

Abstract
We have previously shown that immunoglobulin A1 (IgA1) protease, an exoenzyme of pathogenic neisseriae, can trigger the release of proinflammatory cytokines from human monocytic subpopulations. Here, we demonstrate a dose-dependent T-cell response to recombinant gonococcal IgA1 protease (strain MS11) in healthy human blood donors. This response was delayed in comparison to the immune response against tetanus toxoid. Stimulation with IgA1 protease led to the activation of CD4+ and CD8+ T cells, as well as CD19+ B cells and CD56+ NK cells, indicated by de novo expression of CD69. Only CD4+ T cells proliferated and stained positive for intracellular gamma interferon (IFN-γ). Both proliferation and IFN-γ production were dependent on antigen presentation via major histocompatibility complex class II. Peripheral blood mononuclear cells stimulated with IgA1 protease produce IFN-γ and tumor necrosis factor alpha but no, or very low amounts of, interleukin-10 (IL-10) or IL-4, indicating a Th1-based proinflammatory immune response. These findings support the significance of IgA1 protease as a virulence determinant of bacterial meningitis and its function as a dominant proinflammatory T-cell antigen.
The genus Neisseria includes, in addition to several commensal species, two significant human pathogens, Neisseria gonorrhoeae (gonococci), the causative agent of gonorrhea, and Neisseria meningitidis (meningococci), a major cause of bacterial meningitis. Both pathogens exclusively infect humans as their natural hosts. Gonococci gain access to submucosal tissue after adherence to the urogenital mucosa, where they elicit acute and severe inflammation, primarily in males. In females, infection is mainly associated with a muted or absent inflammatory response (75). In the majority of cases, meningococci colonize the upper respiratory tract, leading to asymptomatic carriage of the bacteria. In some cases, for reasons which are not fully understood, neisseriae invade the colonized tissue, leading to systemic spread of the bacteria (12). Tissue damage is mediated by proinflammatory cytokines, especially tumor necrosis factor alpha (TNF-α), which can be detected at high levels in blood and cerebrospinal fluid (CSF) during meningitis (6, 30, 53, 65). If untreated, meningitis leads to multiorgan failure and septic shock, and mortality rates can be as high as 85% (7).
Various bacterial components are implicated in the pathogenesis of neisserial infections (11, 48, 74). The pili and Opa proteins, for instance, play crucial roles in adhesion to and invasion of different host cells, respectively (15). Neisserial porins could prevent the microbicidal activities of phagocytes (27, 48) and modulate apoptosis in lymphoid cells and epithelial cells (47, 50). Several investigators have also allocated the neisserial immunoglobulin A1 (IgA1) protease to this list of potential virulence factors (see below). IgA1 proteases are produced by a variety of gram-positive and gram-negative human pathogens (35, 55, 71). The neisserial IgA1 proteases are derived from precursor molecules which are secreted through an autocatalytic process (57). The secreted enzyme cleaves human IgA1, but not IgA2, in the hinge region of the heavy chain, a 13-amino-acid, proline-rich consensus cleavage sequence (55, 58). Since IgA1 is thought to be the predominant antibody subclass protecting mucosal tissues against microorganisms, the cleavage of IgA1 by the neisserial enzyme might impair the local mucosal immune function, although no direct in vivo evidence has been supplied so far (31). It has also been hypothesized that such bacterium-induced changes may be a primary event in the pathogenesis of certain inflammatory respiratory diseases and some forms of atopy (1, 35, 36).
The potential role(s) of the IgA1 proteases may extend beyond cleavage of IgA1. Several findings support the role of IgA1 protease in neisserial pathogenesis: its ability to cleave human LAMP1 (hLAMP1), a major integral membrane glycoprotein of lysosomes with an IgA1-like hinge in its luminal domain (3, 29, 43); its roles in gonococcal transepithelial trafficking in T84 monolayers (33) and in the inhibition of TNF-α-mediated apoptosis in human monocytic tumor cells (5); its exclusive association with human pathogens (49, 71, 74); and its ability to exhibit important immunomodulatory properties, in particular the induction of proinflammatory cytokines (46). Very recently, Senior et al. have suggested the hormone human chorionic gonadotropin as the target of the gonococcal IgA1 protease, with major implications for the invasiveness of the pathogen and for the fetus in pregnancy (63).
The immune response to neisserial infections is dominated by immunoglobulin-mediated effector functions, e.g., complement lysis, and most studies have focused on humoral immune responses (8, 18, 41, 56). However, antibody production by B cells against thymus-dependent antigens is regulated by T helper cells, which recognize specific peptide epitopes in the context of major histocompatibility complex (MHC) molecules. In this study we address the to-date-neglected aspect of T-cell responses to neisserial antigens in humans.
A number of neisserial components have been studied as possible vaccine candidates to date. Capsular polysaccharides (CPS), as well as improved CPS-tetanus toxoid conjugates, were thought to induce a CD4+ T-cell-dependent response and led to short-lived, strain-specific protection against serogroups A, C, W135, and Y. CPS did not induce protection against serogroup B meningococci, which cause disease in many countries worldwide (21, 22). Outer membrane proteins, including porin and Opa proteins, are the basis for outer membrane vesicle vaccines, and these are presently under intensive investigation (16, 52). Although data on T-cell recognition of neisserial outer membrane proteins are accumulating (73) and new antigens, such as the recently identified novel neisserial protein TspA (37) and the autotransporter A protein of N. meningitidis (2), have been described, no data are yet available on the T-cell response to neisserial IgA1 protease.
The identification and characterization of neisserial antigens are essential for the optimal design of vaccines. Here, we identify the neisserial IgA1 protease as a T-cell antigen. The IgA1 protease-induced T-cell response was characterized by flow cytometry, and the cytokine production pattern and cell activation status were determined. We report on the interaction of this enzyme with human peripheral blood mononuclear cells (PBMC) and discuss its relevance in infection.
MATERIALS AND METHODS
Purification and characterization of recombinant IgA1 protease.
In this study we used recombinant IgA1 protease expressed in Escherichia coli strain H2053 that harbors plasmid pJP11 containing the iga gene of N. gonorrhoeae MS11 with the temperature-inducible bacteriophage λ promoter PL, as described previously (46). Briefly, the supernatant of a 4-liter stationary-phase culture was recovered after centrifugation. The bacterial supernatant, containing the secreted recombinant IgA1 protease, was applied to a cation exchange column. After being washed, bound protein was eluted with 500 mM potassium phosphate. Fractions (10 ml) were collected and dialyzed into gel filtration buffer (20 mM TRIS [pH 8.0], 300 mM NaCl, 10% gycerol). Gel filtration was performed with a preequilibrated Superdex column (Pharmacia, Erlangen, Germany). Protein-containing eluates were detected by UV absorption, collected, dialyzed against phosphate-buffered saline (PBS) (pH 7.4), and then stored at −20°C. Purified protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and a single band was detected with subsequent Coomassie staining. The protein concentration was determined with a commercial protein determination assay (Bio-Rad, Munich, Germany). The cleavage activity of recombinant IgA1 protease was determined by incubation with human IgA1 (20 ng/μl; Dakopatts, Glostrup, Denmark) in Tris buffer (0.05 M Tris-HCl, 10 mM CaCl2, 10 mM MgCl2 [pH 7.5]) at 37°C according to the method described previously (29).
