

Abstract
VPS4 ATPases function in multivesicular body formation and in HIV-1 budding. Here, we report the crystal structure of monomeric apo human VPS4B/SKD1 (hVPS4B), which is composed of five distinct elements: a poorly ordered N-terminal MIT domain that binds ESCRT-III substrates, large (mixed α/β) and small (α) AAA ATPase domains that closely resemble analogous domains in the p97 D1 ATPase cassette, a three-stranded antiparallel β domain inserted within the small ATPase domain, and a novel C-terminal helix. Apo hVPS4B and yeast Vps4p (yVps4p) proteins dimerized in solution, and assembled into larger complexes (10–12 subunits) upon ATP binding. Human and yeast adaptor proteins (LIP5 and yVta1p, respectively) bound the β domains of the fully assembled hVPS4B and yVps4p proteins. We therefore propose that Vps4 proteins cycle between soluble, inactive low molecular weight complexes and active, membrane-associated double-ring structures that bind ATP and coassemble with LIP5/Vta1. Finally, HIV-1 budding was inhibited by mutations in a loop that projects into the center of the modeled hVPS4B rings, suggesting that hVPS4B may release the assembled ESCRT machinery by pulling ESCRT-III substrates up into the central pore.
Keywords: AAA ATPase, HIV, vacuolar protein sorting, virus budding, X-ray crystallography
Introduction
To infect new cells, retroviruses like HIV-1 must assemble and bud from the limiting membranes of producer cells. Retrovirus budding shares a number of similarities with the formation of intracellular vesicles that bud into late endosomal compartments called multivesicular bodies (MVB). In both processes, proteins are sorted into membrane microdomains, the membrane is distorted away from the cytoplasm, and the vesicle/virion is released via membrane fission. Furthermore, both retroviral budding and MVB vesicle formation utilize a common set of cellular machinery, called the ‘Class E' vacuolar protein sorting (VPS) proteins (reviewed by Katzmann et al, 2002; Gruenberg and Stenmark, 2004; Morita and Sundquist, 2004).
A total of 27 different human Class E proteins have been identified, and most function as subunits of three hetero-oligomeric ESCRT complexes (endosomal complexes required for transport), which are sequentially recruited to sites of vesicle formation. The ESCRT-I and ESCRT-II complexes act early in the pathway, and appear to function primarily as adaptors that recognize protein cargoes and recruit downstream Class E proteins. In contrast, ESCRT-III, Vps4, and LIP5/Vta1p proteins are recruited later and appear to function more directly in protein sorting and vesicle formation. The different Class E complexes may also play analogous roles in virus budding, particularly as not all retroviruses require ESCRT-I proteins to bud, whereas all retroviruses tested to date require ESCRT-III, LIP5, and hVPS4 activities (Garrus et al, 2001; Gottwein et al, 2003; Martin-Serrano et al, 2003a, 2003b; Strack et al, 2003; von Schwedler et al, 2003; Shehu-Xhilaga et al, 2004; Ward et al, 2005).
Current models hold that soluble ESCRT-III proteins are recruited to sites of virus budding and MVB vesicle formation where they coassemble into a membrane-associated lattice or coat that appears to function in protein sorting and perhaps also in membrane deformation and fission (Katzmann et al, 2002; Gruenberg and Stenmark, 2004). Soluble Vps4 ATPases are then recruited and utilize the energy of ATP hydrolysis to release assembled ESCRT complexes from the membrane (Babst et al, 1997, 1998). Thus, Vps4 ATPase activity is required to catalyze multiple rounds of vesicle formation, and may also be coupled directly to protein sorting and membrane fission, although these processes are not yet understood in mechanistic detail.
Vps4 proteins are conserved across eukaryotes, and some species have more than one Vps4 paralog. For example, humans and other mammals have two closely related Vps4 proteins (hVPS4A and hVPS4B/SKD1, 80% identity), whereas Saccharomyces cerevisiae has a single Vps protein (yVps4p) that is ∼60% identical to both human proteins (Supplementary Figure S1). The functional conservation of VPS4 proteins is emphasized by the observation that murine Vps4B can fully complement a yVps4p deletion in yeast (Scheuring et al, 2001). Vps4 proteins contain a single ATPase cassette (Type I ATPases), and they belong to the meiotic clade of AAA ATPases, which is a phylogenetic subset of the very large family of AAA+ ATPases (reviewed by Ogura and Wilkinson, 2001; Frickey and Lupas, 2004). AAA ATPase cassettes typically function as oligomeric rings, usually hexamers, and bind adenine nucleotides within Walker A/B motifs located at the interface between the two subdomains of the ATPase cassette and between adjacent subunits.
Although sequence analyses and functional studies clearly indicate that Vps4 proteins are AAA ATPases, several observations suggest that they may also have unique features not seen in other AAA ATPases. For example, primary sequence alignments break down toward the C-terminal end of the AAA ATPase cassette (Karata et al, 2001; see Supplementary Figure S1), and yVps4p forms higher-order complexes than do other hexameric Type I AAA ATPases (Babst et al, 1998). Here, we report the crystal structure of hVPS4B and describe a series of functional studies that provide a framework for understanding how Vps4 proteins assemble and function in MVB vesicle formation and virus budding.
Results and discussion
Expression of Vps4 proteins and crystallization of hVPS4B
To facilitate biochemical and structural studies, we established systems for expressing and purifying recombinant full-length hVPS4A, hVPS4B, and yVps4p and their constituent AAA ATPase cassettes (VPS4A119–437, hVPS4B126–444, yVps4p122–437). Multimilligram quantities of all proteins were obtained, but yields decreased in the order yVps4p>hVPS4B>hVPS4A, primarily because the human proteins tended to aggregate, particularly hVPS4A.
Full-length SeMet hVPS4B crystallized in space group P65 with one protein molecule per asymmetric unit and the structure was determined at a resolution of 2.8 Å and an Rfree value of 26.0% (Table I). The refined model lacked density for the MIT domain and linker (residues 1–122), the β2/α3 loop (residues 200–211), and the β3/α4 loop (residues 239–247). The His10-hVPS4B126–444 E235Q protein crystallized isomorphously with SeMet hVPS4B and had a very similar structure, consistent with the idea that the N-terminal MIT domain and flexible linker did not make significant lattice contacts. The SeMet hVPS4B structure contained three additional ordered N-terminal residues and is presented here.
Table 1.
hVPS4B crystal structure statistics
| |
SeMet-hVPS4B |
|---|---|
| Data collection statistics | |
| Space group | P65 |
| Cell dimensions (Å) | a=88.1 Å |
| c=112.6 Å | |
| Resolution limit (Å)a | 30.00–2.80 (2.95–2.80) |
| No. of reflections measured | 133 252 (19 859) |
| No. of unique reflections | 12 140 |
| Completeness (%) | 99.3 (99.3) |
| I/σ(I) | 9.7 (2.6) |
| Rmergeb | 0.058 (0.285) |
| Refinement statistics | |
| Resolution (Å) | 2.8 (2.873-2.80) |
| R-valuec | 0.192 (0.303) |
| Rfreed | 0.260 (0.411) |
| R.m.s.d. bonds (Å) | 0.018 |
| R.m.s.d. angles (deg) | 1.962 |
| No. of reflections | 10 917 (822) |
| No. of reflections Free R | 1190 (95) |
| No. of waters/sulfates | 9/2 |
| B-factors (Å2) protein/water/sulfate | 52.9/33.2/45.3 |
| % Residues in most favored phi–psi regionse | 87.8 |
| % Residues in additional phi–psi regionse | 12.2 |
| aThe single molecule in the asymmetric unit lacked defined density for residues 1–122 (omitted from model) and 200–211 and 239–247 (included with zero occupancy). | |
| bRmerge=∑∣I−IAverage∣/∑I, where I is the intensity of an individual measurement and IAverage is the average intensity from multiple observations. | |
| cR-value=100∑∣∣F(obs)∣–∣F(calc)∣∣/∑∣F(obs)∣. | |
| dRfree=R-value for a randomly selected subset (10%; 1190 reflections) of the data that were not used for minimization of the crystallographic residual. | |
| eStereochemistry was assessed with PROCHECK (Laskowski et al, 1993). | |
Overview of the structure
The ordered region of hVPS4B is composed of two central domains that comprise the AAA ATPase cassette, a three-stranded, antiparallel β domain inserted within the small domain of the AAA ATPase cassette, and a C-terminal helix (α10) that wraps underneath the AAA ATPase domain (Figure 1). The β domain insertion was unexpected, and its presence explains why the C-terminal ATPase domain of VPS4 proteins could not be satisfactorily aligned with other AAA ATPases (Karata et al, 2001). Indeed, primary sequence alignments indicate that while all Vps4 proteins contain the β domain insertion, this structural element is absent in the other members of the meiotic clade of AAA ATPases, including figetin, spastin, and katanin A1 (see Supplementary Figure S1).
