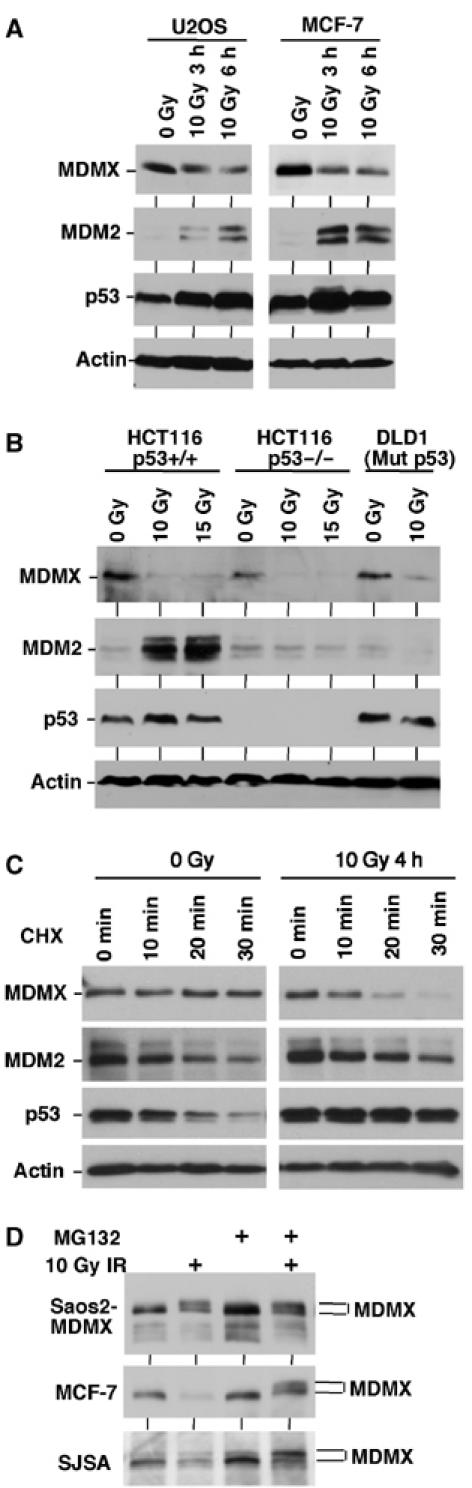
DNA damage induces degradation of MDMX. (A, B) Cell lines with different p53 mutation status were treated with IR for 3–6 h and analyzed for expression levels of indicated markers by Western blot. (C) MCF7 cells were treated with 10 Gy IR and cultured for 4 h. Cyclohexamide (CHX, 100 μg/ml) was added to inhibit protein synthesis and samples were analyzed for the rates of MDMX, MDM2, and p53 degradation. (D) Saos2 cells stably transfected with MDMX expression plasmid (Saos2-MDMX) or MCF7 and SJSA cells expressing endogenous MDMX were treated with IR and MG132 for 4 h and analyzed for MDMX expression by gradient gel electrophoresis and Western blot. IR induces the appearance of a slow migrating form of MDMX.
Previous experiments showed that the proteasome inhibitor MG132 prevents MDMX degradation after DNA damage (Pan and Chen, 2003). Gradient gel analysis of endogenous MDMX from MCF7 and SJSA cells treated with gamma radiation and MG132 revealed that a slower migrating form of MDMX accumulated 3–6 h after DNA damage (Figure 1D). This suggested that a population of MDMX was phosphorylated and then rapidly degraded after ionizing irradiation. When MDMX was ectopically expressed in p53-null H1299 or Saos2 cells with very low levels of MDM2, the accumulation of slow migrating MDMX was evident after gamma irradiation without MG132 treatment, possibly due to saturation of the MDM2 degradation system (Figure 1D).
Phosphorylation of MDMX promotes degradation by MDM2
To determine whether the retardation of MDMX mobility was caused by phosphorylation, MDMX immunoprecipitated from irradiated Saos2-MDMX cells were treated with calf intestinal alkaline phosphatase. This treatment restored the mobility of the slow-migrating MDMX (Figure 2A), suggesting that DNA damage induced phosphorylation of MDMX. To further identify agents that can induce MDMX phosphorylation, Saos2-MDMX cells were treated with a variety of cytotoxic drugs. The results showed that agents that can induce DNA damage were all capable of inducing MDMX phosphorylation. Microtubule targeting agents taxol and nocodazole did not cause MDMX mobility shift (Figure 2B), suggesting that the effect was unique to DNA-damaging drugs.
Figure 2.
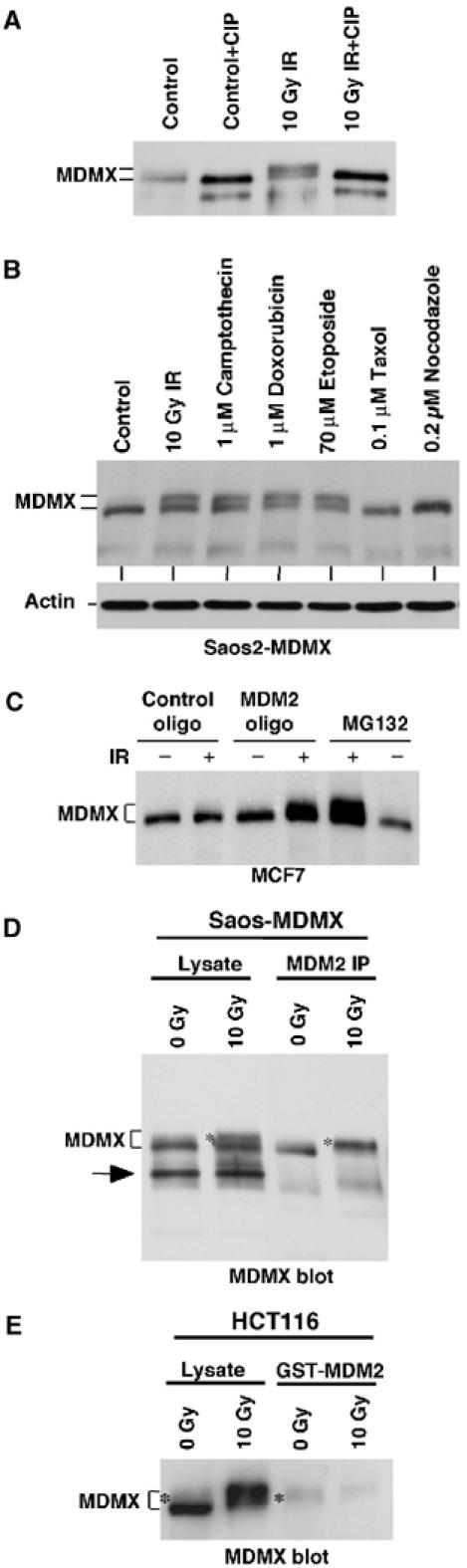
Phosphorylation of MDMX promotes degradation by MDM2. (A) Saos2-MDMX cells were treated with IR and immunoprecipitated using MDMX monoclonal antibody 8C6. The immunoprecipitate was incubated with CIP and changes in MDMX mobility was determined by gradient gel Western blot. (B) Saos2-MDMX cells were treated with indicated agents for 6 h and MDMX was analyzed by gradient gel Western blot. (C) MCF7 cells were treated with 200 nM MDM2 antisense oligonucleotide or control oligonucleotide for 24 h by lipofectin-mediated transfection and then irradiated with 10 Gy IR for 6 h. MDMX was detected by Western blot. Loading was empirically adjusted to compensate for the loss of MDMX and to detect the phosphorylated population. (D) Saos2-MDMX cells were tested for binding efficiencies of MDM2 to different forms of MDMX by MDM2 IP and MDMX Western blot after irradiation in the presence of MG132. The arrow indicates a background band in the whole-cell lysate crossreacting with the rabbit-anti-MDMX serum. (E) Glutathione-agarose beads loaded with GST-MDM2 were incubated with extract of control and irradiated HCT116 cells (treated with MG132). The captured MDMX were analyzed by gradient gel Western blot. The phosphorylated MDMX preferentially bound by MDM2 or GST-MDM2 was marked by *.