Endotoxin assays.
The endotoxin content of IgA1 protease preparations was measured by the Limulus amoebocyte lysate assay (Haemochrom Diagnostica, Essen, Germany) as described by the manufacturer. All preparations contained <15 pg of endotoxin per μg of protein.
Cells.
Healthy adult volunteers with no history of invasive neisserial disease were selected. PBMC were isolated from freshly drawn, heparinized blood by density gradient centrifugation over Ficoll-Isopaque (Pharmacia) and washed twice in Ca2+ Mg2+-free PBS. For some experiments, monocytes were separated by using a monocyte isolation kit (Miltenyi, Bergisch Gladbach, Germany). The purity of monocytes (>95% CD14 positive) was assessed by flow cytometry with a fluorochrome-conjugated anti-CD14 antibody (Coulter Immunotech, Krefeld, Germany).
Cell culture and stimulation of cytokine release.
All cell cultures were performed in RPMI 1640 supplemented with 2 mM L-glutamine, 500 U of penicillin/ml, 500 μg of streptomycin/ml, and 5% autologous human serum. Hereafter, this medium is designated as culture medium. A total of 105 cells were cultured in 200 μl of culture medium in round-bottom 96-well plates at 37°C and in 5% CO2. Cell-free supernatants were harvested at various times (100 μl/well and three to six wells per each experiment), transferred to round-bottom 96-well plates, and frozen at −20°C until assayed.
Cytokine ELISA.
The levels of cytokines released in the culture supernatants were quantified with commercially available sandwich enzyme-linked immunosorbent assay (ELISA) kits (BioSource, Solingen, Germany). The assays were performed as recommended by the manufacturer. In brief, polystyrene microtiter plates were coated with the capturing monoclonal antibody overnight, washed (PBS-0.02% Tween 20), and blocked with PBS-0.5% bovine serum albumin (BSA). Test samples and cytokine standards were diluted, 100 μl was added to wells in triplicate, and the mixtures were incubated at room temperature for 2 h. Next, the detecting monoclonal antibody was added, followed by washing and incubation with streptavidin-peroxidase conjugate for 20 min. After being washed, the chromogenic substrate (tetramethylbenzidine substrate with H2O2; Kierkegaard & Perry) was added to each well, and color development was stopped after 20 min by the addition of 50 μl of 1 M H2SO4. The absorbance of the samples was measured at 450 nm. Cytokine concentrations were determined by extrapolation from the standard curve. The results (in nanograms or picograms per milliliter) are expressed as the means of triplicate determinations. The detection limits of the ELISA kits were ≤10 pg/ml for all cytokines.
IFN-γ ELISPOT.
The number of cells that released gamma interferon (IFN-γ) upon activation was quantified with a commercially available pair of anti-IFN-γ-detecting antibodies (R&D, Wiesbaden, Germany). Enzyme-linked immunospot (ELISPOT) assays were performed as follows. ELISPOT filter plates (Millipore, Eschborn, Germany) were coated with monoclonal IFN-γ-capture antibody overnight, washed extensively with PBS, and blocked with PBS-0.5% BSA for 2 h. PBMC previously stimulated in culture plates were transferred to the ELISPOT filter plates for overnight incubation under standard tissue culture conditions. The plates were washed (PBS-0.05% Tween 20) to remove the cultured cells, and the detecting antibody was added. After being washed, alkaline phosphatase solution was added to each well, and the mixtures were incubated for 30 min. After further washing, alkaline phosphatase substrate (5-bromo-4-chloro-3-indolylphosphate-nitroblue tetrazolium) was added to each well, and color development of the spots was stopped after 15 min by being washed with water. The number of spots resulting from cells activated by previous stimulation was quantified with the spot counter BioReader (BioSys, Karben, Germany).
Flow cytometry.
The following antibody conjugates were used for flow cytometry: fluorescein isothiocyanate (FITC)-conjugated anti-IFN-γ (25723.11, IgG2b; R&D), anti-CD14 phycoerythrin (PE) as a monocytic marker (RMO52, IgG2a), anti-CD3 PE as a T-cell marker (UCHT1, IgG1), anti-CD4 PE as a T helper cell marker (13B8.2, IgG1), anti-CD56 PE as an NK cell marker (N901 [NKH1], IgG1), anti-CD19 PE as a B-cell marker (J4.119, IgG1), and anti-CD69 PE as an early activation marker (TP1.55.3, IgG2b) (all from Coulter Immunotech). Control mouse IgG was also obtained from Coulter Immunotech.
For detection of surface markers, cells were washed, adjusted to 2 × 105 cells/200 μl in PBS with 0.5% BSA, and incubated with the antibodies for 30 min at 4°C. For the detection of intracellular cytokines, 2 × 106 cells were washed and resuspended in 200 μl of Cytofix/Cytoperm solution (Becton Dickinson, Erembodegem-Aalst, Belgium) for fixation (30 min, 4°C). After being washed with Perm/Wash buffer (Becton Dickinson) to retain a permeable state, cells were adjusted to 2 × 105/200 μl in Perm/Wash buffer and incubated with the antibodies for 20 min at 4°C. After being washed, the samples were analyzed on a FACScalibur (Becton Dickinson) with the Cell Quest program. The cells were gated by their forward scatter-side scatter profile. Background levels of immunofluorescence were determined using isotype control IgG.
Confocal laser scanning microscopy.
For immunofluorescence staining we used mouse anti-hLAMP1 (H4A3; Developmental Studies, Hybridoma Bank, University of Iowa, Ames) as a marker for late phagolysosomes, goat anti-mouse Cy3 (Jackson ImmunoResearch Laboratories), and FITC-labeled IgA1 protease.
For detection of intracellular localization of IgA1 protease, CD14+ cells were purified from PBMC. Cells were incubated with FITC-labeled IgA1 protease for 1 h, washed, and fixed on a slide with 2% paraformaldehyde. After permeabilization with PBS-0.5 Triton X-100, cells were incubated with the anti-hLAMP1 antibody (dilution 1:30) for 45 min at 37°C, washed in PBS, and then incubated with secondary antibody for an additional 30 min. Slides were mounted with Mowiol (Merck, Darmstadt, Germany).
Proliferation assay.
Proliferation assays with PBMC (105 cells/well in culture medium) were performed at 37°C in 96-well U-bottom plates in a total of 200 μl per well. The cells were cultured for various times (1 to 10 days) and pulsed with 0.5 μCi of [3H]thymidine per well, followed by overnight incubation. Cells were then harvested on filters and the incorporation of [3H]thymidine (Amersham, Freiburg, Germany) was measured with a micro-beta scintillation counter (Wallac Instruments, Turku, Finland). Unstimulated cells and tetanus toxoid (TT; 2 limit of flocculation units [LF]/ml)-stimulated cells were used as controls.
RESULTS
Induction of T-cell proliferation in human PBMC by IgA1 protease.