Figure 1.
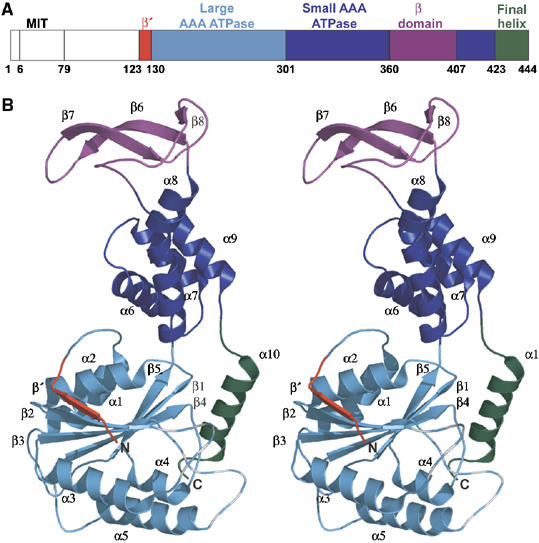
Structure of hVPS4B. (A) Domain map and numbering scheme for hVPS4B. The domain color coding scheme shown here is also used in Figures 2A, B, and 6. (B) Ribbon diagram (stereoview) of the hVPS4B123–444 structure. Note that the β2/α3 and β3/α4 loops were not defined by experimental density (dashed lines).
The AAA ATPase cassette
Like other AAA ATPases, the central ATPase cassette of hVPS4B is composed of two domains: an amino-proximal α/β domain and a smaller four-helix bundle. The large domain is composed of a six-stranded β-sheet sandwiched between five α-helices (Figure 1B). The final five strands (β1–5) form a parallel sheet, and equivalent parallel five-stranded β-sheets are present in all AAA ATPases (Ogura and Wilkinson, 2001). The amino-terminal strand (β′; red in Figures 1, 2A, and B) packs in an antiparallel orientation along the edge of β2 to complete the sheet, and this structural feature appears to be conserved in the meiotic AAA ATPases (Supplementary Figure S1), although it is only found in a few other AAA ATPases, such as FtsH (Niwa et al, 2002).
Figure 2.
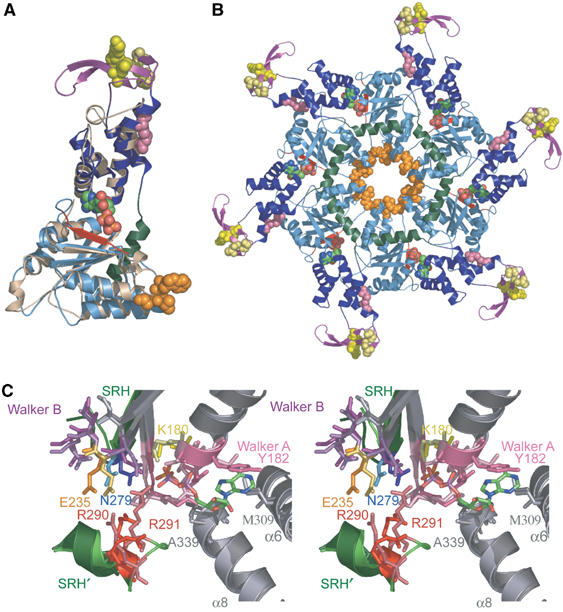
Structural similarities between hVPS4B123–444 and p97 D1. (A) Superposition of hVPS4B123–444 (color coded) onto ADP-bound p97 D1 protein (tan). Note that the individual domains of the AAA ATPase cassettes overlay very well, but that hVPS4B123–444 contains an additional N-terminal β-strand (red), an inserted β domain (purple), and an additional C-terminal helix (green). ADP (CPK atomic coloring) and the following residues are shown explicitly: pink; interface residue R354 (yR352); yellow, β domain residues K364 (yD362), L381 (yL373), S385 (yS377), D388 (yD380); orange, Pore 1 motif, 208WLG210 (y206WLG208). (B) Structural model for the hVPS4B123–444 ring. For clarity, the p97 hexamer used as an overlay template has been omitted, but the bound ADP molecule was retained. Residues in the two disordered loops of hVPS4B123–444 protein were modeled after those of the p97 D1 structure. Color coding matches panel A. (C) Stereoview of the overlaid nucleotide binding sites of p97 D1 (lighter shades) and hVPS4B (darker shades). Secondary structural elements that contribute to the active site are shown (gray), with the following highlights: Walker A motif (pink), Walker B motif (purple), the SRH β-strand (green), and the C-terminal SRH helix from an adjacent protomer (denoted SRH′, green). The positions of the bound ADP molecule from p97 and the sulfate ion from hVPS4B123–444 are shown. Key hVPS4B/p97 D1 residues shown explicitly are listed in the text, and the following residues (hVPS4B numbering) have been highlighted in unique colors: K180 (ATP binding, yellow), E235 (Mg2+ and ATP hydrolysis, gold), N279 (γ-phosphate sensing, blue), and R290–R291 (arginine fingers, red). Key residues in the two active sites are identical, except for a Ser to Thr substitution in the Walker A loop (181S in hVPS4B), and for conservative changes in the residues that stack on either side of the adenine ring (182Y and 309M in hVPS4B versus 253L and 380L in p97). A color-coded list of all active site residues is given in Supplementary Figure S1.
The small ATPase domain forms an antiparallel four-helix bundle. This structure is similar to other AAA ATPases, except that the third helix (α8) is extended and kinked near its C-terminus, which helps to accommodate the inserted β domain. The kink is induced by a proline residue (Pro 352), which, like the β domain itself, is found in all known Vps4 proteins, but not other meiotic AAA ATPases (Supplementary Figure S1).
AAA ATPases typically bind ATP at the interface between the large and small domains, with Walker A, Walker B, second region of homology (SRH), arginine fingers, and sensor 1 motifs all performing critical roles in ATP binding, hydrolysis, and conformational changes (Ogura and Wilkinson, 2001). As discussed below, these elements are all present in hVPS4B, and appear to form a canonical ATP binding site.
The β domain
The unique β domain of hVPS4B (residues 360–406) forms a twisted, three-stranded antiparallel β-sheet inserted between the third (α8) and fourth (α9) helices of the small subunit of the AAA ATPase cassette. An extended loop containing two well-defined type-I turns connects the β domain back to the small helical domain, and the two discrete domains make only modest interactions. Below, we present evidence that the β domains of hVPS4B and yVps4p participate in binding the adaptor proteins, LIP5/Vta1p.
ClpB is the only other structurally characterized AAA+ ATPase with an extra domain inserted in the same place as the β domain of hVPS4B. In that case, however, the ClpB insertion is a long antiparallel coiled-coil. Like the hVPS4B β domain, the ClpB insertion appears to interact with substrates, perhaps helping to recognize and/or pry apart large protein aggregates (Lee et al, 2003).
The C-terminal helix
A final helix (α10; green in Figures 1, 2A, and B) wraps beneath the large AAA ATPase subunit where it packs against β1 and the β4/α5 loop. This helix has not been observed in other AAA+ ATPase structures, but sequence alignments indicate that other meiotic AAA ATPases likely have analogous helices (Supplementary Figure S1). Although helix 10 appears to be well positioned to help stabilize a double-ring structure, we have not been able to obtain evidence in support of this model (discussed below).
