

Abstract
The objective of this study was to determine (i) if complementation of ureB-negative Helicobacter pylori restores colonization and (ii) if urease is a useful reporter for promoter activity in vivo. Strains used were M6, M6ΔureB, and 10 recombinant derivatives of M6 or M6ΔureB in which urease expression was under the control of different H. pylori promoters. Mice were orally inoculated with either the wild type or one of the mutant strains, and colonization, in vivo urease activity, and extent of gastritis were determined. Of eight M6ΔureB recombinants tested, four colonized mice. Of those, three had the highest in vitro urease activity of any of the recombinants, significantly different from that of the noncolonizing mutants. The fourth colonizing recombinant, with ureB under control of the cag-15 promoter, had in vitro urease activity which did not differ significantly from the noncolonizing strains. In vivo, urease activities of the four colonizing transformants and the wild-type control were indistinguishable. There were no differences in gastritis or epithelial lesions between mice infected with M6 and those infected with the transformants. These results demonstrate that recovery of urease activity can restore colonizing ability to urease-negative H. pylori. They also suggest that cag-15 is upregulated in vivo, as was previously suggested by demonstrating that it is upregulated upon contact with epithelial cells. Finally, our results suggest that total urease activity and colonization density do not contribute to gastritis due to H. pylori.
The link between Helicobacter pylori and peptic ulcer, first recognized by Barry Marshall and Robin Warren in 1982 (23, 24), provided major insight into human gastric pathology. In its most severe manifestations H. pylori can be responsible for gastric disease ranging from peptic ulcer to gastric cancer (24, 29, 30). Infection with H. pylori is common worldwide. Colonization ranges from 50 to 100%, making H. pylori the most common infectious agent of humans in the world today.
Because of the prevalence and importance of H. pylori infection, understanding the mechanisms by which it colonizes the gastric mucosa and causes disease has received intense interest. H. pylori possesses several putative colonization factors, including urease (23, 24), various adhesins (13, 14, 28, 35), and flagellar motility (19, 23), some of which have been shown to be necessary for gastric colonization (1, 2, 5, 11, 36). Of these factors, urease was the first described and is probably most widely studied. Strong urease activity was noted in the initial description of H. pylori (23), and subsequent studies have implicated urease activity as an important virulence factor. Urease accounts for about 5% of H. pylori protein (18) and is consistently present in all naturally occurring strains. Further, genetically engineered urease-deficient H. pylori is unable to colonize either germfree piglets (5), ferrets (2), or mice (36). It is thought that urease may contribute to gastric damage due to H. pylori physical injury from ammonia (4, 21, 26), inflammation due to a host immune response to the protein (12, 16), or other means of mucosal damage (33, 34). Taken together, these data indicate an important role for urease both for promoting colonization and in the pathogenesis of gastric disease.
Urease is a nickel metalloenzyme that catalyzes the hydrolysis of urea to ammonia and carbon dioxide. Synthesis of active urease by H. pylori requires the presence of the structural genes ureA and ureB, which associate to form the 550-kDa holoenzyme (18, 22), and the accessory genes, ureIEFGH, which are necessary for full expression of urease activity (3). In addition, nickel transport enzymes such as NixA (27) are required for full expression of urease activity. Animal experiments with urease deletion mutants have demonstrated that ureB, ureG, and ureI are all necessary for gastric colonization (2, 5, 32, 36).
One limitation of H. pylori genetic deletion studies is that, until recently, in vivo genetic complementation has not been possible. Shuttle plasmids that allow expression of genes in H. pylori have been described (17), but these tend to be strain specific and difficult or impossible to maintain in animal-virulent H. pylori strains (unpublished observations). Thus, evaluation of the role of colonization and virulence factors has depended on experiments demonstrating loss of virulence due to loss of the factor to be tested. Here we show that a transcriptional reporter system which utilizes urease production as a measure of gene expression (20) complements a urease-negative, ureB-null mutant strain and restores both urease activity and the ability to colonize mice. This confirms the essential nature of urease activity for colonization and illustrates that H. pylori ureB is an excellent reporter of in vivo gene expression in H. pylori.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
For mouse inoculation experiments H. pylori strain M6 (kindly provided by Steven Czinn) was used. This strain was originally isolated from a human patient, colonizes mice, and is easily transformed in vitro. Table 1 summarizes the constructs and terminology used in this report. Construction of the mutants has been previously described in detail (20). M6ΔureB, a deletion mutant of M6 that fails to express urease activity, contains a kanamycin resistance cassette (aph3′III) that replaces all but the 35 N-terminal nucleotides of the ureB coding sequence (20). A series of seven M6ΔureB derivatives was constructed to contain cag-ureB fusions in which ureB expression was under control of putative promoters derived from the cag pathogenicity island. Fusions were integrated within a putative noncoding region 55 bp downstream of hpn, a nickel-binding protein (15). A transcriptional terminator sequence was integrated just upstream of the cag promoter sequence to block hpn-mediated ureB expression. M6ΔureB(hpn-ureB) is a derivative of M6ΔureB in which ureB was integrated within hpn, inactivating that gene and placing the recombinant ureB under control of the hpn promoter. M6(cag9-ureB) and M6(cag25-ureB) are M6 derivatives in which the native ureB was intact but cag-ureB fusions were integrated downstream of hpn as described above. The last two strains were included as colonization controls. They express normal urease activity but colonize at a density comparable to that of the urease mutants (about 1 to 4% of M6; see below). Therefore, in vivo urease activity in these strains could be compared directly with in vivo urease activity of the mutants without regard to differences in bacterial colonization density.