Slow migrating forms of endogenous MDMX were usually not detectable after DNA damage but accumulate significantly after addition of proteasome inhibitor MG132, suggesting that it is unstable compared to unmodified MDMX. A recent study showed that DNA damage-induced degradation of MDMX is dependent on MDM2 in mouse embryonic fibroblasts (Kawai et al, 2003). To determine whether MDM2 was responsible for the degradation of phosphorylated MDMX after DNA damage, MCF-7 cells were treated with MDM2 antisense oligonucleotide and IR (Chen et al, 1999). Inhibition of MDM2 expression led to the accumulation of slow migrating MDMX after radiation, similar to the effect of proteasome inhibition by MG132 (Figure 2C). Therefore, MDM2 expression was required for the rapid degradation of phosphorylated MDMX after DNA damage.
DNA damage induced MDMX degradation in p53-deficient cells without increasing MDM2 levels (Figure 1b). It is possible that this occurred through enhanced MDM2 binding to phosphorylated MDMX. To test whether this is the case, Saos2-MDMX cells were irradiated in the presence of MG132 and MDM2/MDMX coprecipitation was determined. The results showed that after irradiation, MDM2 co-precipitated more phosphorylated forms of MDMX than the unmodified form, despite both forms were present at comparable levels in the cell lysate (Figure 2D). Incubation of GST-MDM2-loaded beads with HCT116 extract also selectively captured phosphorylated endogenous MDMX (Figure 2E). The selectivity of the binding was most evident when using MDMX from nonirradiated cells as input substrate (only a minor fraction was phosphorylated, Figure 2E). The total amount of MDMX captured by GST-MDM2 did not increase after irradiation, possibly because most of the phosphorylated MDMX was already in a complex with endogenous MDM2. These results showed that phosphorylated MDMX bound to MDM2 more efficiently, which may cause its selective degradation by MDM2.
ATM is required for stimulating MDMX phosphorylation
If MDMX phosphorylation and degradation contributes to p53 activation after DNA damage, it should occur before p53 is stabilized. In a time course experiment, MDMX phosphorylation was detectable 15 min after IR in MCF7 cells (Figure 3A). Interestingly, phosphorylated MDMX was detectable even without MG132 treatment at the earliest time points, but was degraded at the 3 and 6-h time points. Significant stabilization of p53 was detected at the 2 and 4-h time points, correlating with the degradation of phosphorylated forms of MDMX. Therefore, MDMX phosphorylation occurs before p53 stabilization after ionizing irradiation, suggesting that degradation of MDMX contributes to p53 activation.
Figure 3.
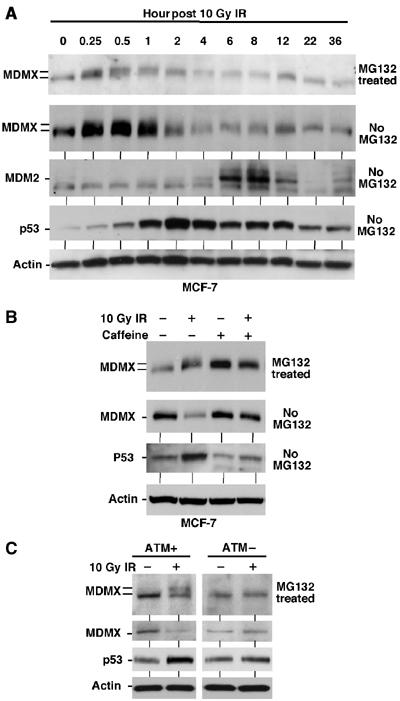
MDMX phosphorylation after DNA damage requires ATM activity. (A) MCF7 cells were irradiated with 10 Gy IR and samples collected at different time points were analyzed by gradient gel Western blot for the indicated markers. (B) MCF-7 cells were irradiated at 10 Gy in the presence of 10 mM caffeine and MG132 and MDMX phosphorylation was determined after 6 h by Western blot in the top panel (loading was empirically adjusted to show phosphorylated forms rather than degradation). Cells in the second panel were treated without MG132 and MDMX degradation was determined by Western blot. (C) Human skin fibroblasts that are wild type or ATM-deficient were irradiated in the presence or absence of MG132 for 4 h. MDMX phosphorylation and degradation were determined by gradient gel electrophoresis and Western blot.
The rapid induction of MDMX phosphorylation was consistent with the kinetics of ATM activation (Bakkenist and Kastan, 2003). Previously c-Abl and ATM have been shown to modify the C-terminal region of MDM2. The targets for these kinases on MDM2 (Y394, S395) are not conserved in the same locations on MDMX, although two of the MDMX phosphorylation sites detected by MS (S391 and S403, see below) are located in the same region. One phosphorylation site (ES403Q) matches the consensus sequence for ATM and ATR (Kastan and Lim, 2000). Therefore, we tested whether ATM plays a role in regulating the phosphorylation of MDMX. MCF7 cells were treated with caffeine to inhibit ATM/ATR. This treatment partially inhibited the mobility shift and degradation of MDMX after radiation (Figure 3B). This was confirmed using phosphorylation-specific antibodies (see Figure 8B). As expected, caffeine also blocked stabilization of p53 and induction of MDM2 and p21 (Figure 3B and data not shown). Therefore, ATM or ATR was important for the induction of MDMX phosphorylation after DNA damage. To further test the role of ATM, human skin fibroblasts from AT and normal individuals were treated with radiation. ATM-deficient fibroblasts failed to induce MDMX mobility shift and degradation after DNA damage (Figure 3C), demonstrating that ATM activity was required for this response.