Similarity to p97 D1
The entire ordered region of hVPS4B was used to search the protein database for proteins of similar structure (Holm and Sander, 1995), and the best match was to the D1 cassette of the Type II AAA ATPase, p97 (PDB 1S3S) (Dreveny et al, 2004). Separate searches were also performed using the isolated large and small ATPase domains and the isolated domains of p97 D1 were again the best matches for both hVPS4B domains. To construct the best possible structural model for the assembled hVPS4B protein, the interdomain angle was closed slightly (∼8°) by adjustments in the flexible interdomain linker so that both domains overlaid optimally with those of p97 D1. The agreement between the two structures throughout the ATPase cassette was excellent, with a Cα RMSD of 1.76 Å, over the 199 ordered residues of the two hVPS4B domains (see Figure 2A). p97 therefore appears to be a good structural model for hVPS4B.
AAA ATPases function as oligomeric rings, usually hexamers, and ring formation is often promoted by adenine nucleotide binding between adjacent subunits (Ogura and Wilkinson, 2001). This is the case for both p97 and Vps4 proteins, as adenine nucleotides promote assembly of p97 D1 (Wang et al, 2003), yVps4p (Babst et al, 1998), and hVPS4B (Supplementary Figure S3). Importantly, the known p97 D1 structures are hexamers, and have been crystallized with bound ADP (Zhang et al, 2000; Dreveny et al, 2004; DeLaBarre and Brunger, 2005). The similarity between hVPS4B and p97 D1 allowed us to model how hVPS4B forms rings upon nucleotide binding. As shown in Figure 2B, simply overlaying hVPS4B subunits onto the six subunits of the p97 hexamer produced a feasible model with no unacceptable steric clashes. We therefore favor the idea that VPS4B forms hexameric rings, although we cannot formally rule out the possibility that hVPS4B could assemble into pentameric rings, as has been suggested based upon chemical crosslinking studies of yeast Vps4p (Babst et al, 1998). In our model, the hVPS4B ATPase ring has an approximate diameter of 110 Å and a height of 35 Å, and the six β domains form a discontinuous outer ring (145 Å in outer diameter). Intersubunit contacts are mediated primarily by the two domains of the hVPS4B ATPase cassette, although the C-terminal helices (which lack a counterpart in p97) are also positioned so that they could touch end-on and form a continuous belt on one side of the ring (green in Figure 2B).
The similarities between hVPS4B and p97 D1 extend to the nucleotide binding sites, where both the identities and positions of all key active site residues are conserved (Figure 2C). As shown in Figure 2A–C, adenosine nucleotides bind at the junction between adjacent domains and adjacent subunits. There is a single sulfate ion in the active site of the hVPS4B structure, which binds in a position equivalent to the β-phosphate in the ADP-bound form of p97 D1 (Zhang et al, 2000; Dreveny et al, 2004; DeLaBarre and Brunger, 2005). The hVPS4B nucleotide binding site is defined by (1) three elements from the large domain (Walker A/P loop: 174GPPGTGKSY182, Walker B: 232FIDEID237, and the C-terminal end of the β4 strand from the SRH: 277ATN279), (2) two elements from the small domain (helix α6: 309M and helix α8: 337SGA339), and (3) two arginine residues from the SRH of the large domain of the adjacent subunit (arginine fingers: 290RR291) (underlined residues are shown explicitly in Figure 2C). Briefly, these different motifs function in phosphate binding (Walker A), magnesium coordination and nucleotide hydrolysis (Walker B), ribose binding (α8), adenine base stacking (Walker A, 182Y, and α6 309M), γ-phosphate sensing (SRH residue 279N), and oligomerization, ATP binding, and hydrolysis (arginine fingers) (reviewed by Ogura and Wilkinson, 2001). Every key residue in all of these motifs is identical between hVPS4B and p97 D1 except for a Ser to Thr substitution in the Walker A loop (181S in hVPS4B), and for conservative changes in the residues that stack on either side of the adenine base (see Figure 2C caption). The intermolecular connections mediated by binding of the arginine fingers rationalize the interdependence of hVPS4B assembly and ATP binding/hydrolysis (Babst et al, 1998).
Available mutational data are consistent with this model for the hVPS4B active site and for its functional importance. Specifically, mutation of a key lysine residue in the Walker A motif (Lys180Gln in hVPS4B) reduces yVps4p nucleotide binding in vitro (Babst et al, 1998), and impairs vacuolar protein sorting (Babst et al, 1998; Bishop and Woodman, 2000) and HIV-1 budding (Garrus et al, 2001; von Schwedler et al, 2003). Similarly, a mutation in the Walker B magnesium binding motif (Glu235Gln in hVPS4B) allows ATP binding and protein assembly (Babst et al, 1998; see Supplementary Figure S3), but inhibits ATP hydrolysis, as well as vacuolar protein sorting (Babst et al, 1998; Bishop and Woodman, 2000; Yoshimori et al, 2000; Scheuring et al, 2001) and HIV-1 budding (Garrus et al, 2001; von Schwedler et al, 2003).
Oligomerization of hVPS4B
Although the human VPS4 proteins can form higher order structures, detailed analyses of self-assembly were complicated by a lack of reversibility and variable yields of higher order products. Detailed oligomerization and protein binding studies were therefore performed using yVps4p, although the two key observations (i.e., ATP-dependent assembly and LIP5/Vta1p binding) were also documented for hVPS4B constructs (see Supplementary Figures S3 and S4, respectively). Oligomerization and binding studies utilized the yVps4p E233Q protein because this mutation inhibits ATP hydrolysis without blocking ATP binding (Babst et al, 1998), and therefore allowed us to test the dependence of ATP binding on yVps4p oligomerization and Vta1p binding.
In the absence of nucleotide, yVps4p E233Q (MW 48 kDa) eluted from a gel filtration column as a single species, which we assign as a dimer (MWest∼94 kDa; see Figure 3, upper panel). This assignment is consistent with equilibrium analytical ultracentrifugation experiments, where the best-fitting single-species model was to a yVps4p dimer, although global fits across a range of concentrations showed systematic bias in the residuals, indicating that lower molecular weight species were also present. Although appreciable concentrations of monomeric yVps4p were not observed at the concentrations used for gel filtration, both monomeric and dimeric species were observed for hVPS4B (see Supplementary Figure S3).
Figure 3.
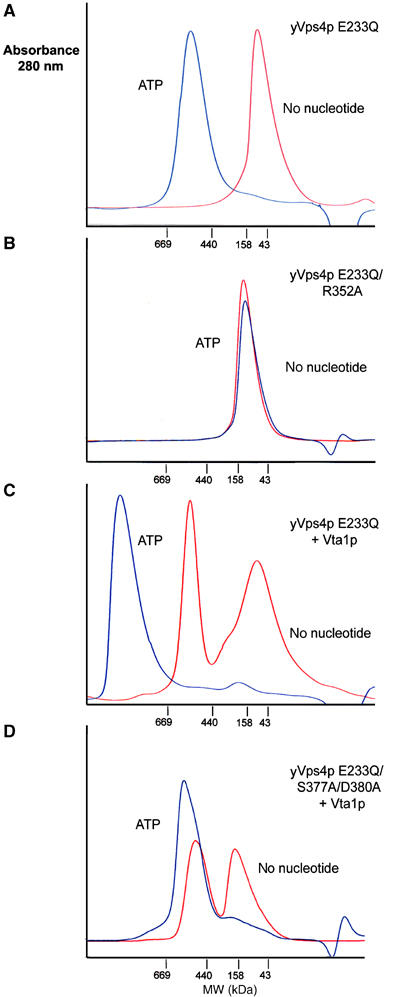
ATP induces yVps4 E233Q assembly and Vta1p binding. (A–D) Gel filtration chromatography of yVps4p proteins (154 μM) in the absence (red) or presence (blue) of ATP. Note that all yVps4p proteins contained the E233Q mutation that allows ATP binding without hydrolysis. (A) WT (E233Q) yVps4p, which assembled in an ATP-dependent fashion. (B) R352A yVps4p, which failed to assemble in the presence of ATP. (C) WT (E233Q) yVps4p+equimolar Vta1p, which forms a high molecular weight Vta1p/yVps4p complex in the presence of ATP. (D) S377A/D380A yVps4p+equimolar Vta1p, which failed to form the ATP-dependent Vta1p complex. Gel filtration chromatography was performed on a Superose HR6 column (Amersham) in 100 mM NaCl, 25 mM Tris–HCl (pH 7.5), 5% glycerol, 1 mM MgCl2, and 1 mM DTT. Samples labeled ATP samples were preincubated for 1 h in 1 mM ATP and chromatographed with 1 mM ATP in the running buffer. Elution positions of molecular weight standards are shown below each chromatogram. Note that the estimated oligomerization state of the lowest molecular weight species (red in Figure 5A) ranged between 2 and 4 subunits in multiple repeats of the experiment.