To assess the antigenic potential of IgA1 protease, PBMC from three healthy human donors (donors 14, 26, and 27) were incubated with the purified enzyme. Proliferation was monitored for 10 days by using different concentrations of IgA1 protease. TT and medium alone were used as positive and negative controls, respectively. IgA1 protease induced proliferation of PBMC from all donors in a dose-dependent manner starting at day 5 and reached levels of 50,000 cpm during the first 10 days of stimulation, as indicated by [3H]thymidine incorporation (Fig. 1). PBMC stimulated with TT began to proliferate earlier (day 4) and reached a maximum of proliferation (up to 90,000 cpm) on day 7, after which proliferation decreased in all samples tested. Unstimulated cells showed proliferative responses of <1,000 cpm. Proliferation of PBMC in one donor was detectable with 0.1 μg of IgA1 protease/ml, whereas PBMC from the other two donors responded to concentrations higher than 1 or 10 μg/ml (Fig. 2).
FIG. 1.

Proliferation of human PBMC in response to IgA1 protease (0.1, 1, 10, and 20 μg/ml), TT (2 LF/ml), and medium alone. PBMC from donors 14, 26, and 27 were cultured with the indicated stimuli for 4, 5, 7, and 10 days. Filled symbols, different concentrations of IgA1 protease used in the experiment; open symbols, controls. The results (means ± standard deviations of six determinations) shown are from donor 27 as representative of the donors tested.
FIG. 2.

Proliferative responses to IgA1 protease of donors differ. The plot shows values from all three donors tested on day 10 of the experiment for which the results are shown in Fig. 1. Proliferation in response to IgA1 protease occurred at different concentrations of enzyme used, depending on the donor. Medium alone and TT (2 LF/ml) were used as controls (data not shown). Means ± standard deviations for six determinations are shown.
To test whether the observed proliferative response is a common feature, PBMC from five additional healthy human donors (donors 8, 12, 25, 42, and 68) were incubated with the purified enzyme (10 μg/ml) and proliferation was determined on day 7. The concentration of the IgA1 protease and the time point were adopted from previous experiments. Medium alone and TT were used as controls. IgA1 protease induced proliferation by PBMC from all donors tested; however, proliferative responses differed between donors, as indicated by various proliferation indices (Table 1). Proliferative responses from unstimulated cells were <1,000 cpm.
TABLE 1.
Proliferation of human PBMCs in response to IgA1 protease (10 μg/ml) and TT (2 LF/ml)
| Donor no. | Proliferation index fora:
|
|
|---|---|---|
| IgA1 protease | TT | |
| 14 | 74 | 200 |
| 26 | 23 | 103 |
| 27 | 132 | 235 |
| 12 | 20 | 76 |
| 25 | 31 | 45 |
| 68 | 10 | 34 |
| 8 | 12 | 43 |
| 42 | 27 | 153 |
Values are the proliferation indices for eight donors tested, representing the ratio between stimulus and medium control (mean values for six determinations).
To determine whether the observed proliferation was induced specifically by IgA1 protease and not by trace amounts of lipopolysaccharide (LPS) remaining in the preparation after purification from E. coli, BSA and LPS were used as controls (Fig. 3). For these and further experiments, two donors with high proliferation indices and one with a low proliferation index (donors 14, 26, and 27) were chosen. The endotoxin content present in the protease preparation of 10 μg/ml did not exceed 150 pg/ml, as determined by Limulus amebocyte lysate assay. Neither BSA (10 μg/ml) nor LPS (200 pg/ml) caused proliferation of PBMC, indicating that the proliferative response was an IgA1 protease-specific effect, although a potentiating effect of residual LPS cannot be excluded.
FIG. 3.

Specific proliferation of PBMC in response to IgA1 protease. To determine whether the proliferative response was IgA1 protease specific, BSA (10 μg/ml) and LPS (200 pg/ml) were used as controls along with medium alone, TT (2 LF/ml), and IgA1 protease (10 μg/ml). PBMC were cultured with the respective stimuli for 3, 5, and 7 days, and proliferation of cells was monitored by incorporation of [3H]thymidine. Results (means ± standard deviations for three determinations) shown here are from donor 14 as representative of the donors tested. The filled triangle accounts for the response to IgA1 protease, whereas the open symbols represent the stimuli which were used as controls.
IgA1 protease is taken up by human monocytes and intracellularly colocalizes with hLAMP1.
To further characterize the cellular interactions involved in this process, in vitro binding of IgA1 protease to different subpopulations of PBMC was investigated. Upon incubation of IgA1 protease with human PBMC, IgA1 protease associated with CD14+ cells (monocytes) within the first few minutes of incubation, a process that could be monitored with FITC-labeled IgA1 protease in flow cytometric analyses. IgA1 protease did not bind to B or T cells (Fig. 4A). Association of IgA1 protease with human monocytes could be prevented by applying the microfilament-disrupting macropinocytosis-blocking agent cytochalasin D or by performing the experiment at 4°C, suggesting that IgA1 protease is internalized by monocytes. These culture conditions modulated the binding pattern of FITC-conjugated BSA (10 μg/ml), which was used as control, in a similar manner (data not shown).
FIG. 4.

(A) IgA1 protease is associated with human CD14+ cells (monocytes). After isolation, PBMC were incubated with FITC-conjugated IgA1 protease (10 μg/ml) for 30 min at 37°C. The cells were washed and stained with anti-CD3 or anti-CD14 antibody to differentiate between cell subpopulations. The results for the interaction of IgA1 protease with monocytes (CD14+), T cells (CD3+) and non-T cells (CD3−) from donor 26 as representative of the donors tested are shown. (B) IgA1 protease colocalizes with hLAMP1 within lysosomal compartments of CD14+ monocytes. After isolation of PBMC from peripheral blood, monocytes were isolated. Cells were then incubated with FITC-labeled IgA1 protease and with anti-hLAMP1 antibody to mark the lysosomal compartments. The figure shows confocal images demonstrating the localization of IgA1 protease within lysosomal compartments of CD14+ monocytes (shown for donor 26 as representative of the donors tested). (I) IgA1 protease is shown in green; (II) lysosomal compartments are shown in red; (III) the overlay in yellow indicates colocalization.
After internalization, antigen-containing macrosolutes usually are concentrated and accumulate in a lysosomal compartment, which is characterized by the presence of hLAMP-1 and contains MHC class II molecules and proteases. To further track the IgA1 protease after uptake by the monocytes, purified CD14+ cells were incubated with FITC-labeled IgA1 protease and confocal laser scanning microscopy revealed the presence of the IgA1 protease within lysosomal compartments, as determined by colocalization with hLAMP-1 (Fig. 4B).
Incubation of PBMC with IgA1 protease results in activation of all lymphocyte subsets.
Next, we investigated whether incubation of PBMC with IgA1 protease has a stimulatory effect on CD19+ B cells, CD4+ and CD8+ T cells, and CD56+ NK cells. The activation status of the cells was determined by detection of CD69 on the cell surface using flow cytometry. Culture of cells with medium alone or with TT was included as a control. Incubation of human PBMC with IgA1 protease led to increased expression of CD69 on the cell surfaces of CD4+ and CD8+ T cells, as well as on B and NK cells (Fig. 5), in comparison to the medium control. TT induced a similar but more intense activation, as evidenced by a higher level of expression of CD69.