Figure 8.
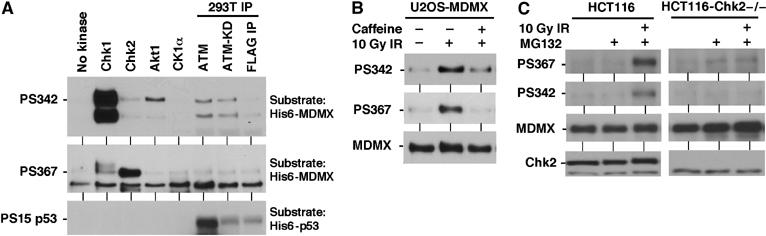
Phosphorylation of MDMX by Chk1 and Chk2 kinases. (A) In vitro phosphorylation of S342 and S367 by Chk1 and Chk2. Recombinant His6-MDMX and His6-p53 purified from E. coli were incubated with indicated kinases in the presence of ATP. The reaction products were analyzed by Western blot using PS342, PS467, and p53PS15 antibodies. (B) U2OS stably transfected with MDMX was treated with gamma irradiation for 2 h in the presence of 10 mM caffeine and analyzed by MDMX IP and PS342 and PS367 Western blot. (C) Chk2-null HCT116 cells were treated with 10 Gy gamma irradiation for 4 h in the presence of MG132, and analyzed for MDMX phosphorylation by IP-Western blot.
Reduction of MDMX level increases p53 response to DNA damage
To test the role of MDMX degradation in p53 activation, U2OS cells were transfected with MDMX siRNA for 48 h, followed by treatment with 10 Gy gamma irradiation. Transient knockdown of MDMX resulted in moderate increase of p53 level in the absence of DNA damage (Figure 4A). However, higher expression levels of p53 target MDM2 and p21 were induced by gamma radiation compared to control siRNA-treated cells. Similar results were also observed using MCF7 cells with stable knockdown of MDMX (data not shown). Therefore, reduction of MDMX level sensitizes p53 to DNA damage.
Figure 4.
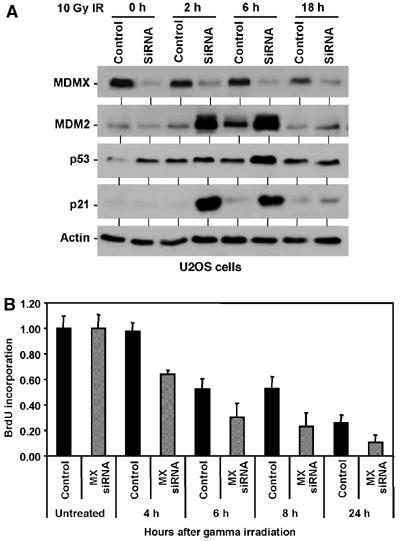
Knockdown of MDMX enhances p53 response to DNA damage. (A) U2OS cells were transfected with control or MDMX siRNA for 48 h, treated with 10 Gy IR, and analyzed for p53 activation at the indicated time points. (B) U2OS cells transfected with control or MDMX siRNA for 48 h were treated with 10 Gy IR, and starting at the indicated time points labeled for 2 h with BrdU. The levels of BrdU incorporation were measured by an ELISA assay (n=4).
P53 activation has important cell cycle checkpoint function in stopping DNA replication and providing time for repair after DNA damage. To test whether MDMX degradation has a physiological impact on cell cycle arrest after DNA damage, the rate of DNA synthesis was analyzed by measuring BrdU incorporation. The results showed that DNA synthesis inhibition after gamma irradiation was significantly more efficient in MDMX knockdown cells compared to controls (Figure 4B). These results suggested that degradation of MDMX after DNA damage facilitates p53 activation and cell cycle arrest.
Identification of phosphorylation sites on MDMX
To determine the types of phosphorylated residues on MDMX, we performed metabolic labeling of transfected MDMX using radioactive 32P-orthophosphate. Phosphorylation of MDMX in vivo was readily detected and additional treatment with gamma irradiation did not result in significant increase in the labeling efficiency (Figure 5A), possibly because the 32P labeling had induced significant DNA damage. Phospho-amino-acid analysis showed that MDMX was mainly phosphorylated on serine in vivo, with minor labeling of threonine and no detectable phosphorylation of tyrosine (Figure 5A).
Figure 5.
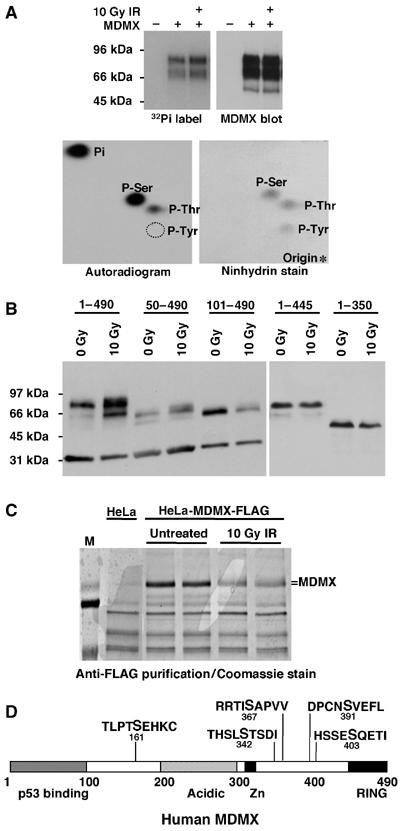
Mapping of phosphorylation sites on MDMX. (A) H1299 cells transiently transfected with MDMX were treated with 10 Gy IR and immediately labeled for 4 h with 32P orthophosphate. MDMX was immunoprecipitated, run on SDS–PAGE, transferred to nylon membrane and detected by autoradiography and Western blot. Radiolabeled MDMX bands were hydrolysed by HCl and analyzed by 2-D electrophoresis. Autoexposure of radiolabeled MDMX residues and ninhydrin stain of phospho-amino-acid standards on the same cellulose plate were shown. (B) Saos2 cells stably transfected with MDMX deletion mutants were treated with radiation and MDMX mobility was determined by gradient gel Western blot. (C) HeLa cells stably transfected with FLAG-tagged MDMX were treated with 10 Gy IR for 6 h, immunoprecipitated using M2-agarose beads, fractionated on SDS–PAGE, and stained by Coomassie blue. Entire MDMX bands from control and irradiated cells were excised and subjected to mass spectrometric analysis. (D) Diagram of MDMX and relative positions of phosphorylation sites identified by mass spectrometry on irradiated MDMX. Bold letter indicate the modified serine.