TABLE 1.
H. pylori urease mutants used in this study
| Strain | Analogous strain used for in vitro evaluationa | Genotype or description |
|---|---|---|
| M6 | C57 | Wild type |
| M6ΔureB | 412 | Replacement of the ureB open reading frame with a kanamycin resistance cassette (ΔureB::kan) |
| M6(cagx-ureB) | NAb | M6{hpn::[Φ(Pcagx-ureBcat)]}, wild-type strain M6 with the recombinant cag-ureB fusion integrated 55 bp downstream of the hpn stop codon; x denotes the specific cag promoter region; cag promoters used were cag9 and cag25 |
| M6ΔureB(cagx-ureB) | 585(cagx-ureB) | M6ΔureB{hpn::[Φ(Pcagx-ureBcat)]}, M6ΔureB with the recombinant cag-ureB fusion integrated 55 bp downstream of the hpn stop codon; x denotes the specific cag promoter region; cag genes used were cag-1, -9, -13, -14, -15, -21, and -25 |
| M6ΔureB(hpn-ureB) | 472 | M6ΔureB{hpn::[Φ(hpn-ureBcat)]}, M6ΔureB with the recombinant cag-ureB fusion integrated within the coding region of hpn |
Detailed descriptions of the mutant constructs and their characteristics in laboratory-passaged H. pylori strain C57 have been published (20).
NA, not applicable.
Mutant constructs were transferred to mouse virulent M6 by natural transformation. For this, chromosomal or plasmid DNA was isolated by lysis and differential solubilization with a commercially available kit (Qiagen). The recipient strain, H. pylori M6 or M6ΔureB, was grown overnight at 37°C in Brucella broth with 10% fetal calf serum in a microaerobic environment with gentle agitation. When bacteria reached mid-log-phase growth (approximately 108 to 109 CFU/ml), they were diluted 1:100 into 10 ml of fresh Brucella broth. Broths were incubated for 0 to 6 h, approximately 20 to 50 ng of donor DNA was added, and incubation was continued overnight. Each 10-ml broth was then diluted 1:4 in Brucella broth containing selective antibiotics (kanamycin, chloramphenicol, or both) (20 μg/ml) as appropriate. After another overnight incubation, 1-ml aliquots were plated on 5% sheep blood agar plates containing 20 μg of antibiotics per ml as described above. After 4 to 5 days 1 to 500 colonies were visible. Colonies were pooled, plated on selective agar plates as described above, and stored at −70°C in Brucella broth with 15% glycerol until mouse inoculation. Proper insertion of the constructs was confirmed by PCR and verified by urease test before and after animal challenge. Primers used to verify correct insertion of constructs are shown in Table 2. PCR was done by routine methods, and correct insertion was inferred by amplification of a band of the expected size. All of the cag-ureB recombinant strains used in animals were derived from the same M6ΔureB isolate by transformation with the relevant DNA constructs. To ensure consistency of colonization data, all transformations were performed at least twice and pools from each transformation were used in animal inoculation.
TABLE 2.
Primers used to verify cag-ureB fusions
| Gene | Direction | Sequence |
|---|---|---|
| ureB | Reverse | TCT AGA GCC TGT AGT AGG ACC ATA CA |
| cag-15 | Forward | ACA CCC ATT TGA AGC AAA GC |
| cag-13 | Forward | GAT ATG GCT TGT TTG GTG GC |
| cag-21 | Forward | ATC AGG CCT TGA GGC AAA TG |
| cag-14 | Forward | TCG TTG GTT TGT GCT ATA CC |
| cag-1 | Forward | TTC ATA GCC AAA TTC TGC GG |
| cag-25 | Forward | ATT GGT TGT TAC CAC TAG CC |
| cag-9 | Forward | CTT TGC TCA ACA CCT TAT CC |
| hpn | Forward | CGA GGA TCC GTT GGT TTT AAT CAA GCG |
Urease testing.