FIG. 5.

IgA1 protease leads to an upregulation of CD69 on the cell surfaces of lymphocytes. Human PBMC were incubated with IgA1 protease (10 μg/ml) overnight. Nonstimulated cells and TT-stimulated cells (2 LF/ml) were used as controls. Cells were harvested and stained with a monoclonal antibody against the early activation marker CD69 on the cell surface of T lymphocytes (CD4+ or CD8+), B lymphocytes (CD19+), or NK cells (CD56+) for flow cytometry. The results (donor 26 as representative of the donors tested) shown are the percentages of CD69+ cells of each cell subpopulation investigated.
Incubation of PBMC with IgA1 protease induces cytokine production.
Stimulation and proliferation of leukocytes are usually accompanied by cytokine production. To assess the profile and concentration of cytokines produced in response to IgA1 protease, supernatants from IgA1 protease-stimulated PBMC were collected at different times throughout the 10 days of cell culture, and IFN-γ, TNF-α, interleukin-10 (IL-10), and IL-4 were quantified by ELISA (Fig. 6). The levels of secreted IFN-γ were dose dependent and increased with time, reaching concentrations of up to 5 ng/ml. Cytokine levels in unstimulated cell cultures were below the limit of detection. Similar to the levels of IFN-γ, the levels of TNF-α increased in a dose-dependent manner. When low concentrations of IgA1 protease were used (1 and 10 μg/ml), IL-10 and IL-4, respectively, were detectable at low levels. IL-10 was detectable at day 3 of culture. IL-10 concentrations reached a maximum around day 4, decreased after day 6, and were no longer detectable on day 9. In contrast to IL-10, IL-4 was detected later (beginning at day 4) but, similarly to IL-10, decreased after day 6. Interestingly, the highest concentration of IgA1 protease used (20 μg/ml) completely suppressed IL-10 and IL-4, whereas no such effect was observed for IFN-γ or TNF-α.
FIG. 6.

Purified IgA1 protease induces dose-dependent release of the cytokines IFN-γ, TNF-α, IL-10, and IL-4 from PBMC. PBMC from donors 14, 26, and 27 were cultured with different concentrations of IgA1 protease. Cell-free supernatants were harvested, and cytokine levels were determined by ELISA. Filled symbols, different concentrations of IgA1 protease used in the experiment; open symbols, control nonstimulated cells (medium) and TT (2 LF/ml)-stimulated cells. Results (means ± standard deviations for six determinations) for donor 27 as representative of the donors tested are shown.
CD4+ T cells are the source of IFN-γ produced upon incubation of PBMC with IgA1 protease.
We demonstrated by confocal laser scanning microscopy that IgA1 protease is internalized by monocytes, probably via macropinocytosis, and is directed to lysosomal compartments. Antigen presentation via MHC class II molecules leads to the specific activation of CD4+ T cells and to their clonal expansion. On the other hand, other lymphocytic cell subsets become activated upon incubation with IgA1 protease, as indicated by increased expression of CD69. To determine the source of cytokine release, IFN-γ-producing cell subsets of human PBMC were identified by intracellular staining and flow cytometry. Intracellular cytokines were first detected after 6 days (Fig. 7A). The lymphocytes that stained positive for IFN-γ expressed the early activation marker CD69 (data not shown). Additional surface staining with either anti-CD4, anti-CD8, anti-CD56, or anti-CD19 revealed that the main producers of IFN-γ were CD4+ T cells (Fig. 7B).
FIG. 7.
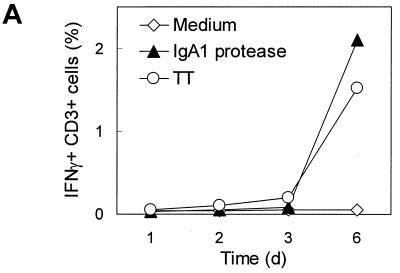
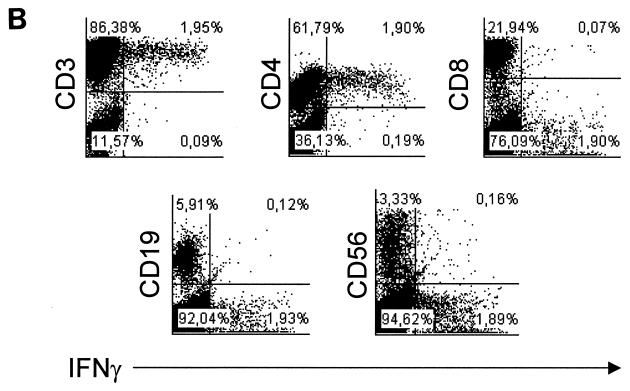
(A) IFN-γ production can be detected intracellularly by flow cytometry in CD3+ lymphocytes after stimulation of human PBMC with IgA1 protease. Human PBMC were cultured with IgA1 protease (10 μg/ml) for different lengths of time. Nonstimulated cells and TT-stimulated cells (2 LF/ml) were used as controls. After 1, 2, 3, and 6 days, cells were harvested and stained with monoclonal antibodies against CD3 on the cell surface and against IFN-γ intracellularly. The results (donor 27 as representative of the donors tested) are the percentage of double-positive cells. (B) CD4+ T cells are mainly responsible for the IFN-γ produced upon incubation of human PBMC with IgA1 protease. In the same experiment for which the results are shown in panel A, cells were harvested and stained with monoclonal antibodies against lymphocytic surface markers and IFN-γ intracellularly, of T lymphocytes (CD4+ or CD8+), B lymphocytes (CD19+), or NK cells (CD56+), respectively, for flow cytometry. The results are the percentages of double-positive cells of each cell subpopulation investigated.
To examine whether IFN-γ production depends on IgA1 protease processing and presentation in the context of MHC class II, the monoclonal antibody L243 was used to inhibit CD4+ T-cell activation by blocking the peptide-binding groove and thus interfering with MHC class II presentation. PBMC were cultured for 2 days with IgA1 protease in the presence or absence of the monoclonal anti-MHC class II antibody L243 or an isotype control. Unstimulated cells and TT-stimulated cells were treated in the same manner (data not shown). After 2 days of culture, cells were transferred to ELISPOT filter plates and cultured for another 18 h. The results show that blocking MHC class II presentation leads to decreased IFN-γ production, indicating that CD4+ T cells become specifically activated and are primarily responsible for the secretion of IFN-γ (Fig. 8). Similarly, TT induced IFN-γ production from CD4+ T cells, which also was inhibited by the use of L243 (data not shown). Unstimulated cells did not produce IFN-γ.
FIG. 8.

Production of IFN-γ by CD4+ T cells is inhibited by blocking antigen presentation via MHC class II molecules. Human PBMC were cultured for 2 days with IgA1 protease (10 μg/ml) in the presence or absence of the monoclonal anti-MHC class II antibody L243 (10 μg/ml) or an isotype control (10 μg/ml). Unstimulated cells and TT-stimulated cells were treated in the same manner (data not shown). The results (donor 14 as representative of the donors tested) are the means ± standard deviations representing the number of spots from three wells.