In order to locate the region of MDMX important for phosphorylation after DNA damage, Saos2 cells stably transfected with different MDMX deletion mutants were analyzed for mobility retardation. The results indicated that deletion of the MDMX C-terminal RING domain caused a complete loss of mobility shift after DNA damage, whereas deletion of the N-terminal p53-binding domain did not prevent phosphorylation (Figure 5B). Therefore, the C-terminal region of MDMX contains sequence important for phosphorylation. This is consistent with the significant effect of phosphorylation on binding to MDM2 but not p53, since MDM2 binding involves the MDMX C-terminal RING domain.
To map precisely the phosphorylation sites, we employed sensitive tandem mass spectrometric analysis. FLAG-tagged MDMX expressed in HeLa cells also undergo mobility shift after irradiation (Figure 5C). Therefore, FLAG-MDMX was immunoprecipitated from pre- and post-irradiated HeLa cells, entire MDMX bands were purified by SDS–PAGE and subjected to protease digestion and MS/MS peptide sequence analysis. Approximately 77% coverage of the MDMX peptide sequence was achieved. Analysis of the MS results led to detection of phosphorylation on S161 (post-IR), S342 (pre- and post-IR), S367 (pre- and post-IR), S391 (post-IR), and S403 (post-IR). Phosphorylation signals were detectable in the nonirradiated MDMX sample for S342 and S367, suggesting the presence of basal phosphorylation. This was confirmed using phosphorylation-specific antibodies (see below). Four of the five sites were near the C-terminus as expected from the deletion analysis.
Phosphorylation site mutations reduce ubiquitination and degradation by MDM2
To determine the role of different phosphorylation sites detected by MS in the regulation of MDMX degradation, these sites were mutated to alanine to block phosphorylation. Single mutants of MDMX were stably transfected into H1299 cells and pooled colonies were tested for mobility change after IR. The results showed that mutation of S342, S367, and S403 significantly blocked mobility shift after DNA damage (Figure 6A), suggesting that phosphorylation of these serine residues contributed to the mobility shift.
Figure 6.
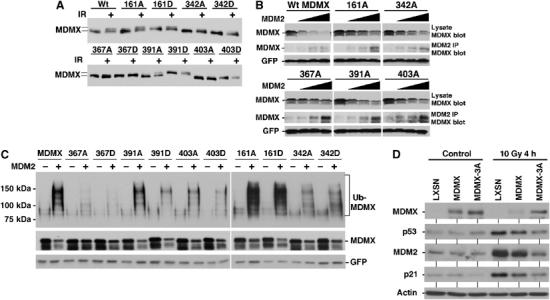
Effects of MDMX phosphorylation site mutations. (A) H1299 cells stably transfected with MDMX point mutants were irradiated at 10 Gy and MDMX mobility shift was analyzed after 6 h by Western blot. The 342A, 367A, and 403A mutants significantly reduced MDMX mobility shift. (B) MDMX mutant plasmids (4 μg) were cotransfected with 0, 0.5, 1, 2 μg of MDM2 plasmid into H1299 cells and MDMX expression levels were determined by Western blot 48 h after transfection. The same cell lysate were also analyzed by MDM2 IP and MDMX Western blot to detect MDM2–MDMX binding. The 367A mutant retained MDM2 binding function but was resistant to degradation by MDM2. (C) MDMX mutants were cotransfected with His6-ubiquitin and MDM2 into H1299 cells. Ubiquitinated MDMX were detected by Western blot after purification using Ni-NTA beads 48 h after transfection. (D) U2OS cells stably infected with retrovirus vector (LXSN) expressing MDMX or MDMX-342A/367A/403A triple mutant (MDMX-3A) was treated with 10 Gy gamma irradiation for 4 h and analyzed for expression of indicated markers of p53 activation by Western blot.
To test the effect of the mutations on degradation by MDM2, the mutants were cotransfected with increasing doses of MDM2 plasmid into H1299 cells and MDMX expression level was determined by Western blot. The results showed that within the sensitivity limit of this assay, the 367A mutant was significantly more resistant to degradation by MDM2 (Figure 6B), suggesting that phosphorylation of S367 may be critical for this function. Analysis of the cotransfected cells by MDM2 IP-MDMX Western blot showed that the 367A mutation did not prevent MDM2 binding (Figure 6B). Therefore, phosphorylation of S367 was not necessary for MDM2 binding, but may further stimulate MDM2 binding or induce a conformational change that favor degradation by MDM2.
To further analyze the functional significance of the phosphorylation sites on MDMX ubiquitination, the MDMX mutants were cotransfected with His6-ubiquitin into H1299 cells. MDMX conjugated to His6-ubiquitin was purified by Ni2+-NTA beads and detected by MDMX Western blot (Pan and Chen, 2003). The results showed that mutating S342, S367, and S403 each resulted in reduced poly-ubiquitination by MDM2, with S367 having the most significant effect (Figure 6C). Mutations on S161 and S391 had little effect on MDMX ubiquitination. Although mutation to aspartic or glutamic acid can sometimes mimic the effect of phosphorylation, we did not observe increased ubiquitination or degradation of MDMX with aspartic acid mutations. This was not entirely unexpected since aspartic acid may not truly mimic phosphorylation (e.g., does not recruit 14-3-3). MDMX also undergoes mono-ubiquitination that is MDM2-independent (Pan and Chen, 2003); this modification was not affected by the mutations (Figure 6C). Therefore, phosphorylation of S342, S367, and S403 may each contribute to stimulating poly-ubiquitination and degradation by MDM2, with S367 having the most dominant role.
To test the effects of the mutants on p53 activation after DNA damage, the phosphorylation site mutants were expressed in U2OS cells by retrovirus. Compared to wild-type MDMX retrovirus, single-point mutants of S342A, S367A, and S403A only showed minor differences (data not shown). However, the triple alanine compound mutant (MDMX-3A) showed significant resistance to degradation after DNA damage, and inhibited p53 activation more efficiently than wild-type MDMX (Figure 6D). This result suggested that multiple phosphorylation events cooperate to regulate MDMX degradation after DNA damage. More rigorous test of this notion will require gene replacement to avoid MDMX overexpression, which could not be prevented despite the use of retrovirus delivery.