For quantitation of urease in vitro, the phenol-hypochlorite method was used (25, 37). Briefly, bacteria grown on plates or in Brucella broth were resuspended in 50 mM HEPES, pH 7.5, and diluted (1:20 to 1:1,000, depending on urease activity) into 50 mM HEPES containing 25 mM urea. The dilutions were chosen to ensure that urease activity would be linear for at least 20 min. Suspensions were incubated at 37°C for 20 min, and phenol-nitroprusside solution and hypochlorite solution were added in sequence. The reactions were incubated for 30 min at 37°C and A625 was determined. Ammonia was quantified using a standard curve. Urease activity was expressed as nanomoles of ammonia produced per microgram of bacterial protein per minute. Bacterial protein concentration was determined by the Lowry method using a commercially available kit (Sigma). The urease activity of each mutant was based on the average of five to eight separate assays performed with different bacterial preparations on different days.
For quantitation of urease activity in vivo, a modification of the more sensitive ammonia assay kit (Boehringer-Mannheim) was used as previously described (5). Ammonia determination is based on the following linked reactions:
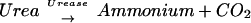 |
 |
The amount of ammonia produced by urease is proportional to the amount of NAD+ produced. The assay was modified such that glutamate dehydrogenase was in excess and urease activity was the rate-limiting step (5). Thus, the urease activity was proportional to the rate of NAD+ production and was measured by a change in optical density at 340 nm. To evaluate urease activity in stomach tissue, gastric mucosal homogenate was washed twice in 50 mM PIPES (piperazine-N,N"-biz(2-ethanesulfonic acid) buffer, pH 6.8, containing a proteinase inhibitor cocktail (Roche) (1 tablet per ml). The homogenate was resuspended in PIPES and urea was added to a final concentration of 200 μM. Samples were incubated at room temperature for 120 min, and the homogenate was removed by sedimentation in a microcentrifuge. Ammonia concentration in the supernatant was measured by enzymatic assay as described above. Controls contained homogenate with buffer alone (no urea), urea with buffer alone (no homogenate), and uninfected homogenate with buffer (no bacteria). Tissue urease was measured in mice killed 2 weeks after inoculation.
Animals.
Female 4- to 6-week-old Helicobacter-free C57BL/6 and C57BL/6-Prkdcscid (severe combined immunodeficient [SCID]) mice were purchased from Jackson laboratory and were maintained in microisolator cages and fed sterile lab chow and water ad libitum. Germfree 129RAGI−/− (recombinase activating gene I knockout [RAG-KO]) mice came from our own colony. C57BL/6 mice and germfree mice were orally inoculated with 107 CFU of broth-cultured H. pylori and killed 2 or 8 weeks after inoculation. SCID mice were inoculated as described above, reconstituted with congenic splenocytes 2 weeks after inoculation, and killed 6 weeks after transfer. RAG-KO mice support high-density colonization by H. pylori and were used to determine the in vivo urease level (10). Reconstituted SCID mice develop rapidly progressive severe gastritis in response to infection with H. pylori (9, 10), and this model was used to determine the role of urease and bacterial density in eliciting these lesions. The number of animals in each group is given in Table 3.
TABLE 3.
Colonization by M6 strains and mutants
| Bacterial strain | No. of mice colonized/total in each inoculation group
|
|||
|---|---|---|---|---|
| C57BL/6 mice killed 2 wks after inoculation | Germfree RAG-KO mice | C57BL/6 mice killed 8 wks after inoculation | Reconstituted SCID mice | |
| M6 | 10/10 | 5/5 | 11/1 | 2/5 |
| M6ΔureB | 0/3 | 0/4 | 0/5 | NDa |
| M6 transformants | ||||
| M6(cag9-ureB) | 5/5 | 4/4 | ND | ND |
| M6(cag25-ureB) | 5/5 | ND | ND | ND |
| M6ΔureB transformants | ||||
| M6ΔureB(hpn-ureB) | ND | 6/6 | ND | ND |
| M6ΔureB(cag1-ureB) | 11/13 | 5/5 | 5/5 | 2/5 |
| M6ΔureB(cag9-ureB) | 0/8 | 0/5 | ND | ND |
| M6ΔureB(cag13-ureB) | 0/8 | ND | ND | ND |
| M6ΔureB(cag14(ureB) | 0/8 | ND | ND | ND |
| M6ΔureB(cag15-ureB) | 11/12 | 5/5 | 4/4 | ND |
| M6ΔureB(cag21-ureB) | 0/12 | ND | ND | ND |
| M6ΔureB(cag25-ureB) | 8/8 | 5/5 | ND | ND |
ND, not done.