DISCUSSION
To assess the antigenic potential of neisserial IgA1 protease, we initially screened healthy volunteers (n = 8) for the presence of T cells that responded to IgA1 protease. We found that neisserial IgA1 protease could indeed induce proliferative responses in vitro and IFN-γ production by CD4+ T cells in a dose-dependent manner in all donors tested. Incubation of PBMC with IgA1 protease resulted in de novo expression of CD69 on CD4+ and CD8+ T cells, as well as CD19+ B cells and CD56+ NK cells. Together, these data suggest that neisserial IgA1 protease is a potent human CD4+ T-cell antigen, stimulating a Th1-type immune response in vitro.
PBMC from all tested donors showed in vitro proliferative responses to neisserial IgA1 protease accompanied by IFN-γ production, although to different extents. In addition, in subsequent experiments, donors with high proliferation indices elicited response patterns similar to those with low indices. The sera of all donors tested contained antibodies against IgA1 protease; however, the antibody levels did not correspond to the intensity of the proliferative responses observed. IgA1 protease-specific antibodies are stimulated by clinical infection as well as by asymptomatic carriage (9). Antibody titers against IgA1 protease are present at high levels in sera from acute- and convalescent-phase meningitis patients (19). This was also shown in vaccination trials where antibody titers remained constant or even increased over a 5-year period (67). Our findings may represent variations of a primary response or a memory response following carriage of pathogenic neisseriae (24) or Haemophilus influenzae, which expresses a closely related IgA1 protease (38, 45). The differences in stimulation observed with different donors may reflect HLA polymorphism or exposure to cross-reactive antigens of other organisms (23).
T cells recognizing the peptide-MHC complex on the surface of antigen-presenting cells (APC) through their T-cell receptor (TCR) become activated and proliferate. The activation can be monitored by the detection of specific activation markers expressed on the cell surface. An early activation marker, CD69, was chosen to demonstrate the activation status of CD4+ T cells upon incubation of PBMC with IgA1 protease. In contrast to the untreated control, CD69 could be detected on the cell surfaces of IgA1 protease-treated CD4+ T cells. Interestingly, several other cell types, including CD19+ B cells, CD8+ T cells, and CD56+ NK cells, were found to express CD69. The IL-2 receptor CD25, a late activation marker and indicator for proliferation, was also upregulated upon incubation of PBMC with IgA1 protease (data not shown).
It has been shown that exogenous protein antigens can be internalized (especially by dendritic cells as professional APCs) and presented to CD8+ T cells through MHC class I molecules by so-called cross-presentation (10, 40, 42). We found IFN-γ production associated with only CD4+ T cells. Neither B cells, CD8+ cells, nor NK cells participate in IFN-γ production, suggesting that the activation of CD8+ T cells seen in this study is a consequence of cytokine-mediated bystander events rather than through specific engagement of the TCR. It is also known that high concentrations of LPS (>100 ng/ml) can induce IFN-γ production and proliferation of NK cells and may lead to an activation of T cells, B cells, and monocytes (25, 32). Using nonpathogenic bacteria, NK cells were shown to be primary targets for LPS-induced proliferation, but neither activation nor proliferation of purified CD4+, CD8+, or CD19+ cells has been reported (28). In our study, IgA1 protease was produced as a recombinant protein in E. coli, and the preparations contained <15 pg of LPS/μg of protein. Equivalent amounts of purified LPS could neither induce proliferation nor stimulate IFN-γ production (data not shown).
Cytokines play an essential role in regulating immune responses. The presence of specific cytokines can influence the development of the immune response generated (Th1 or Th2) and can contribute to the virulence and putative clearance of a pathogen (26, 51). It has been described that during systemic meningococcal infection, high levels of proinflammatory cytokines are present in blood and CSF, contributing to severe tissue damage and facilitating brain infection by increasing permeability of the blood-brain barrier. In particular, TNF-α and IL-1β have been shown to be key mediators of meningococcal sepsis (17, 69, 72). These effects mediated by endotoxin may be potentiated by IgA1 protease itself, as previously demonstrated by our group (46). In this study, we found high levels of IFN-γ and the proinflammatory cytokine TNF-α in supernatants of PBMC cultured with IgA1 protease whereas the levels of IL-10 and IL-4 were very low, consistent with a Th1-like profile. IFN-γ, which is mainly induced by IL-12, is also detected in plasma of humans with bacterial meningitis (39). Nevertheless, the type of the immune response in bacterial meningitis is not yet clear. In some studies, type 1 cytokines were detected during bacterial meningitis (20, 62), whereas other studies report type 2 cytokines (61). Recent work describes age-dependent humoral as well as cellular immune responses to N. meningitidis (59, 60). Neonatal naive T cells respond differently to cytokine stimulation than do adult T cells. Neonatal CD45RA cells are induced to develop an IL-4 producing a Th2 phenotype (with production of IgG1 and IgG3 antibodies), whereas adult cells develop a Th1 phenotype (production of IgG2) following stimulation with IL-12 (66). These low levels of IgG2 in young children are thought to be an important factor reflected in the age-dependent incidence of disease, since a deficiency in IgG2 causes enhanced susceptibility to encapsulated bacteria, including meningococci (4, 64, 68). The relatively low levels of the Th2-type cytokines observed in this study could result from the increased levels of IFN-γ, which is known to inhibit IL-10 production (13), especially since the decrease of both cytokines, IL-10 and IL-4, correlates with the increase of IFN-γ at day 6 of culture. Interestingly, high concentrations of antigen (>20 μg/ml) resulted in a lack of cytokine production corresponding to the Th2 type, whereas it had no effect on IFN-γ production, indicating that a supression of a Th2 response may have occurred initially. IL-10, however, is crucial in limiting damage in bacterial meningitis by controlling TNF-α levels (54, 70). The reduction that we observed would promote infection rather than bacterial clearance.
This study demonstrates that healthy individuals generate a Th1-type-dominated immune response against gonococcal IgA1 protease in vitro and that IgA1 protease-specific T cells can be detected in peripheral blood. These T cells might have been locally stimulated and might further traffic to mucosal sites (14, 34). Extensive sharing of neutralizing epitopes was found between N. meningitidis and N. gonorrhoeae IgA1 proteases, making them potentially attractive vaccine components (44). Based on these homologies, the observed T-cell response to gonococcal IgA1 protease might also be relevant for the meningococcal enzyme in meningitis, where the occurrence of an inflammatory response with strong production of proinflammatory cytokines occurs frequently as a pathogenic factor. Although it is possible that Th2 type cytokines reduce inflammation through reduction of TNF-α by IL-10, it is also likely that a Th1-dominated immune response and subsequent production of IgG2 antibodies might have a protective effect in meningitis. Altogether, these findings invite further studies on the role of neisserial IgA1 protease as an immunologically important antigen.
Acknowledgments
We thank Frauke Kühl and Ralf Winter for technical assistance. We are also grateful to A. Walduck, T. Aebischer, and A. Szczepek for critically reading the manuscript.
This work was supported by a grant from the Fonds der Chemischen Industrie.