Detection of MDMX phosphorylation by peptide-specific antibodies
We attempted to generate phosphopeptide-specific polyclonal antibodies against three functionally relevant sites: S342, S367, and S403. To date, we have succeeded in obtaining specific antibodies against PS342 and PS367. The specificity of the antibodies was confirmed by the lack of binding to un-phosphorylated His6-MDMX produced in Escherichia coli (Figure 7A and B). Phosphorylation of endogenous MDMX in MCF7 cells and transfected MDMX in U2OS cells were analyzed by antiphosphorylation Western blot of immunoprecipitated MDMX. The results showed that low levels of MDMX phosphorylation on S342 and S367 were detected in the absence of DNA damage (Figure 7A and B), consistent with MS results. The presence of basal phosphorylation on S342 was verified by treatment of immunoprecipitated MDMX with alkaline phosphatase (Figure 7C). Interestingly, dephosphorylation of PS367 by calf intestine phosphatase (CIP) was very inefficient compared to PS342 (Figure 7C), indicating that this site was not accessible for the phosphatase due to unfavorable conformation or protection by another protein. Gamma irradiation significantly increased the phosphorylation levels of S342 and S367 in MCF7 cells in the presence of MG132 (Figure 7A and B). These results demonstrated that the levels of modification on these residues were strongly stimulated by DNA damage. Furthermore, endogenous MDMX phosphorylated on S342 or S367 were rapidly degraded after DNA damage and only accumulate after MG132 treatment.
Figure 7.
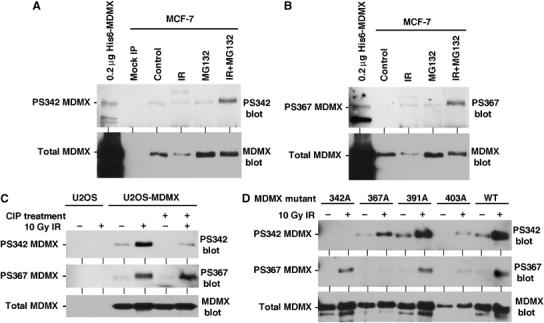
Detection of MDMX phosphorylation by peptide-specific antibodies. (A, B) MCF-7 cells treated with 10 Gy IR for 4 h in the presence of MG132 were immunoprecipitated using 8C6 antibody and probed with anti-phospho S342 or S367 peptide antibodies. His6-MDMX expressed in E. coli was used as a control for crossreactivity to nonphosphorylated MDMX. The same membranes were striped and probed using MDMX polyclonal antibody to determine the level of total MDMX. (C) U2OS cells stably transfected with MDMX were irradiated with 10 Gy IR and MDMX was immunoprecipitated after 2 h and probed with PS342 and PS367 antibodies. The presence of basal level S342 phosphorylation on unirradiated MDMX was confirmed by treatment with CIP. The membranes were reprobed with anti-MDMX polyclonal antibody to confirm loading. (D) U2OS stably transfected with MDMX point mutants were treated with 10 Gy IR for 2 h and analyzed by MDMX IP and PS342 and PS367 Western blot.
Next, we used the phosphorylation-specific antibodies to examine the relationship between each phosphorylation site after DNA damage. U2OS cells stably transfected with 342A, 367A, 391A, and 403A MDMX point mutants were treated with gamma radiation and analyzed for S342 and S367 phosphorylation levels. The results showed that mutating the adjacent sites had no effect on the phosphorylation of S342 and S367 (Figure 7D). As expected, 342A and 367A mutations abrogated detection by PS342 and PS367 antibodies, respectively, confirming their specificity. This result suggested that the modification at each site was independently regulated after DNA damage. The mutations did not cause a global structural change that prevents modification of other sites.
Phosphorylation of MDMX by Chk2
Activation of ATM by DNA damage leads to phosphorylation and activation of downstream kinases Chk1 and Chk2 (Bartek and Lukas, 2003). Chk1 and Chk2 have overlapping substrate specificity and also have distinct functions. Chk1 has house-keeping activity in the absence of DNA damage and is required for cell viability. Chk2 is activated after DNA damage and is thought to function as a signal amplifier for ATM. To determine whether kinases in the DNA damage response pathway can directly target MDMX, His6-MDMX was incubated with ATM, Chk1, and Chk2 in vitro in the presence of ATP. Western blot showed that Chk1 modified S342 and S367, but with strong preference for S342. Chk2 modified both S342 and S367, but with strong preference for S367 (Figure 8A).
Although S367 is in an optimal sequence context for Akt1, it was not modified by Akt1 in vitro (Figure 8A). Weak modification of S342 by Akt1 was detected, the significance of this reaction is not clear at present. The casein kinase 1 alpha isoform that binds and phosphorylates MDMX in the central region in such reactions (Chen et al, 2005) did not target S342 and S367 (Figure 8A). ATM immunoprecipitated from transfected cells phosphorylated S15 of p53 but did not show specific modification of MDMX on S342 and S367. Lack of a PS403 antibody precluded analysis of this site for direct modification by ATM in our experiments. A recent study was able to demonstrate the phosphorylation of S403 by ATM using a phosphorylation-specific antibody (Pereg et al, 2005).
Since the amount of endogenous MDMX in ATM-deficient fibroblasts was too low for phosphorylation analysis, we used pharmacological inhibition of ATM in U2OS-MDMX cells. Treatment of U2OS-MDMX cells with caffeine blocked induction of S342 and S367 phosphorylation after gamma irradiation (Figure 8B). This is consistent with the interpretation that ATM is required for activation of Chk2, which mediates S342 and S367 phosphorylation. To directly test the role of Chk2 in the phosphorylation of MDMX, HCT116-Chk2−/− cells were examined (Jallepalli et al, 2003). Loss of Chk2 abrogated the induction of S342 and S367 phosphorylation after irradiation (Figure 8C), suggesting that Chk2 is required for the phosphorylation of both sites in vivo. Cotransfection of MDMX and Chk2 plasmid into HCT116-Chk2−/− cells restored the phosphorylation of MDMX S367 after gamma irradiation, whereas kinase-dead Chk2-A347 mutant, activated myr-Akt1 and CK1α had no effect (Figure 9A). In wild-type HCT116 cells, Chk2-A347 reduced S367 phosphorylation, possibly through dominant-negative effect (Figure 9A). In cotransfection experiments, MDMX ubiquitination and degradation by MDM2 can occur in both wild-type and Chk2-null HCT116 cells, suggesting that Chk2 was not absolutely necessary under these conditions. However, expression of MDM2 stimulated MDMX ubiquitination more significantly in wild-type HCT116 compared to the Chk2-null cells (Figure 9B), consistent with Chk2 being a facilitator. As expected, cotransfection of Chk2 strongly stimulated MDMX ubiquitination and degradation (Figure 9B).
Figure 9.