For animal challenge, bacteria were grown in Brucella broth with 10% fetal calf serum with selective antibiotics as appropriate. Animals were inoculated by oral gavage with 107 CFU of mid-log-phase bacteria (about 108 to 109 CFU of rapidly motile organisms per ml). At sacrifice, stomachs were removed and divided in half longitudinally. One half of each stomach was homogenized in Brucella broth, colonization was determined by plate dilution, and the remainder of the homogenate was used for tissue urease determination. The other half of each stomach from C57BL/6 mice killed 8 weeks after inoculation and from recipient SCID mice was immersed in 10% neutral buffered formalin and embedded in paraffin for histologic examination. Six-micrometer hematoxylin-and-eosin-stained sections were examined and gastritis, neutrophilic infiltration, polymorphonuclear leukocytes (PMN), and metaplasia were quantified as previously described (10). Because gastritis takes at least 8 weeks to develop in H. pylori-infected C57BL/6 mice, mice killed 2 weeks after inoculation were not examined histologically.
Statistics.
Means were compared by Student's t test or Fisher's protected least significant difference to correct for multiple comparisons. Error bars in graphs indicate standard errors of the means.
RESULTS
Urease activity of bacterial mutants in vitro.
Urease activity of the bacterial strains in vitro is shown in Fig. 1. The activities of strains with wild-type urease activity [M6, M6(cag9-ureB), and M6(cag25-ureB)] were not significantly different from each other or from M6ΔureB(hpn-ureB). Comparison by pairwise t test indicated that the urease activity of all the strains was significantly greater than that of the urease-negative strain M6ΔureB. However, correction for multiple comparisons (Fisher's protected least significant difference) revealed significant differences only between M6ΔureB and the three mutants with the highest urease activity, M6ΔureB(cag1-ureB), M6ΔureB(cag25-ureB), and M6ΔureB(hpn-ureB), in addition to the control strains expressing wild-type urease. Thus, the strains could be classified based on in vitro urease activity into three groups as follows: (i) strains with wild-type urease activity, (ii) mutants with recombinant urease activity that was significantly greater than zero, and (iii) strains with detectable urease activity but for which a statistically significant difference from zero could not be demonstrated.
FIG. 1.

In vitro urease activity of M6 and mutants. cag designations denote the promoter used to drive recombinant ureB expression. Strains with wild-type urease activity [M6, M6(cag9-ureB), and M6(cag25-ureB)] did not differ significantly from each other or from M6ΔureB(hpn-ureB). The urease activity of strains M6ΔureB(cag1-ureB) and M6ΔureB(cag25-ureB) was significantly less than that of the wild type and was significantly greater than zero. Urease activity of the other strains did not differ significantly from that of urease-negative M6ΔureB (see text). ∗, significantly different from M6, P < 0.05. †, significantly different from M6ΔureB, P < 0.05.
Bacterial colonization by wild-type and urease-deficient H. pylori.
Bacterial colonization of mice by H. pylori strain M6 and its mutants and recombinants is shown in Fig. 2. Wild-type strain M6 colonized all mice at a density of 106 to 107 CFU/g of gastric mucosa, and urease-negative M6ΔureB failed to colonize (not shown). Colonization by mutant strains varied. Of the 11 mutants tested, only the M6 derivatives M6(cag9-ureB) and M6(cag25-ureB), which expressed wild-type urease, and 4 of the 8 M6ΔureB derivatives colonized mice, as indicated in Fig. 2. All mutant strains, regardless of urease activity or genotype, colonized at a similar density, between 1 and 4% of M6.
FIG. 2.

Colonization density by M6 and colonizing mutants. The colonization densities of all mutants differed significantly from M6 but not from each other (P < 0.05).
Of the four colonizing M6ΔureB transformants, three expressed in vitro urease activity that was significantly different from zero while none of the noncolonizing strains did. The association between high urease production and colonization was significant at a P value of 0.0476 (Fisher's exact test). The fourth colonizing strain, M6ΔureB(cag15-ureB), had in vitro urease activity that clustered with the noncolonizing strains. These strains had urease activity which was detectable but was too low to be significantly different from zero using our methods. Thus, strain M6ΔureB(cag15-ureB) was in a category by itself. It expressed minimal urease activity in vitro, yet unlike the other low-urease strains, it colonized mice.
In vivo urease activity of wild-type and urease-deficient H. pylori.
In contrast to differing in vitro urease activity, in vivo urease did not differ between the colonizing strains (Fig. 3). The level of ammonia production by tissue homogenates revealed that urease activity in homogenates colonized by M6 was higher than in homogenates colonized by the mutant strains, as would be expected based on the higher bacterial colonization density. Lower colonization density in the urease-positive colonization control strains was associated with lower in vivo urease activity due to the smaller number of bacteria present. When homogenates with similar bacterial colonization density were compared, however, there were no detectable differences in urease activity between strains. Surprisingly, in vivo urease activity of M6ΔureB(cag15-ureB), the mutant with low to undetectable in vitro urease activity, could not be distinguished from that of the other colonizing mutants. In fact, when corrected for colonization density, the in vivo urease activity of M6ΔureB(cag15-ureB) was one of the higher activities of the four colonizing mutants (Fig. 4).