Upon addition of 1 mM ATP, the yVps4p dimer was converted entirely into a higher order complex (MWest∼540 kDa). These data are in excellent agreement with a previous study of the yVps4p protein by Emr and colleagues (Babst et al, 1998). The mobility of the ATP-bound complex suggested that it contained ∼10–12 subunits, although we were unable to assign the oligomeric state unambiguously because gel filtration retention times lacked the required precision. Attempts to determine the precise oligomeric state using equilibrium analytical ultracentrifugation gave best fits to models with 12 subunits, although we were again unable to fit data from multiple concentrations without systematic bias in the residuals (data not shown). Overall, our data indicate that ATP binding promotes assembly of Vps4 proteins from dimers into higher order complexes that correspond to some type of double-ring structure, and the most likely oligomeric state is a dodecamer.
As discussed above, we modeled hexameric rings of Vps4 based upon sequence and structural similarities with the hexameric p97 D1 ring. Indeed, many residues that make up the protein–protein interfaces are conserved between p97 and Vps4 proteins, indicating that details of the two hexameric assemblies are likely to be conserved. As an initial test of our structural model for the Vps4 ring, we examined the effect on yVps4p assembly of a substitution mutation in a conserved interface residue (Arg352Ala) predicted to make important intermolecular contacts in both p97 and Vps4. As shown in Figure 3B, the apo yVps4p E233Q/R352A mutant dimerized just like the wild-type (WT) protein in the absence of ATP, but failed to assemble into the higher order structure upon addition of ATP. We therefore conclude that this mutation blocks yVps4p ring formation by destabilizing the interface between adjacent subunits, supporting the idea that the p97 and Vps4 rings have similar structures. However, a point mutation in an adjacent interface residue (R323A) did not inhibit higher order Vps4p assembly at the concentrations tested (not shown), so not all putative interface mutants blocked assembly. We note that a number of the interface residues found in p97 and Vps4 are also found in the meiotic AAA ATPase, spastin, and that hereditary spastic paraplesia results from mutations in six of these putative spastin interface residues, including the yVps4p R352 position (see Supplementary Figure S2). We therefore suggest that, at least in some cases, the hereditary mutations block protein function by inhibiting spastin assembly.
Our oligomerization studies, as well as previous chemical crosslinking and gel filtration studies on yVps4p (Babst et al, 1998), indicate that fully assembled Vps4 proteins contain ∼10–12 subunits, which suggests a double-ring structure. The C-terminal helix of VPS4B, which is missing in other AAA+ ATPases, forms an exposed annulus on one side of the Vps4 ring and is an obvious candidate to mediate ring dimerization and thereby stabilize a double-ring structure. Unfortunately, however, deletion of the C-terminal helix of yVps4p caused the protein to form very large assemblies/aggregates (>1000 kDa) that could not be interpreted structurally. Moreover, a series of alanine substitution mutations at exposed surface residues in the C-terminal helix of yVps4p (E419/L423, E426/R430, Q427/D431) did not measurably affect yVps4p E233Q dimerization or higher order assembly. Hence, our mutational analyses did not support the idea that the C-terminal helix of yVps4p mediates formation of the double-ring structure. At this point, we also do not know whether the two rings are oriented tail-to-tail, which would allow formation of two-fold symmetrical dimer interactions, or head-to-tail, which would place the two rings in the same orientation and allow their ATPase pumps to work in concert. There is precedent for both types of arrangement in the related Type II AAA ATPases, NSF (Furst et al, 2003) and p97 (DeLaBarre and Brunger, 2005).
Vta1p/LIP5 proteins bind assembled Vps4 proteins
The Vps4 proteins are unusual in that, in addition to binding ESCRT-III substrates via their N-terminal MIT domains, they also interact with a second type of Class E protein, LIP5 (yeast Vta1p) (Yeo et al, 2003; Fujita et al, 2004). Vta1p and yVps4p were previously reported to interact in a yeast two-hybrid assay (Yeo et al, 2003), and we were able to confirm this interaction, demonstrate an orthologous interaction between LIP5 and hVPS4B, and demonstrate the analogous interspecies interactions (i.e., LIP5/yVps4p and Vta1p/hVPS4B) (not shown).
Gel filtration and GST pulldown experiments were used to demonstrate that the interaction between yVps4p and Vta1p was direct, and to characterize the interaction further (Figures 3C, D, and 4). As shown in Figure 4, pure recombinant yVps4p E233Q formed a complex with WT GST-Vta1, but not with a GST control (lane 1 and data not shown). The analogous interaction was again observed for the human orthologs, as hVPS4B126–444 also formed a specific complex with GST-LIP5 (Fujita et al, 2004; see Supplementary Figure S4).
Figure 4.
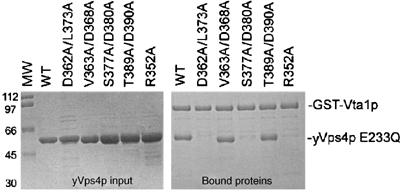
GST-Vta1p binds the β domain of assembled yVps4p. Left panel: Control experiment showing equivalent input levels of different yVps4p E233Q proteins. The ‘WT' sample (lane 2) had only the E233Q mutation, whereas other yVps4p proteins (lanes 3–7) had the additional mutations listed above. Molecular weight (MW) markers are shown in lane 1. Right panel: GST pulldown experiment showing the binding of the different yVps4p proteins to GST-Vta1p.
Stable complexes between yVps4p and Vta1p were also observed using gel filtration chromatography, but only in the presence of ATP. As shown in Figure 3C, pure recombinant Vta1p formed a high molecular weight complex that corresponded to approximately 10–11 subunits (MWest∼430 kDa, MWmonomer=40 kDa). Others have previously shown that Vta1p forms an oligomer, although our estimate of the molecular weight is slightly higher than earlier estimates (∼300 kDa; Shiflett et al, 2004). In the absence of ATP, the Vta1p oligomer did not form a complex with yVps4p E233Q that was stable to gel filtration chromatography, although the peak for the yVps4p E233Q dimer was slightly broadened and shifted to higher molecular weight, consistent with the idea that Vta1p and yVps4p E233Q interacted weakly. This weak interaction was also consistent with GST pulldown experiments, where yVps4p/Vta1p complexes were observed even in the absence of ATP (Yeo et al, 2003, and data not shown).
In the presence of ATP, yVps4p E233Q and Vta1p formed a single complex of very high molecular weight (>1000 kDa) that remained intact during gel filtration chromatography, and presumably consisted of the fully assembled yVps4p E233Q protein in complex with the Vta1p oligomer (Figure 3C). The ATP requirement for stable complex formation can be rationalized by the fact that ATP-dependent oligomerization of yVps4p is likely to increase the avidity of Vta1p binding. Consistent with this idea, the R352A mutation, which blocked formation of the higher order yVps4p oligomer (Figure 3B), also inhibited Vta1p binding in the GST pulldown assay (Figure 4, right panel, lane 6). In summary, our gel filtration and binding data indicate that fully assembled yVps4p E233Q forms stable complexes with oligmeric Vta1p, and the apparent size of this complex suggests that it could contain 12 copies of both yVps4p E233Q and Vta1, although this stoichiometry remains to be tested rigorously.