Editor: E. I. Tuomanen
REFERENCES
- 1.Ahl, T., and J. Reinholdt. 1991. Subclass distribution of salivary secretory immunoglobulin A antibodies to oral streptococci. Infect. Immun. 59: 3619–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ait-Tahar, K., K. G. Wooldridge, D. P. Turner, M. Atta, I. Todd, and D. A. Ala’Aldeen. 2000. Auto-transporter A protein of Neisseria meningitidis: a potent CD4+ T-cell and B-cell stimulating antigen detected by expression cloning. Mol. Microbiol. 37: 1094–1105. [DOI] [PubMed] [Google Scholar]
- 3.Ayala, P., L. Lin, S. Hopper, M. Fukuda, and M. So. 1998. Infection of epithelial cells by pathogenic neisseriae reduces the levels of multiple lysosomal constituents. Infect. Immun. 66: 5001–5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bass, J. L., R. Nuss, K. A. Mehta, P. Morganelli, and L. Bennett. 1983. Recurrent meningococcemia associated with IgG2-subclass deficiency. N. Engl. J. Med. 309: 430. [DOI] [PubMed] [Google Scholar]
- 5.Beck, S. C., and T. F. Meyer. 2000. IgA1 protease from Neisseria gonorrhoeae inhibits TNF alpha-mediated apoptosis of human monocytic cells. FEBS Lett. 472: 287–292. [DOI] [PubMed] [Google Scholar]
- 6.Bogdan, I., S. L. Leib, M. Bergeron, L. Chow, and M. G. Tauber. 1997. Tumor necrosis factor-alpha contributes to apoptosis in hippocampal neurons during experimental group B streptococcal meningitis. J. Infect. Dis. 176: 693–697. [DOI] [PubMed] [Google Scholar]
- 7.Brandtzaeg, P., P. Kierulf, P. Gaustad, A. Skulberg, J. N. Bruun, S. Halvorsen, and E. Sorensen. 1989. Plasma endotoxin as a predictor of multiple organ failure and death in systemic meningococcal disease. J. Infect. Dis. 159: 195–204. [DOI] [PubMed] [Google Scholar]
- 8.Brooks, G. F., and C. J. Lammel. 1989. Humoral immune response to gonococcal infections. Clin. Microbiol. Rev. 2(Suppl.): S5–S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks, G. F., C. J. Lammel, M. S. Blake, B. Kusecek, and M. Achtman. 1992. Antibodies against IgA1 protease are stimulated both by clinical disease and asymptomatic carriage of serogroup A Neisseria meningitidis. J. Infect. Dis. 166: 1316–1321. [DOI] [PubMed] [Google Scholar]
- 10.Brossart, P., and M. J. Bevan. 1997. Presentation of exogenous protein antigens on major histocompatibility complex class I molecules by dendritic cells: pathway of presentation and regulation by cytokines. Blood 90: 1594–1599. [PMC free article] [PubMed] [Google Scholar]
- 11.Cannon, J. G., and P. F. Sparling. 1984. The genetics of the gonococcus. Annu. Rev. Microbiol. 38: 111–133. [DOI] [PubMed] [Google Scholar]
- 12.Cartwright, K. A., and D. A. Ala’Aldeen. 1997. Neisseria meningitidis: clinical aspects. J. Infect. 34: 15–19. [DOI] [PubMed] [Google Scholar]
- 13.Chomarat, P., M. C. Rissoan, J. Banchereau, and P. Miossec. 1993. Interferon gamma inhibits interleukin 10 production by monocytes. J. Exp. Med. 177: 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davenport, M. P., M. C. Grimm, and A. R. Lloyd. 2000. A homing selection hypothesis for T-cell trafficking. Immunol. Today 21: 315–317. [DOI] [PubMed] [Google Scholar]
- 15.Dehio, C., S. D. Gray-Owen, and T. F. Meyer. 2000. Host cell invasion by pathogenic neisseriae. Subcell. Biochem. 33: 61–96. [DOI] [PubMed] [Google Scholar]
- 16.de Kleijn, E. D., R. de Groot, J. Labadie, A. B. Lafeber, G. van den Dobbelsteen, L. van Alphen, H. van Dijken, B. Kuipers, G. W. van Omme, M. Wala, R. Juttmann, and H. C. Rumke. 2000. Immunogenicity and safety of a hexavalent meningococcal outer-membrane-vesicle vaccine in children of 2–3 and 7–8 years of age. Vaccine 18: 1456–1466. [DOI] [PubMed] [Google Scholar]
- 17.de Kleijn, E. D., J. A. Hazelzet, R. F. Kornelisse, and R. de Groot. 1998. Pathophysiology of meningococcal sepsis in children. Eur. J. Pediatr. 157: 869–880. [DOI] [PubMed] [Google Scholar]
- 18.Densen, P. 1989. Interaction of complement with Neisseria meningitidis and Neisseria gonorrhoeae. Clin. Microbiol. Rev. 2(Suppl.): S11–S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devenyi, A. G., A. G. Plaut, F. J. Grundy, and A. Wright. 1993. Post-infectious human serum antibodies inhibit IgA1 proteinases by interaction with the cleavage site specificity determinant. Mol. Immunol. 30: 1243–1248. [DOI] [PubMed] [Google Scholar]
- 20.Diab, A., J. Zhu, L. Lindquist, B. Wretlind, M. Bakhiet, and H. Link. 1997. Haemophilus influenzae and Streptococcus pneumoniae induce different intracerebral mRNA cytokine patterns during the course of experimental bacterial meningitis. Clin. Exp. Immunol. 109: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finne, J., D. Bitter-Suermann, C. Goridis, and U. Finne. 1987. An IgG monoclonal antibody to group B meningococci cross-reacts with developmentally regulated polysialic acid units of glycoproteins in neural and extraneural tissues. J. Immunol. 138: 4402–4407. [PubMed] [Google Scholar]
- 22.Frasch, C. E. 1989. Vaccines for prevention of meningococcal disease. Clin. Microbiol. Rev. 2(Suppl.): S134–S138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glode, M. P., J. B. Robbins, T. Y. Liu, E. C. Gotschlich, I. Orskov, and F. Orskov. 1977. Cross-antigenicity and immunogenicity between capsular polysaccharides of group C Neisseria meningitidis and of Escherichia coli K92. J. Infect. Dis. 135: 94–104. [DOI] [PubMed] [Google Scholar]
- 24.Goldschneider, I., E. C. Gotschlich, and M. S. Artenstein. 1969. Human immunity to the meningococcus. II. Development of natural immunity. J. Exp. Med. 129: 1327–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodier, M. R., and M. Londei. 2000. Lipopolysaccharide stimulates the proliferation of human CD56+CD3− NK cells: a regulatory role of monocytes and IL-10. J. Immunol. 165: 139–147. [DOI] [PubMed] [Google Scholar]
- 26.Guler, M. L., J. D. Gorham, C. S. Hsieh, A. J. Mackey, R. G. Steen, W. F. Dietrich, and K. M. Murphy. 1996. Genetic susceptibility to Leishmania: IL-12 responsiveness in TH1 cell development. Science 271: 984–987. [DOI] [PubMed] [Google Scholar]