FIG. 3.

Urease activity in gastric homogenates from mice colonized by wild-type H. pylori strain M6 and colonizing mutants, compared to baseline. Results are expressed as the change in A340 over 2 h (see text). All mutant strains are significantly different from M6. Activities of strains M6(cag9-ureB), M6ΔureB(cag15-ureB), M6ΔureB(cag25-ureB), and M6ΔureB(hpn-ureB) are significantly greater than those of uninfected gastric homogenate (none) (P < 0.05).
FIG. 4.

Urease activity in gastric homogenates corrected for colonization density. Correcting for the number of bacteria per gram of gastric mucosa did not reveal any significant differences in the in vitro urease activity of the different bacterial mutants.
Gastritis due to wild-type and urease-deficient H. pylori.
Gastritis in mice infected with H. pylori strain M6 and colonizing mutants was typical of H. pylori gastritis in mice. In C57BL/6 mice killed 8 weeks after inoculation, lesions were moderate in extent, most commonly involving less than 20% of the gastric mucosa (Fig. 5). They were characterized by lymphocytic, plasmacytic, and neutrophilic infiltrates which were multifocal in distribution and mild to moderate in severity (Fig. 6). Foci of severe inflammatory infiltrates were accompanied by gastric epithelial metaplasia and were characterized by loss of normal fundic gland morphology and replacement by undifferentiated glands lined by mucus-type epithelium. In C57BL/6 mice, gastric lesions were most extensive in mice colonized by strain M6 but were also present in mice colonized by the two urease transformants. Differences in the extent of lesions between groups did not reach statistical significance (Fig. 5).
FIG. 5.

Extent of gastric lesions in C57BL/6 mice killed 8 weeks after infection (scored as described in Materials and Methods). None, uninfected mice; ∗, significantly different from uninfected mice, P < 0.05. There were no significant differences between the three infected groups.
FIG. 6.

(A) Fundic gastric mucosa from a C57BL/6 mouse infected with H. pylori strain M6ΔureB(cag15-ureB) and killed 8 weeks after inoculation. Mild gastritis characterized by mixed lymphocytes and neutrophils with a few plasma cells is present at the base of the gastric glands. (B) Uninfected control mouse. No inflammatory cells are present.
In recipient SCID mice killed 6 weeks after transfer, gastritis was more extensive than in C57BL/6 mice. Up to 100% of the gastric mucosa was affected and metaplasia was common (Fig. 7). Like C57BL/6 mice, there was no difference in the extent of gastritis or neutrophilic infiltrate between mice infected with M6 and those infected with M6ΔureB(cag1-ureB), in spite of a difference of up to 100-fold in bacterial density and 35-fold in in vivo urease activity (see Fig. 2 and 3). Histologic lesions in recipient SCID mice were similar to those in C57BL/6 mice but were more severe (Fig. 8).
FIG. 7.

Gastric lesions in recipient SCID mice were more extensive than in C57BL/6 mice, but like C57BL/6 mice there were no significant differences in the extent of lesions in the two infected groups. None, uninfected mice; ∗, significantly different from uninfected mice, P < 0.05.
FIG. 8.

Fundic gastric mucosa from a recipient SCID mouse infected with H. pylori strain M6 and killed 6 weeks after transfer. Marked gastritis characterized by mixed lymphocytes, neutrophils, plasma cells, and histiocytes is accompanied by loss of the normal fundic glands and replacement by less-differentiated mucus-type glands.
DISCUSSION
The results of this study demonstrate that restoration of urease activity to urease-deficient H. pylori can restore colonization ability. This constitutes the first definitive proof of the essential role of urease in colonization by H. pylori. In addition, these results suggest that there is a minimum threshold of urease activity necessary for colonization. Of the four recombinants that colonized mice, three had the highest in vitro urease activity, which was significantly greater than that of the urease-negative mutant, although it was less than that of the wild type. The five noncolonizing recombinants all had low in vitro urease activity. In these strains urease activity was detectable, but our assays were insufficiently sensitive to distinguish these strains statistically from the urease-negative mutant.