The yVps4p β domain contributes to Vta1p binding
In our model of the Vps4 hexamer, the six β domains project outward on the external surface of the ring, where they are well positioned to interact with other proteins such as Vta1p. This, together with the fact that a C-terminal fragment of yVps4p (residues 351–437) spanning the β domain can bind Vta1p in a yeast two-hybrid experiments (Yeo et al, 2003), prompted us to test whether the β domain of yVps4p functioned in Vta1p binding. To test this model, we created four sets of paired alanine scanning mutations designed to remove the side chains of adjacent surface residues without disrupting the overall β domain structure. Two of the mutations, S377A/D380A and D362A/L373A, efficiently inhibited the yVps4p/Vta1p interaction, as assayed in both by gel filtration (Figure 3D and data not shown) and GST pulldown assays (Figure 4B, right panel, lanes 2 and 4). The remaining two mutations (V363A/D368A and T389A/D390A) did not affect Vta1p binding (Figure 4B, right panel, lanes 3 and 5, and data not shown). Hence, mutations on the yVps4p β domain surface can inhibit Vta1p binding, implying that this domain is directly involved in Vta1p binding. This interaction may provide another connection between the enzyme and its substrates because previous studies have shown that LIP5/Vta1p can interact directly with ESCRT-III proteins (Bowers et al, 2004; Shiflett et al, 2004; Ward et al, 2005).
Functional importance of the VPS4B Pore 1 motif
It is not yet clear how proteins like p97 and NSF unfold/translocate their protein substrates. However, there is increasing evidence that many other AAA+ ATPases act by translocating substrates through the central pore (e.g., see Horwich, 2004). An elegant mechanistic model has recently been proposed by Stuart and colleagues to explain how RNA strands are translocated through the central pore of the hexameric ATPase P4 of the bacteriophage φ12 (Mancini et al, 2004). Briefly, they propose that six β-hairpin ‘levers' project into the protein's central cavity and act sequentially to pull the RNA strand through the pore. Their structural studies support a model in which the levers bind RNA in an ‘up' position in the presence of ATP, pull downward during a power stroke that accompanies ATP hydrolysis, and then ‘prime' the adjacent subunit to hydrolyze ATP and continue the cycle. A related but concerted model was recently proposed to explain the DNA helicase activity of the SV40 large T antigen (Gai et al, 2004).
By analogy, we hypothesized that individual ESCRT-III proteins might be pulled into the constricted central pore of the assembled VPS4 rings and thereby removed from the assembled ESCRT-III protein lattice/cage. In our model of the active hVPS4B enzyme, a conserved protein motif (the ‘Pore 1' motif (Wang et al, 2001; Yamada-Inagawa et al, 2003), 208WLG210 in hVPS4B) is an excellent candidate for a polypeptide interacting lever because (1) it is located in the β2/α3 loop that projects into the central cavity of each ring (highlighted in orange in Figure 2B), (2) analogous Pore 1 motifs corresponding to the consensus sequence ϕϕG (where ϕ represents large hydrophobic residues) are conserved across AAA+ ATPases that act on peptide substrates, but are not found in any AAA+ ATPases that act on nucleic acid substrates (Wang et al, 2001; Weibezahn et al, 2004; see Figure 5A), and (3) Pore 1 residues, especially the first bulky hydrophobic residue, are important for the protein unfolding/translocation activities of the AAA+ ATPases, HslU (Song et al, 2000), ClpB (Weibezahn et al, 2004), ClpX (Siddiqui et al, 2004), and FtsH (Yamada-Inagawa et al, 2003).
Figure 5.
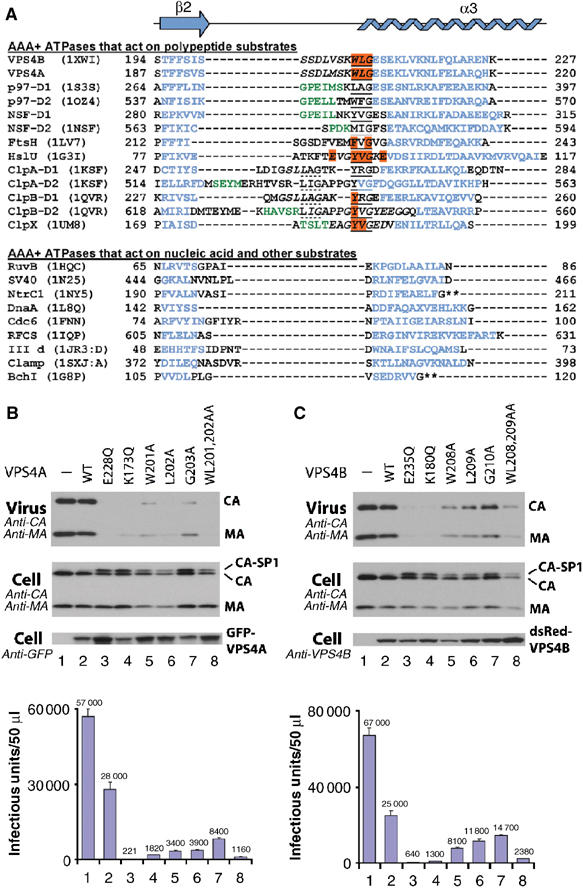
The Pore 1 motif is required for VPS4 function and HIV-1 replication. (A) Structure-based sequence alignment of the Pore 1 regions of all structurally characterized AAA+ ATPases (PDB entries and residue numbers are shown), with VPS4A and NSF-D2 AAA ATPase sequences also included for reference. The aligned region spans hVPS4B strand β2 (purple) and helix α3 (blue). Note that the Pore 1 motif (underlined) is found in all AAA+ ATPase cassettes that work on polypeptide substrates (above the break), but not in any AAA+ ATPases that work on nucleic acid or other substrates (below). Orange residues impair AAA+ ATPase function when mutated (Song et al, 2000; Yamada-Inagawa et al, 2003; Siddiqui et al, 2004; Weibezahn et al, 2004), italicized residues are disordered, green residues form a short helix in some AAA ATPases, dotted underlines denote a second Pore 1 consensus motif, and asterisks denote sites where helix α3 is interrupted by an insert. (B) Pore mutations in GFP-VPS4A dominantly inhibit HIV-1 release and infectivity. Shown are Western blots of pelleted virions (Virus) and cytoplasmic extracts (Cell), and viral titers from 293T cells that coexpressed WT HIV-1NL4-3 and various GFP-VPS4A proteins (histogram, below). Coexpressed proteins were as follows: GFP alone (negative control, lanes 1); WT GFP-Vps4A (negative control, lanes 2); GFP-Vps4A E228Q (positive control, lanes 3); GFP-Vps4A K173Q (positive control, lanes 4); GFP-Vps4A W201A (lanes 5); GFP-Vps4A L202A (lanes 6); GFP-Vps4A G203A (lanes 7); or GFP-Vps4A WL201,202AA (lanes 8). (C) Pore 1 mutations in dsRed-hVPS4B also dominantly inhibit HIV-1 release and infectivity. The experiment is analogous to that shown in panel B except that (1) the equivalent mutations in dsRed-hVPS4B were E235Q and K180Q (positive controls), and W208A, L209A, G210A, and WL208,209AA (Pore 1 mutants), and (2) levels of dsRed-hVPS4B were detected using an affinity-purified anti-hVPS4B antibody.
The three hVPS4B Pore 1 motif residues are poorly ordered in the crystal structure, perhaps because the protein is not fully assembled or engaging substrate. In the ADP-bound p97 D1 hexamer, however, the terminal glycine residue is part of a reverse turn and adopts a positive ϕ angle (+95), which apparently explains the preference for a glycine in this position. In contrast, the two hydrophobic Pore 1 side chains (278LA279 in p97 D1) project into the central cavity of the ring and make no significant intra- or intermolecular contacts. We confirmed that the hydrophobic Vps4 Pore 1 residues apparently do not make important assembly contacts by showing that a mutation in the first Pore 1 residue, W206A, did not affect yVps4p E233Q assembly in vitro (not shown).