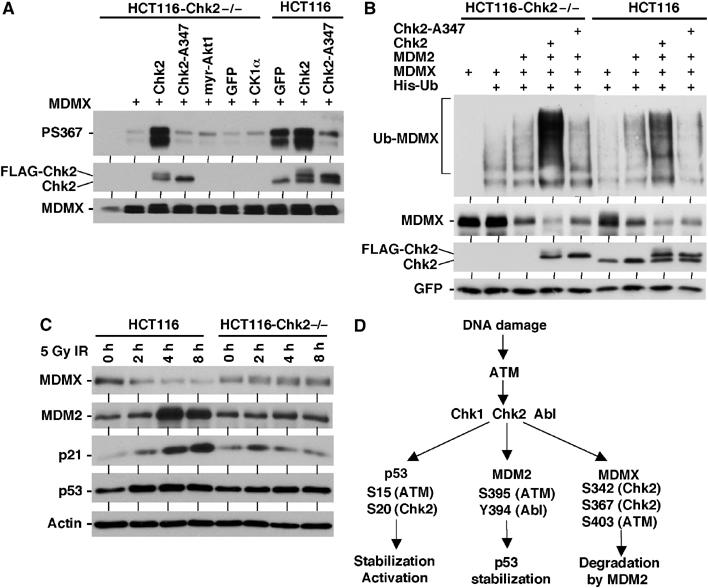
Regulation of MDMX degradation by Chk2. (A) Chk2-null HCT116 cells were transiently transfected with MDMX, FLAG-Chk2, or inactive FLAG-Chk2-A347 mutant. Cells were treated with 10 Gy gamma irradiation for 2 h in the presence of MG132 and analyzed for MDMX S367 phosphorylation by IP-Western blot. The Chk2 blot used a Chk2-specific antibody that detects both endogenous and transfected Chk2. (B) Chk2-null HCT116 cells were transfected with indicated plasmids and analyzed for MDMX ubiquitination and degradation after 48 h. (C) Chk2-null HCT116 cells were treated with 5 Gy gamma irradiation and analyzed for MDMX degradation, induction of MDM2, and stabilization of p53 at indicated time points. (D) DNA damage activates p53 through multiple ATM-dependent mechanisms.
When the HCT116-Chk2−/− cells were compared to wild-type HCT116 after gamma irradiation (5 Gy), we reproducibly found that MDMX degradation was much less efficient (Figure 9C). MDM2 and p21 induction and p53 stabilization were also reduced compared to wild-type cells (Figure 9C). Interestingly, a slight shift in MDMX mobility was observed in irradiated Chk2−/− cells, possibly the result of phosphorylation by ATM. These results showed that Chk2 plays a role in regulating p53 response to gamma irradiation, possibly through promoting degradation of MDMX. The results in Figure 9C were unexpected since a previous study using the same HCT116-Chk2−/− cell line reported that Chk2 knockout did not affect p53 response to higher doses (12 Gy) of ionizing irradiation (Jallepalli et al, 2003). We speculate that differences in experimental conditions may be responsible for the discrepancy. Irradiation dosage alone is probably not the cause, since the deficiency we observed in the HCT116-Chk2−/− cells were reproducible in a range between 5 and 20 Gy (data not shown).
Discussion
Results described above showed that MDMX undergoes significant phosphorylation and degradation after DNA damage in an ATM and Chk2-dependent fashion. Elimination of MDMX would facilitate p53 activation by relieving p53 from MDMX binding, and reducing the E3 ligase activity of MDM2. In vivo metabolic labeling revealed that MDMX is predominantly phosphorylated on serine, with minor modifications on threonine and no detectable modification on tyrosine. This suggests that unlike MDM2, MDMX may not be directly modified by the c-Abl tyrosine kinase. Most of the phosphorylation sites identified on MDMX are localized near the C-terminal RING domain, consistent with the observed impact on MDM2 binding and degradation. Of the three functionally relevant phosphorylation sites identified in this study, S342 and S367 are targets of Chk2 in vitro and in vivo. S403 is in a favorable context for ATM and a recent study by Pereg et al (2005) provided direct evidence that it is an ATM target site.
The current results suggest that DNA damage activates ATM and subsequently Chk2. ATM modifies S403 and Chk2 modifies S342 and S367 on MDMX. Multiple phosphorylation of MDMX C-terminal region may lead to conformational changes in the MDMX RING domain, increasing the affinity for MDM2 binding, and leading to ubiquitination and degradation of MDMX. Chk1 also efficiently modifies S342 in vitro, it is likely that Chk1 plays a role in the basal phosphorylation of MDMX in the absence of DNA damage. However, DNA damage-induced phosphorylation of S342 and S367 strictly requires Chk2. At present, the lack of a viable Chk1-null mammalian cell line, and the strong toxicity of Chk1 siRNA hamper the investigation of its role in MDMX phosphorylation.
Although a role of ATM in p53 response is well established, the significance of Chk2 is still a matter of debate. Studies using Chk2-null mice, Chk2-null HCT116 cells, and Chk2 siRNA resulted in different conclusions (Zhang et al, 1998; Chehab et al, 2000; Shieh et al, 2000; Ahn et al, 2003; Jallepalli et al, 2003). We show here that Chk2 is directly involved in the phosphorylation of MDMX on S342 and S367. Although multiple phosphorylations are necessary for a complete phenotype, our results showed that knockout of Chk2 alone can have measurable effects on MDMX degradation and p53 activation under certain conditions such as gamma irradiation. In fact, mutation of S367 has the most dramatic effect on MDMX ubiquitination and stability (Figure 6). In vivo 32P phosphate metabolic labeling and phosphopeptide analysis of MDMX revealed that S367 is the most heavily phosphorylated residue on MDMX after DNA damage (LeBron and Chen, unpublished observation). Therefore, Chk2 contributes to p53 regulation in part through phosphorylation of MDMX. However, a single defect in Chk2 alone may have subtle, cell line and damage dose-dependent effects on p53 response that often elude detection.
The mechanism by which phosphorylation enhances MDMX–MDM2 binding and MDMX ubiquitination by MDM2 remains to be further investigated. A recent study showed that DNA damage also accelerates MDM2 degradation through a mechanism that requires its own RING domain (Stommel and Wahl, 2004). It is possible that phosphorylation of MDMX induces changes of its C-terminal conformation and the functional characteristics of the MDM2-MDMX heterodimer, facilitating activation of p53 and degradation of MDMX and MDM2. Phosphorylation may also recruit adaptor molecules that facilitate MDMX–MDM2 heterodimerization and induce MDMX conformational change. It is noteworthy that phosphorylated S367 of MDMX is a potential 14-3-3 binding site (Tzivion et al, 2001). We have recently found that 14-3-3 copurifies with MDMX (Chen et al, 2005). Experiments are underway to investigate whether 14-3-3 plays a role in MDMX degradation after DNA damage.
These observations add to our knowledge of an intricate and tightly coordinated signaling mechanism that is important for p53 function (Figure 9D). Previous studies showed that phosphorylation of S15 and S20 of p53 by ATM and Chk2 stimulate p53 transcription activation function and reduce MDM2 binding. Phosphorylation of MDM2 Y394 by c-Abl and S395 by ATM reduce its ability to promote p53 degradation. We show here that phosphorylation of MDMX S342 and S367 by Chk2 promote its ubiquitination and degradation by MDM2. Our results of S403 phosphorylation also corroborate recent findings from Pereg et al (2005) on ATM modification of this site. It is apparent that combination of these and possibly additional changes on multiple targets in the p53 pathway lead to timely activation of p53 and G1 arrest in response to DNA damage.