A surprising exception to this observation was strain M6ΔureB(cag15-ureB). In vitro, the urease activity of this mutant clustered with that of the noncolonizing mutants. However, the strain was able to colonize and urease assays of gastric homogenates infected with M6ΔureB(cag15-ureB) were indistinguishable from those of homogenates infected with the other colonizing strains. In fact, all five colonizing mutants tested, M6(cag9-ureB), M6ΔureB(hpn-ureB), M6ΔureB(cag1-ureB), M6ΔureB(cag15-ureB), and M6ΔureB(cag25-ureB), had similar in vivo urease activities. Taken together, these findings strongly suggest that urease activity in M6ΔureB(cag15-ureB) is upregulated in vivo, thus allowing sufficient urease expression to support colonization. These results are further supported by previously reported in vitro findings indicating that cag-15 is upregulated in response to contact with epithelial cells in vitro (20). The function of Cag-15 remains unknown and the protein lacks known homology. However, both in vitro and in vivo evidence of upregulation suggests that it may represent an important H. pylori colonization or virulence factor.
Interestingly, colonization density by all the mutants, including the positive control strains [M6(cag9-ureB) and M6(cag25-ureB)], was lower than colonization density by M6. We previously determined that insertional mutagenesis of hpn itself decreases colonization density by H. pylori (not shown), accounting for the decreased colonization by strain M6ΔureB(hpn-cag), but the other mutants expressed hpn and the diminished colonization could not be attributed to effects on hpn itself. The loss of colonization ability with insertion of the cag-ureB fusions is most likely due to the location of insertion (downstream of hpn). According to the published H. pylori gene sequences (35), the insertion did not interrupt any known open reading frame (20), but it is possible that M6 differs from published strains or that this chromosomal region may contain a regulatory or other sequence important to H. pylori survival in vivo. Diminished colonization density could not be attributed to loss of the wild-type ureB because even strains with wild-type urease activity expressed lower colonization in the presence of the cag-ureB insertion. In addition, in vivo manipulation is unlikely to account for differences in colonization by the mutants. We and others have shown that some H. pylori mutants colonize with an efficiency equal to that of the wild type in spite of in vitro manipulations which were similar to the ones used in this study (6, 7, 31). In addition, we used pools of mutants and repeated the transformations at least twice to ensure that random genetic events in a single clone would not result in spurious loss of colonization potential. Whatever the pathogenesis, the decrease in colonization density was consistent and similar for all strains which contained fusion constructs and thus was likely unrelated to urease activity.
In addition to their implications for the role of urease and cag-15 in colonization, the results reported here also have implications for the study of H. pylori in areas other than urease pathogenesis. First, this is the first demonstration of in vivo complementation of any H. pylori virulence factor. We have demonstrated that in vivo complementation can be successful in restoring enzyme activity and is a good method to demonstrate unequivocal colonization dependence of a specific bacterial factor. Previous studies showing that urease-negative mutants fail to colonize (2, 5, 8, 36) provided strong evidence that urease was needed for growth in vivo. However, definitive proof awaited the demonstration that loss of colonization could be restored specifically by replacement of urease activity. Success in complementation of urease activity in vivo indicates that this method can be useful in evaluating the role of other putative colonization factors for H. pylori. In addition, we have shown that urease is an excellent in vivo reporter gene. Expression of ureB under control of H. pylori cag promoters indicates not only that recombinant ureB can restore colonization ability but also that some cag genes are expressed in vivo at a level sufficient to allow colonization. Further, in combination with the previously published in vitro observations (20), the results are highly suggestive that at least one gene, cag-15, is upregulated in vivo.
We did not demonstrate differences in gastritis associated with urease activity in this study. In C57BL/6 mice this was not surprising given the relatively mild gastritis characteristic of H. pylori infection of these mice 8 weeks after inoculation (10). However, the similarity in lesions in recipient SCID mice colonized by the different strains was striking. We have shown that recipient SCID mice rapidly develop severe chronic active gastritis with epithelial metaplasia in response to infection by H. pylori (10). In these animals up to 100% of the gastric mucosa is inflamed 4 to 6 weeks after transfer, and in some mice normal gastric fundic mucosa is virtually absent. This model represents a robust method of evaluating the host response to H. pylori antigens and therefore was used in this study. Even in this strong host response model, however, there was no difference between mice infected with wild-type and urease-deficient H. pylori, in spite of differences in in vitro urease activity, absolute urease activity in vivo, and colonization density. This is strong evidence that neither urease activity nor bacterial colonization density are primary inducers of gastritis and gastric epithelial damage due to H. pylori.
Because of the similarity of urease activity in vivo in all the colonizing mutants we could not definitively distinguish the effects of urease activity per bacterium from those of decreased activity per gram of gastric mucosa. However, clearly total urease activity does not influence severity of gastritis in this model whether the difference is due to inherent urease activity of the colonizing strain or bacterial burden alone. We have previously shown that there is no direct toxic effect of large numbers of colonizing urease-positive bacteria in the absence of a host response (10). Taken together, these studies strongly suggest that while urease is an essential colonization factor for H. pylori it does not contribute to severity of disease.