To test whether Pore 1 residues were required for hVPS4A and hVPS4B functions in vivo, we assayed the efficiency of HIV-1 release and infectivity from cells expressing exogenous WT or Pore 1 mutant VPS4 proteins (Figure 5B and C). The mutants tested included single alanine scanning mutations in every Pore 1 residue, as well as double mutations (WL to AA) in the first two hydrophobic residues of the Pore 1 motif. hVPS4 mutations known to inhibit ATP binding (hVPS4A K173Q and hVPS4B K180Q, respectively) or ATP hydrolysis (hVPS4A E228Q and hVPS4B E235Q) were employed as positive controls, as these mutations have previously been shown to dominantly inhibit HIV-1 release (Garrus et al, 2001; von Schwedler et al, 2003). Protein levels and viral infectivity were analyzed by Western blotting (Figure 5B and C, upper panels) and in single-cycle (MAGIC) assays (lower panels).
In every case, overexpression of Pore 1 mutant GFP-hVPS4A or dsRed-hVPS4B proteins inhibited HIV-1 release, as reflected by reductions in the levels of the viral MA and CA proteins released into the supernatant and in viral infectivity. As expected, HIV-1 release and infectivity were also inhibited by overexpression of the VPS4 ATP binding and hydrolysis mutants, whereas overexpression of the WT hVPS4 proteins caused only a modest (∼2-fold) decrease in particle release and infectivity. Cellular expression levels of all hVPS4 proteins were generally similar within each experiment, and none of the hVPS4 proteins altered cell viability or Gag protein synthesis/stability significantly. Not surprisingly, the different hVPS4 Pore 1 mutations inhibited viral infectivity to different degrees, with reductions ranging between two- (hVPS4B G210A) and 25-fold (hVPS4A WL201,202AA) as compared to the WT controls. Nevertheless, levels of virus release and infectivity always correlated well, and the relative effects of the different Pore 1 mutations were similar for both hVPS4A and hVPS4B (WL/AA>W/A>L/A>G/A), although hVPS4A Pore 1 mutations typically inhibited HIV-1 infectivity about two-fold better than their hVPS4B counterparts. It was also notable that all of the hVPS4A and hVPS4B Pore 1 and ATPase mutant proteins induced accumulation of the HIV-1 Gag CA-SP1 processing intermediate (Figure 5B and C, panel 2). This is a hallmark of arrested virus budding and occurs because Gag processing and viral budding are linked events (Göttlinger et al, 1991).
Although the precise mechanistic role(s) of the Pore 1 motif in hVPS4 proteins and other AAA ATPases require further study, it seems likely that they may contact substrates as they pass up into the central pore of the protein. Indeed, the first Pore 1 residue (Tyr653) of the Escherichia coli ClpB D2 ATPase cassette has been crosslinked directly to translocating substrates (Weibezahn et al, 2004). Thus, our data demonstrate that alanine substitution mutations in all hVPS4A or hVSP4B Pore 1 residues impair protein function and HIV-1 release in vivo, and support the idea that ESCRT-III substrates are translocated up into the central channel of the hVPS4 proteins.
Working model for the functional cycle of VPS4 proteins
The hVPS4B structure, together with available biochemical data, suggests a functional enzymatic cycle (see Figure 6). Under steady-state conditions, human VPS4B can be isolated from the cytoplasm as a soluble monomeric protein (Fujita et al, 2004) and this state presumably corresponds to the crystal structure reported here. The enzyme is then recruited to the membrane where it functions in ESCRT protein release and possibly also in protein sorting and vesicle formation. Recruitment of VPS4 proteins to endosomal membranes requires both the C-terminal domains of ESCRT-III proteins (Lin et al, 2005) and the MIT domains of VPS4 proteins (Babst et al, 1998), and we have recently shown that these two domains bind one another (Scott et al, 2005), indicating that this interaction mediates Vps4 recruitment and substrate recognition.
Figure 6.
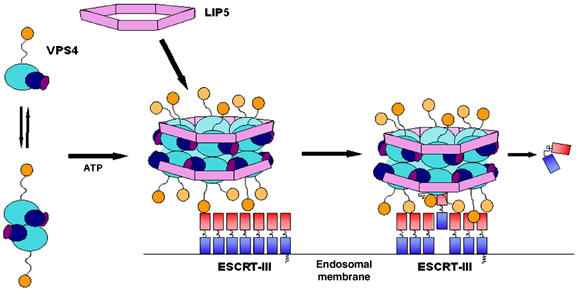
Model for the functional cycle of VPS4 proteins. Left: At steady state, hVPS4B is primarily a monomeric cytoplasmic protein (Fujita et al, 2004), and exhibits a monomer–dimer equilibrium in the absence of bound nucleotide (Babst et al, 1998; Supplementary Figure S3). LIP5/Vta1p is an oligomer of uncertain stoichiometry. Middle: Vps4 proteins are recruited to sites of vesicle formation at the endosomal membrane by interactions between the N-terminal MIT domain and the C-proximal domains of assembled ESCRT-III lattice/cage (Babst et al, 2002; Lin et al, 2005; Scott et al, 2005). The assembled Vps4 proteins can also bind ATP and LIP5/Vta1p oligomers via β domain interactions to form an enzymatically active complex. Note that a head-to-tail orientation of the two Vps4 rings (not shown) is equally consistent with our data. Right: We propose that bound ESCRT-III subunits are freed from the assembled lattice/cage and released into the cytoplasm as they are pulled up into the narrow central chamber of the hVPS4B ring.
At present, there is no indication that Vps4 proteins oligomerize or bind LIP5/Vta1p proteins in the cytosol, and it is therefore likely that assembly of hVPS4/LIP5 complexes occurs when hVPS4 proteins are recruited by the ESCRT-III lattice (Babst et al, 2002; Fujita et al, 2004). VPS4 assembly could be promoted by the increases in local protein concentrations that accompany recruitment or by more active mechanisms. Regardless, Vps4 assembly, ATP binding, and LIP5/Vta1p binding will be mutually cooperative events that can convert the enzyme to its active state. LIP5/Vta1p may also modulate or regulate the ATPase activity of the assembled VPS4 complex (Yeo et al, 2003), and/or participate in substrate selection, as it binds ESCRT-III proteins (Bowers et al, 2004; Shiflett et al, 2004; Ward et al, 2005).
Once assembled on the membrane, Vps4 proteins act to disassemble the ESCRT-III lattice (Katzmann et al, 2002). Vps4 proteins apparently perform this function by binding to site(s) downstream of coiled-coiled structures predicted to be present in the ESCRT-III substrates (Lin et al, 2005; Scott et al, 2005), pulling them into the constricted central channel of the enzyme, and releasing them into the cytoplasm. Thus, the mechanism of Vps4 proteins may be analogous to the chaperone activities of other more general AAA ATPase chaperones like ClpB, except that the Vps4 proteins have evolved to recognize their ESCRT-III substrates via specific interactions with the MIT domain. Important, but still unresolved, questions include the relative orientation of the two Vps4 rings, how the ESCRT-III proteins exit the central channel, and whether the removal of ESCRT-III subunits is directly coupled to the energy-requiring processes of cargo sorting and/or vesicle formation (e.g., ESCRT-III subunit removal could act to cinch the assembled lattice and thereby help to ‘extrude' a vesicle). Finally, removing a sufficient number of ESCRT-III subunits apparently also destabilizes the associated ESCRT-I and ESCRT-II complexes, thereby completing the cycle and freeing the Class E machinery to catalyze multiple rounds of protein sorting and vesicle formation.
Materials and methods
Cloning
DNA encoding hVPS4A and hVPS4B was amplified from EST #ATCC 81449 and #ATCC 6216963, respectively, and cloned into the BamHI/NdeI sites of modified pET16b (Novagen) and pGEX (Amersham) vectors that contained TEV protease cleavage sites. LIP5 (Ward et al, 2005) was subcloned and expressed from a modified pET16b vector. DNA encoding yVps4p and Vta1p proteins was amplified from yeast genomic DNA and expressed from pET151 (Invitrogen). Mammalian expression vectors for WT, and ATPase mutants of GFP-VPS4A and dsRed-hVPS4B proteins have been described (Garrus et al, 2001; von Schwedler et al, 2003). Constructs expressing mutant proteins were created by Quickchange mutagenesis (Stratagene) and verified by DNA sequencing.