- 27.Haines, K. A., J. Reibman, X. Y. Tang, M. Blake, and G. Weissmann. 1991. Effects of protein I of Neisseria gonorrhoeae on neutrophil activation: generation of diacylglycerol from phosphatidylcholine via a specific phospholipase C is associated with exocytosis. J. Cell Biol. 114: 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haller, D., S. Blum, C. Bode, W. P. Hammes, and E. J. Schiffrin. 2000. Activation of human peripheral blood mononuclear cells by nonpathogenic bacteria in vitro: evidence of NK cells as primary targets. Infect. Immun. 68: 752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hauck, C. R., and T. F. Meyer. 1997. The lysosomal/phagosomal membrane protein h-lamp-1 is a target of the IgA1 protease of Neisseria gonorrhoeae. FEBS Lett. 405: 86–90. [DOI] [PubMed] [Google Scholar]
- 30.Hazelzet, J. A., E. van de Voort, J. Lindemans, P. G. ter Heerdt, and H. J. Neijens. 1994. Relation between cytokines and routine laboratory data in children with septic shock and purpura. Intensive Care Med. 20: 371–374. [DOI] [PubMed] [Google Scholar]
- 31.Hedges, S. R., M. S. Mayo, L. Kallman, J. Mestecky, E. W. Hook III, and M. W. Russell. 1998. Evaluation of immunoglobulin A1 (IgA1) protease and IgA1 protease-inhibitory activity in human female genital infection with Neisseria gonorrhoeae. Infect. Immun. 66: 5826–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinzelmann, M., H. C. Polk, A. Chernobelsky, T. P. Stites, and L. E. Gordon. 2000. Endotoxin and muramyl dipeptide modulate surface receptor expression on human mononuclear cells. Immunopharmacology 48: 117–128. [DOI] [PubMed] [Google Scholar]
- 33.Hopper, S., B. Vasquez, A. Merz, S. Clary, J. S. Wilbur, and M. So. 2000. Effects of the immunoglobulin A1 protease on Neisseria gonorrhoeae trafficking across polarized T84 epithelial monolayers. Infect. Immun. 68: 906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kantele, A., J. Zivny, M. Hakkinen, C. O. Elson, and J. Mestecky. 1999. Differential homing commitments of antigen-specific T cells after oral or parenteral immunization in humans. J. Immunol. 162: 5173–5177. [PubMed] [Google Scholar]
- 35.Kilian, M., J. Reinholdt, H. Lomholt, K. Poulsen, and E. V. Frandsen. 1996. Biological significance of IgA1 proteases in bacterial colonization and pathogenesis: critical evaluation of experimental evidence. APMIS 104: 321–338. [DOI] [PubMed] [Google Scholar]
- 36.Kilian, M., J. Reinholdt, S. B. Mortensen, and C. H. Sorensen. 1983. Perturbation of mucosal immune defence mechanisms by bacterial IgA proteases. Bull. Eur. Physiopathol. Respir. 19: 99–104. [PubMed] [Google Scholar]
- 37.Kizil, G., I. Todd, M. Atta, S. P. Borriello, K. Ait-Tahar, and D. A. Ala’Aldeen. 1999. Identification and characterization of TspA, a major CD4(+) T-cell- and B-cell-stimulating Neisseria-specific antigen. Infect. Immun. 67: 3533–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klauser, T., J. Kramer, K. Otzelberger, J. Pohlner, and T. F. Meyer. 1993. Characterization of the Neisseria Iga beta-core. The essential unit for outer membrane targeting and extracellular protein secretion. J. Mol. Biol. 234: 579–593. [DOI] [PubMed] [Google Scholar]
- 39.Kornelisse, R. F., C. E. Hack, H. F. Savelkoul, T. C. van der Pouw Kraan, W. C. Hop, G. van Mierlo, M. H. Suur, H. J. Neijens, and R. de Groot. 1997. Intrathecal production of interleukin-12 and gamma interferon in patients with bacterial meningitis. Infect. Immun. 65: 877–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurts, C. 2000. Cross-presentation: inducing CD8 T cell immunity and tolerance. J. Mol. Med. 78: 326–332. [DOI] [PubMed] [Google Scholar]
- 41.Lammel, C. J., R. L. Sweet, P. A. Rice, J. S. Knapp, G. K. Schoolnik, D. C. Heilbron, and G. F. Brooks. 1985. Antibody-antigen specificity in the immune response to infection with Neisseria gonorrhoeae. J. Infect. Dis. 152: 990–1001. [DOI] [PubMed] [Google Scholar]
- 42.Limmer, A., J. Ohl, C. Kurts, H. G. Ljunggren, Y. Reiss, M. Groettrup, F. Momburg, B. Arnold, and P. A. Knolle. 2000. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 6: 1348–1354. [DOI] [PubMed] [Google Scholar]
- 43.Lin, L., P. Ayala, J. Larson, M. Mulks, M. Fukuda, S. R. Carlsson, C. Enns, and M. So. 1997. The Neisseria type 2 IgA1 protease cleaves LAMP1 and promotes survival of bacteria within epithelial cells. Mol. Microbiol. 24: 1083–1094. [DOI] [PubMed] [Google Scholar]
- 44.Lomholt, H., and M. Kilian. 1994. Antigenic relationships among immunoglobulin A1 proteases from Haemophilus, Neisseria, and Streptococcus species. Infect. Immun. 62: 3178–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lomholt, H., K. Poulsen, and M. Kilian. 1995. Comparative characterization of the iga gene encoding IgA1 protease in Neisseria meningitidis, Neisseria gonorrhoeae and Haemophilus influenzae. Mol. Microbiol. 15: 495–506. [DOI] [PubMed] [Google Scholar]
- 46.Lorenzen, D. R., F. Dux, U. Wolk, A. Tsirpouchtsidis, G. Haas, and T. F. Meyer. 1999. Immunoglobulin A1 protease, an exoenzyme of pathogenic neisseriae, is a potent inducer of proinflammatory cytokines. J. Exp. Med. 190: 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Massari, P., Y. Ho, and L. M. Wetzler. 2000. Neisseria meningitidis porin PorB interacts with mitochondria and protects cells from apoptosis. Proc. Natl. Acad. Sci. USA 97: 9070–9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer, T. F., J. Pohlner, and J. P. van Putten. 1994. Biology of the pathogenic neisseriae. Curr. Top. Microbiol. Immunol. 192: 283–317. [DOI] [PubMed] [Google Scholar]
- 49.Mulks, M. H., and A. G. Plaut. 1978. IgA protease production as a characteristic distinguishing pathogenic from harmless Neisseriaceae. N. Engl. J. Med. 299: 973–976. [DOI] [PubMed] [Google Scholar]
- 50.Muller, A., D. Gunther, F. Dux, M. Naumann, T. F. Meyer, and T. Rudel. 1999. Neisserial porin (PorB) causes rapid calcium influx in target cells and induces apoptosis by the activation of cysteine proteases. EMBO J. 18: 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nabors, G. S., and J. P. Farrell. 1994. Depletion of interleukin-4 in BALB/c mice with established Leishmania major infections increases the efficacy of antimony therapy and promotes Th1-like responses. Infect. Immun. 62: 5498–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naess, L. M., F. Oftung, A. Aase, L. M. Wetzler, R. Sandin, and T. E. Michaelsen. 1998. Human T-cell responses after vaccination with the Norwegian group B meningococcal outer membrane vesicle vaccine. Infect. Immun. 66: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nassif, X., J. C. Mathison, E. Wolfson, J. A. Koziol, R. J. Ulevitch, and M. So. 1992. Tumour necrosis factor alpha antibody protects against lethal meningococcaemia. Mol. Microbiol. 6: 591–597. [DOI] [PubMed] [Google Scholar]