In summary, we have confirmed through complementation that urease is essential for colonization and that urease is an excellent reporter gene for in vivo expression of H. pylori promoters. In addition, our results suggest that at least one of these promoters, cag-15, is upregulated in vivo. Finally, we have demonstrated that, at least in mice, neither total urease activity nor bacterial colonization density contributes to gastritis due to H. pylori.
Acknowledgments
This work was supported in part by PHS grants R01 AI43643, R29 DK-45340, R01 DK-53702, and R01 CA67498-01 and the GRASP Digestive Diseases Research Center (Tufts University) grant, P30 DK34928, all from the NIH.
Editor: B. B. Finlay
REFERENCES
- 1.Andrutis, K. A., J. G. Fox, D. B. Schauer, R. P. Marini, X. T. Li, L. L. Yan, C. Josenhans, and S. Suerbaum. 1997. Infection of the ferret stomach by isogenic flagellar mutant strains of Helicobacter mustelae. Infect. Immun. 65:1962-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrutis, K. A., J. G. Fox, D. B. Schauer, R. P. Marini, J. C. Murphy, L. L. Yan, and J. V. Solnick. 1995. Inability of an isogenic urease-negative mutant strain of Helicobacter mustelae to colonize the ferret stomach. Infect. Immun. 63:3722-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cussac, V., R. L. Ferrero, and A. Labigne. 1992. Expression of Helicobacter pylori urease genes in Escherichia coli grown under nitrogen-limiting conditions. J. Bacteriol. 174:2466-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desai, M. A., and P. M. Vadgama. 1993. An in vitro study of enhanced H+ diffusion by urease action on urea. Implications for Helicobacter pylori-associated peptic ulceration. Scand. J. Gastroenterol. 28:915-919. [DOI] [PubMed] [Google Scholar]
- 5.Eaton, K. A., C. L. Brooks, D. R. Morgan, and S. Krakowka. 1991. Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 59:2470-2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eaton, K. A., T. L. Cover, M. K. R. Tummuru, M. J. Blaser, and S. Krakowka. 1997. Role of vacuolating cytotoxin in gastritis due to Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 65:3462-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eaton, K. A., D. Kersulyte, M. Mefford, S. J. Danon, S. Krakowka, and D. E. Berg. 2001. Role of Helicobacter pylori cag region genes in colonization and gastritis in two animal models. Infect. Immun. 69:2902-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eaton, K. A., and S. Krakowka. 1994. Effect of gastric pH on urease-dependent colonization of gnotobiotic piglets by Helicobacter pylori. Infect. Immun. 62:3604-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eaton, K. A., and M. E. Mefford. 2001. Cure of Helicobacter pylori infection and resolution of gastritis by adoptive transfer of splenocytes in mice. Infect. Immun. 69:1025-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton, K. A., S. R. Ringler, and S. J. Danon. 1999. Murine splenocytes induce severe gastritis and delayed-type hypersensitivity and suppress bacterial colonization in Helicobacter pylori-infected SCID mice. Infect. Immun. 67:4594-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eaton, K. A., S. Suerbaum, C. Josenhans, and S. Krakowka. 1996. Colonization of gnotobiotic piglets by Helicobacter pylori deficient in two flagellin genes. Infect. Immun. 64:2445-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ermak, T. H., R. Ding, B. Ekstein, J. Hill, G. A. Myers, C. K. Lee, J. Pappo, H. K. Kleanthous, and T. P. Monath. 1997. Gastritis in urease-immunized mice after Helicobacter felis challenge may be due to residual bacteria. Gastroenterology 113:1118-1128. [DOI] [PubMed] [Google Scholar]
- 13.Evans, D. G., T. K. Karjalainen, D. J. Evans, D. Y. Graham, and C. H. Lee. 1993. Cloning, nucleotide sequence, and expression of a gene encoding an adhesin subunit protein of Helicobacter pylori. J. Bacteriol. 175:674-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falk, P., K. A. Roth, T. Boren, T. U. Westblom, J. I. Gordon, and S. Normark. 1993. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc. Natl. Acad. Sci. USA 90:2035-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert, J. V., J. Ramakrishna, F. W. Sunderman, A. Wright, and A. G. Plaut. 1995. Protein Hpn: cloning and characterization of a histidine-rich metal-binding polypeptide in Helicobacter pylori and Helicobacter mustelae. Infect. Immun. 63:2682-2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris, P. R., H. L. T. Mobley, G. I. Perezperez, M. J. Blaser, and P. D. Smith. 1996. Helicobacter pylori urease is a potent stimulus of mononuclear phagocyte activation and inflammatory cytokine production. Gastroenterology 111:419-425. [DOI] [PubMed] [Google Scholar]