Protein expression and purification
Methods for expressing and purifying the different Vps4 proteins and Vta1p were similar, and are described in detail for full-length hVPS4B. hVPS4B was expressed from the modified pET16b vector in BL21 (DE3) E. coli cells (Novagen) (IPTG 0.5 mM, A600=0.8, 2 h, 21°C). yVps4p/pET151 and Vta1p/pET151 expression was autoinduced in BL21 RP Codon+ E. coli cells (Stratagene) in ZY media (37°C for 6 h and 21°C overnight). Cells were harvested by centrifugation and lysed using 10 mg/ml lysozyme (40 min, 4°C; 50 mM imidazole, 500 mM NaCl, 50 mM Tris pH 8.0+protease inhibitor (Roche)), followed by sonication. The lysate was clarified by centrifugation (45 min, 35 000 g) and the soluble protein was purified by Ni2+ sepharose chromatography (Amersham) (50–750 mM imidazole pH 8.0). Fractions containing hVPS4B (∼500 mM imidazole) were assayed by SDS–PAGE, pooled and dialyzed sequentially into 150 mM NaCl, 20 mM Tris pH 8.0, and 5 mM EDTA (4°C), and then into a buffer containing 1 mM DTT in place of EDTA (21°C).
The histidine tag was removed by incubation with TEV protease (∼1 mg/100 mg protein, 12 h at 21°C). Cleaved hVPS4B was dialyzed into 20 mM MOPS pH 5.4, 100 mM NaCl, and 5 mM BME at 4°C and purified by cation exchange chromatography (SP sepharose, Amersham, linear 0.1–2 M NaCl gradient in 20 mM MOPS, pH 5.4). hVPS4B fractions were dialyzed into 150 mM NaCl, 20 mM Tris pH 6.0, and 5 mM BME (S75 buffer), concentrated, purified by gel filtration (S75, Amersham), and concentrated to 10 mg/ml. Yields were typically 5 mg hVPS4B/6 l culture. The protein was verified by N-terminal sequencing (G-H-M-S-S-T-S) and ESI mass spectrometry (MWcalc=49 496 Da, MWexp=49 494 Da).
Selenomethionine-substituted (SeMet) hVPS4B, VPS4A, VPS4A119–437, and hVPS4B126–444 E235Q were expressed and purified as above except that (1) SeMet hVPS4B cells were grown in M9 minimal media containing selenomethionine (50 mg/l) and (2) hVPS4B126–444 E235Q cleavage was inefficient, and so was performed with ∼5 mg protease/100 mg fusion protein. The mixture of cleaved and uncleaved hVPS4B126–444 E235Q was separated by anion exchange chromatography (Q sepharose, Amersham). The cleaved protein was collected in the flow through, dialyzed into 20 mM HEPES pH 7.6, 100 mM potassium acetate, 5 mM magnesium acetate, and 2 mM DTT (HR6 buffer), concentrated, and used directly in gel filtration experiments. Typical yields were 1 mg hVPS4B126–444 E235Q/6 l culture, and the TEV protease was not completely removed. For crystallization, His10-hVPS4B126–444 E235Q was purified by Ni2+ chromatography, the uncleaved protein was dialyzed into 150 mM NaCl, 20 mM Tris pH 8.0, 1 mM MgCl2, and 2 mM DTT (SD200 buffer), concentrated and purified by gel filtration (SD200, Amersham). Low molecular weight (dimer) fractions were pooled and concentrated to 7 mg/ml. Yields were typically 10 mg/6 l culture.
GST pulldown experiments
A 30 ml portion of E. coli cultures expressing either GST or GST-Vta1p was pelleted, resuspended in 2 ml buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, 5 mM BME, 0.02% NP-40, and 5% glycerol), and lysed by addition of 35 μl of 10 mg/ml lysozyme (20 min, 4°C), followed by 30 μl of 5% deoxycholate (20 min) and sonication. Soluble proteins were collected by centrifugation (30 min, 13 200 g). A 50 μl portion of glutathione agarose (Amersham) was preincubated with GST alone or GST-Vta1p (∼20 μM, 30 min, 4°C, buffer/2 mM ATP). Binding reactions (600 μl) were performed in buffer/2 mM ATP, prebound GST- or GST-Vta1p-glutathione agarose, and 8 μM purified Vps4p E233Q or Vps4p E233Q mutants (30 min, 4°C). Unbound proteins were removed by washing with buffer/2 mM ATP (3 × 1 ml). Bound proteins were eluted by boiling in 50 μl 2 × SDS–PAGE buffer and detected by SDS–PAGE.
Crystallography
Full-length hVPS4B was crystallized by hanging drop vapor diffusion on a glass coverslip at 21°C with 2 μl of 10 mg/ml hVPS4B in S75 buffer mixed with 3 μl reservoir solution (1.6 M ammonium sulfate, 0.1 M HEPES pH 7.5, and 0.1 M NaCl). Hexagonal (40 × 40 × 50 μm) crystals grew after ∼30 days. Crystals were briefly transferred to cyroprotectant (mother liquor supplemented with glycerol to 20%), suspended in a nylon loop, and cooled by plunging into liquid nitrogen. SeMet hVPS4B crystals were grown from 5 mg/ml protein solutions, cyroprotected, and frozen as above.
Native crystals of His10-hVPS4B126–444 E235Q were also grown (3 days) by hanging drop vapor diffusion at 21°C. A 2 μl portion of 7 mg/ml protein solution in SD200 buffer with 1 mM ATP was mixed with 3 μl of reservoir solution (1.0 M ammonium sulfate, 0.1 M Bis–Tris pH 5.5, 1% PEG 3350, and 0.1 M lithium chloride). Crystals were cyroprotected in mother liquor supplemented with glycerol to 25% and flash-frozen in liquid nitrogen.
Data collection and structure determination
Diffraction data from SeMet hVPS4B (single wavelength anomalous) and native His10-hVPS4B126–444 E235Q crystals were collected on Beamline 8.3.1 at the ALS in Berkeley, CA, using CCD ADSC Quantum 210 and Quantum 315 detectors. The SeMet hVPS4B data were processed with MOSFLM (Leslie, 1992) and SCALA (Evans, 1993) and seven of the eight selenium sites were found using SOLVE (Terwilliger, 2002) as implemented in the Elves package (Holton and Alber, 2004) and refined in MLPHARE (Otwinowski, 1991). Model building was performed using O (Jones et al, 1991) and the structure refined using REFMAC (Murshudov et al, 1999). Data processing and refinement statistics are given in Table I.
Assays for HIV expression, release, and infectivity
293T cells (six-well plates) were transfected using 9 μl Lipofectamine 2000 (Invitrogen) with 0.5 μg proviral HIV-R9+0.25 μg GFP-VPS4A or dsRed-hVPS4B expression vectors and analyzed 24 h later. Protocols for Western blots and single-cycle MAGI cell viral infectivity assays have been described (Garrus et al, 2001; von Schwedler et al, 2003).
Supplementary Material
Supplementary Figures
Acknowledgments
We thank Markus Babst and Diane Ward for helpful discussions and Heidi Schubert for crystallographic advice. This work was supported by NIH PO1 GM66521 (to CPH) and NIH RO1 AI 51174 (to WIS). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division, of the US DOE under Contract No. DE-AC03-76SF00098 at Lawrence Berkeley National Laboratory. Data deposition: Model coordinates and structure factors were deposited in the Protein Data Bank, http://www.pdb.org (PDB ID code 1XWI).