In summary, we have shown that DNA damage induces phosphorylation of MDMX through an ATM and Chk2-dependent pathway, resulting in the degradation of MDMX by MDM2. This event contributes to p53 activation after DNA damage. This work and our previous study also reveal an important function of MDM2 in regulating MDMX stability in response to mitogenic stress and DNA damage. The ubiquitin E3 ligase activity of MDM2 can be switched between targeting p53 or MDMX by stress signals, thus having both negative and positive effects on p53 function. The intricate interplay between MDM2 and MDMX and their connection to stress signaling pathways are likely to be critical for the dynamic regulation of p53.
Materials and methods
Cell lines and plasmids
Cells were maintained in DMEM medium with 10% fetal bovine serum. HCT116-p53+/+, HCT116-p53−/−, HCT116-Chk2−/− cells were kindly provided by Dr Bert Vogelstein and maintained in McCoy 5A medium with 10% fetal bovine serum. ATM-deficient human skin fibroblast Hs235.Sk was purchased from the ATCC. Normal human skin fibroblast HFF was provided by Dr Jack Pledger. Human MDMX cDNA was kindly provided by Dr Donna George (Sharp et al, 1999). A version without epitope tags was used for all transfection experiments. FLAG-ATM expression plasmids were provided by Dr Michael Kastan. FLAG tagged Chk2 wild-type and A347 mutant were provided by Dr Thanos Halazonetis (Chehab et al, 2000). For affinity purification and mass spectrometric analysis, a FLAG epitope tag was added to the C-terminus of myc-MDMX to create double-tagged myc-MDMX-Flag. MDMX point mutants were generated by site-directed mutagenesis using the QuickChange kit (Stratagene). All MDM2 and MDMX constructs used in this study were based on human cDNA clones. To inhibit MDMX expression, U2OS cells were transfected with 200 nM control siRNA (AATTCTCCGAACGTGTCACGT) and MDMX siRNA (AGATTCAGCTGGTTATTAA) using Oligofectamine (Invitrogen) according to instruction from the supplier. After 48 h of transfection, cells were irradiated and analyzed for protein expression or BrdU incorporation.
Protein analysis
To detect proteins by Western blot, cells were lysed in lysis buffer (50 mM Tris–HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5% NP40, 1 mM PMSF, 50 mM NaF) and centrifuged for 5 min at 10 000 g. Cell lysate (10–50 μg protein) was fractionated by SDS–PAGE using gradient gel and transferred to Immobilon P filters (Millipore). The filter was blocked for 1 h with phosphate-buffered saline (PBS) containing 5% nonfat dry milk, 0.1% Tween-20. MDMX binding to MDM2 was detected by immunoprecipitation of 200–500 μg total proteins using 2A9, followed by Western blot using rabbit anti-MDMX antibody. Alternatively, glutathione-agarose beads loaded with ∼5 μg GST-MDM2 were incubated with 500 μg cell lysate for 3 h at 4°C, washed with lysis buffer, and the captured MDMX was detected by 8C6 Western blot. Dephosphorylation of MDMX in vitro was performed by immunoprecipitation of MDMX, followed by treatment of the precipitate in 150 mM NaCl, 50 mM Tris pH 8.5, 5 mM MgCl2, 1 mM DTT with 2 unit Calf intestine phosphatase (CIP) for 1 h at 32°C. The following monoclonal antibodies were used: 3G9 and 2A9 for MDM2 Western blot and IP (Chen et al, 1993); DO-1 (Pharmingen) p53 Western blot; 8C6 monoclonal or a rabbit polyclonal serum for MDMX Western blot and IP (Li et al, 2002). The filter was developed using ECL-plus reagent (Amersham). Inhibition of MDM2 expression was achieved by transfection of an MDM2 antisense oligonucleotide (200 nM) using Lipofectin for 24 h as described previously (Chen et al, 1999). Gradient gel SDS–PAGE was performed using precast 4–12% gels (BioRad). Phosphorylation-specific rabbit antibodies were raised against phosphorylated MDMX peptides HSL(pS)TSDIT (PS342) and RTI(pS)APVVR (PS367) (GenScript, NJ) and affinity purified.
Affinity purification of MDMX and phosphorylation site mapping
HeLa cells stably transfected with FLAG-tagged MDMX (∼2 × 108 cells) were lysed in 10 ml lysis buffer (50 mM Tris–HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5% NP40, 1 mM PMSF, 200 nM Okadaic acid). The lysate was precleared with 100 μl bed volume of protein A sepharose beads for 30 min, and then incubated with 50 μl bed volume of M2-agarose bead (Sigma) for 4 h at 4°C. The beads were washed and MDMX was eluted with 70 μl of 20 mM Tris pH 8.0, 2% SDS, 200 μg/ml FLAG epitope peptide for 15 min. The eluted proteins were fractionated on SDS–PAGE and stained with Coomassie Blue.
Ion trap MS/MS was used to determine in vivo phosphorylation sites of MDMX. MDMX isolated by SDS/PAGE was subjected to reduction, carboxyamidomethylation, and digestion with trypsin. The peptides were analyzed by microcapillary reverse-phase HPLC nanoelectrospray MS/MS on a LCQ DECA XP Plus quadrupole ion trap MS (ThermoFinnigan, San Jose, CA). The ion trap repetitively surveyed m/z 395–1600, acquiring data-dependent MS/MS spectra for peptide sequence information on the four most abundant precursor ions in the survey scan. A normalized collision energy of 30% and isolation width of 2.5 Da were used, with recurring ions dynamically excluded. Preliminary mapping of peptide sequences was accomplished with the SEQUEST algorithm. The discovery of peptides carrying phosphate and manual interpretation of the MS/MS spectra was facilitated with the in-house programs MUQUEST and FUZZYIONS, respectively.
Metabolic labeling and phospho-amino-acid analysis
To detect MDMX phosphorylation by endogenous kinases, H1299 cells in 10 cm plates were transiently transfected with 5 μg MDMX expression plasmid. At 40 h after transfection, cells were washed with DMEM without phosphate and incubated with 32P-phosphate (0.2 mCi/ml) in DMEM without phosphate for 4 h. Cell lysate was immunoprecipitated with 8C6, washed, and analyzed by SDS–PAGE and autoradiography. An identical set of 8C6 IP samples was analyzed by MDMX Western blot to confirm protein expression level. Nylon membrane containing radiolabeled MDMX band was excised and incubated with 5.7 N HCl at 110°C for 1 h. The hydrolyzed MDMX residues were lyophilized, mixed with phospho-amino-acid standards (Sigma) and analyzed by two-dimensional (2-D) electrophoresis on thin layer cellulose plate using a pH 1.9 buffer for the first dimension and pH 3.5 buffer for the second dimension on a HTLE-7002 apparatus (CBS Scientific).