- 54.Paris, M. M., S. M. Hickey, M. Trujillo, A. Ahmed, K. Olsen, and G. H. McCracken. 1997. The effect of interleukin-10 on meningeal inflammation in experimental bacterial meningitis. J. Infect. Dis. 176: 1239–1246. [DOI] [PubMed] [Google Scholar]
- 55.Plaut, A. G., J. V. Gilbert, M. S. Artenstein, and J. D. Capra. 1975. Neisseria gonorrhoeae and Neisseria meningitidis: extracellular enzyme cleaves human immunoglobulin A. Science 190: 1103–1105. [DOI] [PubMed] [Google Scholar]
- 56.Plummer, F. A., J. N. Simonsen, H. Chubb, L. Slaney, J. Kimata, M. Bosire, J. O. Ndinya-Achola, and E. N. Ngugi. 1989. Epidemiologic evidence for the development of serovar-specific immunity after gonococcal infection. J. Clin. Invest 83: 1472–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pohlner, J., R. Halter, K. Beyreuther, and T. F. Meyer. 1987. Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature 325: 458–462. [DOI] [PubMed] [Google Scholar]
- 58.Pohlner, J., T. Klauser, E. Kuttler, and R. Halter. 1992. Sequence-specific cleavage of protein fusions using a recombinant Neisseria type 2 IgA protease. Bio/Technology 10: 799–804. [DOI] [PubMed] [Google Scholar]
- 59.Pollard, A. J., R. Galassini, E. M. Rouppe van der Voort, M. Hibberd, R. Booy, P. Langford, S. Nadel, C. Ison, J. S. Kroll, J. Poolman, and M. Levin. 1999. Cellular immune responses to Neisseria meningitidis in children. Infect. Immun. 67: 2452–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pollard, A. J., R. Galassini, E. M. Rouppe van der Voort, R. Booy, P. Langford, S. Nadel, C. Ison, J. S. Kroll, J. Poolman, and M. Levin. 1999. Humoral immune responses to Neisseria meningitidis in children. Infect. Immun. 67: 2441–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raziuddin, S., M. E. el Awad, A. W. Telmesani, N. E. Bilal, and M. al Janadi. 1995. CD4+ Th2 cell response cytokine production in bacterial meningitis. J. Clin. Immunol. 15: 338–348. [DOI] [PubMed] [Google Scholar]
- 62.Raziuddin, S., N. A. Mir, M. E. el Awad, A. W. Telmesani, and M. al Janadi. 1994. Gamma delta T lymphocytes and proinflammatory cytokines in bacterial meningitis. J. Allergy Clin. Immunol. 93: 793–798. [DOI] [PubMed] [Google Scholar]
- 63.Senior, B. W., W. W. Stewart, C. Galloway, and M. A. Kerr. 2001. Cleavage of the hormone human chorionic gonadotropin, by the type 1 IgA1 protease of Neisseria gonorrhoeae, and its implications. J. Infect. Dis. 184: 922–925. [DOI] [PubMed] [Google Scholar]
- 64.Shackelford, P. G., D. M. Granoff, S. H. Polmar, M. G. Scott, M. C. Goskowicz, J. V. Madassery, and M. H. Nahm. 1990. Subnormal serum concentrations of IgG2 in children with frequent infections associated with varied patterns of immunologic dysfunction. J. Pediatr. 116: 529–538. [DOI] [PubMed] [Google Scholar]
- 65.Sharief, M. K., M. Ciardi, and E. J. Thompson. 1992. Blood-brain barrier damage in patients with bacterial meningitis: association with tumor necrosis factor-alpha but not interleukin-1 beta. J. Infect. Dis. 166: 350–358. [DOI] [PubMed] [Google Scholar]
- 66.Shu, U., C. E. Demeure, D. G. Byun, F. Podlaski, A. S. Stern, and G. Delespesse. 1994. Interleukin 12 exerts a differential effect on the maturation of neonatal and adult human CD. J. Clin. Investig. 94: 1352–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thiesen, B., B. Greenwood, N. Brieske, and M. Achtman. 1997. Persistence of antibodies to meningococcal IgA1 protease versus decay of antibodies to group A polysaccharide and Opc protein. Vaccine 15: 209–219. [DOI] [PubMed] [Google Scholar]
- 68.Trollfors, B., T. Lagergard, B. A. Claesson, E. Thornberg, J. Martinell, and R. Schneerson. 1992. Characterization of the serum antibody response to the capsular polysaccharide of Haemophilus influenzae type b in children with invasive infections. J. Infect. Dis. 166: 1335–1339. [DOI] [PubMed] [Google Scholar]
- 69.van Furth, A. M., J. J. Roord, and R. van Furth. 1996. Roles of proinflammatory and anti-inflammatory cytokines in pathophysiology of bacterial meningitis and effect of adjunctive therapy. Infect. Immun. 64: 4883–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Furth, A. M., E. M. Seijmonsbergen, J. A. Langermans, P. H. Groeneveld, C. E. de Bel, and R. van Furth. 1995. High levels of interleukin 10 and tumor necrosis factor alpha in cerebrospinal fluid during the onset of bacterial meningitis. Clin. Infect. Dis. 21: 220–222. [DOI] [PubMed] [Google Scholar]
- 71.Vitovski, S., R. C. Read, and J. R. Sayers. 1999. Invasive isolates of Neisseria meningitidis possess enhanced immunoglobulin A1 protease activity compared to colonizing strains. FASEB J. 13: 331–337. [DOI] [PubMed] [Google Scholar]
- 72.Waage, A., P. Brandtzaeg, A. Halstensen, P. Kierulf, and T. Espevik. 1989. The complex pattern of cytokines in serum from patients with meningococcal septic shock: association between interleukin 6, interleukin 1, and fatal outcome. J. Exp. Med. 169: 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wiertz, E. J., A. Delvig, E. M. Donders, H. F. Brugghe, L. M. van Unen, H. A. Timmermans, M. Achtman, P. Hoogerhout, and J. T. Poolman. 1996. T-cell responses to outer membrane proteins of Neisseria meningitidis: comparative study of the Opa, Opc, and PorA proteins. Infect. Immun. 64: 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wolff, K., and A. Stern. 1995. Identification and characterization of specific sequences encoding pathogenicity associated proteins in the genome of commensal Neisseria species. FEMS Microbiol. Lett. 125: 255–263. [DOI] [PubMed] [Google Scholar]
- 75.Zenilman, J. M. 1993. Gonorrhea: clinical and public health issues. Hosp. Pract. 28: 29–40, 43. [DOI] [PubMed] [Google Scholar]