- 17.Heuermann, D., and R. Haas. 1998. A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Mol. Gen. Genet. 257:519-528. [DOI] [PubMed] [Google Scholar]
- 18.Hu, L. T., and H. L. Mobley. 1990. Purification and N-terminal analysis of urease from Helicobacter pylori. Infect. Immun. 58:992-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones, D. M., A. Curry, and A. J. Fox. 1985. An ultrastructural study of the gastric campylobacter-like organism Campylobacter pyloridis. J. Gen. Microbiol. 131:2335-2341. [DOI] [PubMed] [Google Scholar]
- 20.Joyce, E. A., J. V. Gilbert, K. A. Eaton, A. Plaut, and A. Wright. 2001. Differential gene expression from two transcription units in the cag pathogenicity island of Helicobacter pylori. Infect. Immun. 69:4202-4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawano, S., M. Tsujii, H. Fusamoto, N. Sato, and T. Kamada. 1991. Chronic effect of intragastric ammonia on gastric mucosal structures in rats. Dig. Dis. Sci. 36:33-38. [DOI] [PubMed] [Google Scholar]
- 22.Labigne, A., V. Cussac, and P. Courcoux. 1991. Shuttle cloning and nucleotide sequences of Helicobacter pylori genes responsible for urease activity. J. Bacteriol. 173:1920-1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall, B. 1983. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet i:1273-1275. [PubMed]
- 24.Marshall, B. J., and J. R. Warren. 1984. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet i:1311-1315. [DOI] [PubMed]
- 25.McGee, D. J., C. A. May, R. M. Garner, J. M. Himpel, and H. L. Mobley. 1999. Isolation of Helicobacter pylori genes that modulate urease activity. J. Bacteriol. 181:2477-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Megraud, F., S. V. Neman, and D. Brugmann. 1992. Further evidence of the toxic effect of ammonia produced by Helicobacter pylori urease on human epithelial cells. Infect. Immun. 60:1858-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mobley, H. L. T., R. M. Garner, and P. Bauerfeind. 1995. Helicobacter pylori nickel-transport gene nixA: synthesis of catalytically active urease in Escherichia coli independent of growth conditions. Mol. Microbiol. 16:97-109. [DOI] [PubMed] [Google Scholar]
- 28.O’toole, P. W., L. Janzon, P. Doig, J. Z. Huang, M. Kostrzynska, and T. J. Trust. 1995. The putative neuraminyllactose-binding hemagglutinin HpaA of Helicobacter pylori CCUG 17874 is a lipoprotein. J. Bacteriol. 177:6049-6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parsonnet, J., G. D. Friedman, D. P. Vandersteen, Y. Chang, J. H. Vogelman, N. Orentreich, and R. K. Sibley. 1991. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 325:1127-1131. [DOI] [PubMed] [Google Scholar]
- 30.Parsonnet, J., S. Hansen, L. Rodriguez, A. B. Gelb, R. A. Warnke, E. Jellum, N. Orentreich, J. H. Vogelman, and G. D. Friedman. 1994. Helicobacter pylori infection and gastric lymphoma. N. Engl. J. Med. 330:1267-1271. [DOI] [PubMed] [Google Scholar]
- 31.Raudonikiene, A., N. Zakharova, W. W. Su, J. Y. Jeong, L. Bryden, P. S. Hoffman, D. E. Berg, and K. Severinov. 1999. Helicobacter pylori with separate beta- and beta′-subunits of RNA polymerase is viable and can colonize conventional mice. Mol. Microbiol. 32:131-138. [DOI] [PubMed] [Google Scholar]
- 32.Skouloubris, S., J. M. Thiberge, A. Labigne, and H. Dereuse. 1998. The Helicobacter pylori UreI protein is not involved in urease activity but is essential for bacterial survival in vivo. Infect. Immun. 66:4517-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smoot, D. T. 1997. How does Helicobacter pylori cause mucosal damage? Direct mechanisms. Gastroenterology 113:S31-S34. [DOI] [PubMed]
- 34.Smoot, D. T., H. L. Mobley, G. R. Chippendale, J. F. Lewison, and J. H. Resau. 1990. Helicobacter pylori urease activity is toxic to human gastric epithelial cells. Infect. Immun. 58:1992-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomb, J. F., O. White, A. R. Kerlavage, R. A. Clayton, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539-547. [DOI] [PubMed] [Google Scholar]
- 36.Tsuda, M., M. Karita, M. G. Morshed, K. Okita, and T. Nakazawa. 1994. A urease-negative mutant of Helicobacter pylori constructed by allelic exchange mutagenesis lacks the ability to colonize the nude mouse stomach. Infect. Immun. 62:3586-3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weatherburn, M. W. 1967. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 39:971-974. [Google Scholar]