In vitro kinase reactions
For each phosphorylation reaction in vitro, 5 × 107 293T cells transiently transfected with 20 μg FLAT-ATM expression plasmid were immunoprecipitated using M2-agarose beads and washed as previously described (Kim et al, 1999). His6-MDMX (200 ng) purified from E. coli was incubated with M2 beads containing FLAG-ATM, or 20 ng purified Chk1 (recombinant enzyme expressed in insect cells, Upstate) and Chk2 (His6-tagged enzyme expressed in E. coli, Upstate) in kinase buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 10 mM MgCl2, 1 mM DTT, and 10 mM MnCl2) with 5 mM ATP for 1 h at 30°C. The samples were boiled and fractionated on SDS–PAGE and subjected to Western blot analysis using phosphorylation-specific antibodies.
Acknowledgments
We thank the Moffitt Molecular Biology Core for DNA sequence analyses. We also thank Dr Michael Kastan for ATM plasmids and Dr Thanos Halazonetis for Chk2 plasmids. This work was supported by grants from the American Cancer Society and National Institutes of Health to J Chen.
References
- Ahn J, Urist M, Prives C (2003) Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J Biol Chem 278: 20480–20489 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S-Y, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281: 1674–1677 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J. (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–449 [DOI] [PubMed] [Google Scholar]
- Chao C, Hergenhahn M, Kaeser MD, Wu Z, Saito S, Iggo R, Hollstein M, Appella E, Xu Y (2003) Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J Biol Chem 278: 41028–41033 [DOI] [PubMed] [Google Scholar]
- Chehab NH, Malikzay A, Appel M, Halazonetis TD (2000) Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev 14: 278–288 [PMC free article] [PubMed] [Google Scholar]
- Chen J, Marechal V, Levine AJ (1993) Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 13: 4107–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li C, Pan Y, Chen J (2005) Regulation of p53-MDMX interaction by casein kinase 1 alpha. Mol Cell Biol 25: 6509–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Lu W, Agrawal S, Zhou W, Zhang R, Chen J (1999) Ubiquitous induction of p53 in tumor cells by antisense inhibition of MDM2 expression. Mol Med 5: 19–32 [PMC free article] [PubMed] [Google Scholar]
- De Graaf P, Little NA, Ramos YF, Meulmeester E, Letteboer SJ, Jochemsen AG (2003) Hdmx protein stability is regulated by the ubiquitin ligase activity of mdm2. J Biol Chem 278: 38315–38324 [DOI] [PubMed] [Google Scholar]
- Finch RA, Donoviel DB, Potter D, Shi M, Fan A, Freed DD, Wang CY, Zambrowicz BP, Ramirez-Solis R, Sands AT, Zhang N (2002) mdmx is a negative regulator of p53 activity in vivo. Cancer Res 62: 3221–3225 [PubMed] [Google Scholar]
- Goldberg Z, Vogt Sionov R, Berger M, Zwang Y, Perets R, Van Etten RA, Oren M, Taya Y, Haupt Y (2003) Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J 21: 3715–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM (2002) Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem 277: 19251–19254 [DOI] [PubMed] [Google Scholar]
- Jallepalli PV, Lengauer C, Vogelstein B, Bunz F (2003) The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem 278: 20475–20479 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS (2000) The many substrates and functions of ATM. Nat Rev Mol Cell Biol 1: 179–186 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ (1993) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM (2003) DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem 278: 45946–45953 [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 274: 37538–37543 [DOI] [PubMed] [Google Scholar]
- Li C, Chen L, Chen J (2002) DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol Cell Biol 22: 7562–7571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, Kastan MB, Katzir E, Oren M (2001) ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev 15: 1067–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, Helin K, Pelicci PG, Marine JC (2002) Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol 22: 5527–5538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oca Luna RM, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378: 203–206 [DOI] [PubMed] [Google Scholar]
- Pan Y, Chen J (2003) MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol 23: 5113–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G. (2002) Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 29: 92–95 [DOI] [PubMed] [Google Scholar]
- Pereg Y, Shkedy D, De Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, Biton S, Teunisse AF, Lehmann WD, Jochemsen AG, Shiloh Y (2005) Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci USA 102: 5056–5061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prives C, Hall PA (1999) The p53 pathway. J Pathol 187: 112–126 [DOI] [PubMed] [Google Scholar]
- Ramos YF, Stad R, Attema J, Peltenburg LT, van der Eb AJ, Jochemsen AG (2002) Aberrant expression of HDMX proteins in tumor cells correlates with wild-type p53. Cancer Res 61: 1839–1842 [PubMed] [Google Scholar]
- Sharp DA, Kratowicz SA, Sank MJ, George DL (1999) Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 274: 38189–38196 [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai k, Taya Y, Prives C (2000) The human homologs of checkpoint kinases chk1 and cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 14: 289–300 [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Ikeda M, Taya Y, Prives C (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91: 325–334 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Shvarts A, Steegenga WT, Riteco N, van Larr T, Dekker P, Bazuine M, van Ham RCA, van Oordt WVDH, Hateboer G, van der Eb AJ, Jochemsen AG (1996) MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J 15: 5349–5357 [PMC free article] [PubMed] [Google Scholar]
- Sluss HK, Armata H, Gallant J, Jones SN (2004) Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol Cell Biol 24: 976–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stad R, Little NA, Xirodimas DP, Frenk R, van der Eb AJ, Lane DP, Saville MK, Jochemsen AG (2001) Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep 2: 1029–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stommel JM, Wahl GM (2004) Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J 23: 1547–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M (1999) MDM2 interacts with MDMX through their RING finger domains. FEBS Lett 447: 5–9 [DOI] [PubMed] [Google Scholar]
- Tzivion G, Shen YH, Zhu J (2001) 14-3-3 proteins; bringing new definitions to scaffolding. Oncogene 20: 6331–6338 [DOI] [PubMed] [Google Scholar]
- Vousden KH (2000) p53: death star. Cell 103: 691–694 [DOI] [PubMed] [Google Scholar]
- Wu Z, Earle J, Saito S, Anderson CW, Appella E, Xu Y (2002) Mutation of mouse p53 Ser23 and the response to DNA damage. Mol Cell Biol 22: 2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y (2001) Control of p53 Ubiquitination and Nuclear Export by MDM2 and ARF. Cell Growth Differ 12: 175–186 [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG (1998) ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92: 725–734 [DOI] [PubMed] [Google Scholar]