References
- Babst M, Katzmann D, Estepa-Sabal E, Meerloo T, Emr S (2002) Escrt-III. An endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell 3: 271–282 [DOI] [PubMed] [Google Scholar]
- Babst M, Sato TK, Banta LM, Emr SD (1997) Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO J 16: 1820–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M, Wendland B, Estepa EJ, Emr SD (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J 17: 2982–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop N, Woodman P (2000) ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol Biol Cell 11: 227–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K, Lottridge J, Helliwell SB, Goldthwaite LM, Luzio JP, Stevens TH (2004) Protein–protein interactions of ESCRT complexes in the yeast Saccharomyces cerevisiae. Traffic 5: 194–210 [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT (2005) Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol 347: 437–452 [DOI] [PubMed] [Google Scholar]
- Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS (2004) Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J 23: 1030–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, Data reduction (1993) Proceedings of CCP4 Study Weekend, 1993, on Data Collection & Processing 114–122
- Frickey T, Lupas AN (2004) Phylogenetic analysis of AAA proteins. J Struct Biol 146: 2–10 [DOI] [PubMed] [Google Scholar]
- Fujita H, Umezuki Y, Imamura K, Ishikawa D, Uchimura S, Nara A, Yoshimori T, Hayashizaki Y, Kawai J, Ishidoh K, Tanaka Y, Himeno M (2004) Mammalian class E Vps proteins, SBP1 and mVps2/CHMP2A, interact with and regulate the function of an AAA-ATPase SKD1/Vps4B. J Cell Sci 117: 2997–3009 [DOI] [PubMed] [Google Scholar]
- Furst J, Sutton RB, Chen J, Brunger AT, Grigorieff N (2003) Electron cryomicroscopy structure of N-ethyl maleimide sensitive factor at 11 Å resolution. EMBO J 22: 4365–4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai D, Zhao R, Li D, Finkielstein CV, Chen XS (2004) Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119: 47–60 [DOI] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107: 55–65 [DOI] [PubMed] [Google Scholar]
- Göttlinger HG, Dorfman T, Sodroski JG, Haseltine WA (1991) Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci USA 88: 3195–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein E, Bodem J, Muller B, Schmechel A, Zentgraf H, Krausslich HG (2003) The Mason-Pfizer monkey virus PPPY and PSAP motifs both contribute to virus release. J Virol 77: 9474–9485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H (2004) The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol 5: 317–323 [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C (1995) Dali: a network tool for protein structure comparison. Trends Biochem Sci 20: 478–480 [DOI] [PubMed] [Google Scholar]
- Holton J, Alber T (2004) Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci USA 101: 1537–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwich AL (2004) Chaperoned protein disaggregation—the ClpB ring uses its central channel. Cell 119: 579–581 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Karata K, Verma CS, Wilkinson AJ, Ogura T (2001) Probing the mechanism of ATP hydrolysis and substrate translocation in the AAA protease FtsH by modelling and mutagenesis. Mol Microbiol 39: 890–903 [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol 3: 893–905 [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the sterochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, Tsai FT (2003) The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115: 229–240 [DOI] [PubMed] [Google Scholar]
- Leslie AGW, Recent changes to the MOSFLM package for processing film and image plate data (1992) Joint CCP4+ESF-EAMCB Newsletter on Protein Crystallography, No. 26
- Lin Y, Kimpler LA, Naismith TV, Lauer JM, Hanson PI (2005) Interaction of the mammalian endosomal sorting complex required for transport (ESCRT) III protein hSnf7-1 with itself, membranes, and the AAA+ ATPase SKD1. J Biol Chem 280: 12799–12809 [DOI] [PubMed] [Google Scholar]
- Mancini EJ, Kainov DE, Grimes JM, Tuma R, Bamford DH, Stuart DI (2004) Atomic snapshots of an RNA packaging motor reveal conformational changes linking ATP hydrolysis to RNA translocation. Cell 118: 743–755 [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J, Yaravoy A, Perez-Caballero D, Bieniasz PD (2003a) Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci USA 100: 12414–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD (2003b) Role of ESCRT-I in retroviral budding. J Virol 77: 4794–4804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI (2004) Retrovirus budding. Annu Rev Cell Dev Biol 20: 395–425 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ (1999) Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr D 55 (Part 1): 247–255 [DOI] [PubMed] [Google Scholar]
- Niwa H, Tsuchiya D, Makyio H, Yoshida M, Morikawa K (2002) Hexameric ring structure of the ATPase domain of the membrane-integrated metalloprotease FtsH from Thermus thermophilus HB8. Structure (Camb) 10: 1415–1423 [DOI] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure—diverse function. Genes Cells 6: 575–597 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z (1991) Maximum likelihood refinement of heavy atom parameters. In Isomorphous Replacement and Anomalous Scattering, Wolf W, Evans PR, Leslie AGW (eds) pp 80–86. Warrington, UK: Darsbury Laboratory [Google Scholar]
- Scheuring S, Rohricht RA, Schoning-Burkhardt B, Beyer A, Muller S, Abts HF, Kohrer K (2001) Mammalian cells express two VPS4 proteins both of which are involved in intracellular protein trafficking. J Mol Biol 312: 469–480 [DOI] [PubMed] [Google Scholar]
- Scott A, Gaspar J, Stuchell-Brereton M, Alam SL, Skalicky JJ, Sundquist WI (2005) Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc Natl Acad Sci USA (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu-Xhilaga M, Ablan S, Demirov DG, Chen C, Montelaro RC, Freed EO (2004) Late domain-dependent inhibition of equine infectious anemia virus budding. J Virol 78: 724–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiflett SL, Ward DM, Huynh D, Vaughn MB, Simmons JC, Kaplan J (2004) Characterization of Vta1p, a class E Vps protein in S. cerevisiae. J Biol Chem 279: 10982–10990 [DOI] [PubMed] [Google Scholar]
- Siddiqui SM, Sauer RT, Baker TA (2004) Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes Dev 18: 369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HK, Hartmann C, Ramachandran R, Bochtler M, Behrendt R, Moroder L, Huber R (2000) Mutational studies on HslU and its docking mode with HslV. Proc Natl Acad Sci USA 97: 14103–14108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG (2003) AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114: 689–699 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC (2002) Automated structure solution, density modification and model building. Acta Crystallogr D 58: 1937–1940 [DOI] [PubMed] [Google Scholar]
- von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI (2003) The protein network of HIV budding. Cell 114: 701–713 [DOI] [PubMed] [Google Scholar]
- Wang J, Song JJ, Franklin MC, Kamtekar S, Im YJ, Rho SH, Seong IS, Lee CS, Chung CH, Eom SH (2001) Crystal structures of the HslVU peptidase–ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure (Camb) 9: 177–184 [DOI] [PubMed] [Google Scholar]
- Wang Q, Song C, Li CC (2003) Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem Biophys Res Commun 300: 253–260 [DOI] [PubMed] [Google Scholar]
- Ward DM, Vaughn MB, Shiflett SL, White PL, Pollock AL, Hill J, Schnegelberger R, Sundquist WI, Kaplan J (2005) The role of LIP5 and CHMP5 in multivesicular body formation and HIV-1 budding in mammalian cells. J Biol Chem 280: 10548–10555 [DOI] [PubMed] [Google Scholar]
- Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, Zentgraf H, Weber-Ban EU, Dougan DA, Tsai FT, Mogk A, Bukau B (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119: 653–665 [DOI] [PubMed] [Google Scholar]
- Yamada-Inagawa T, Okuno T, Karata K, Yamanaka K, Ogura T (2003) Conserved pore residues in the AAA protease FtsH are important for proteolysis and its coupling to ATP hydrolysis. J Biol Chem 278: 50182–50187 [DOI] [PubMed] [Google Scholar]
- Yeo SC, Xu L, Ren J, Boulton VJ, Wagle MD, Liu C, Ren G, Wong P, Zahn R, Sasajala P, Yang H, Piper RC, Munn AL (2003) Vps20p and Vta1p interact with Vps4p and function in multivesicular body sorting and endosomal transport in Saccharomyces cerevisiae. J Cell Sci 116: 3957–3970 [DOI] [PubMed] [Google Scholar]
- Yoshimori T, Yamagata F, Yamamoto A, Mizushima N, Kabeya Y, Nara A, Miwako I, Ohashi M, Ohsumi M, Ohsumi Y (2000) The mouse SKD1, a homologue of yeast Vps4p, is required for normal endosomal trafficking and morphology in mammalian cells. Mol Biol Cell 11: 747–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, Gorman MA, Kondo H, Dokurno P, Lally J, Leonard G, Meyer H, van Heel M, Freemont PS (2000) Structure of the AAA ATPase p97. Mol Cell 6: 1473–1484 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures