

Abstract
Although caspase-2 is believed to be involved in death receptor-mediated apoptosis, the exact function, mode of activation, and regulation of caspase-2 remain unknown. Here we show that protein kinase (PK) CK2 phosphorylates procaspase-2 directly at serine-157. When intracellular PKCK2 activity is low or downregulated by specific inhibitors, procaspase-2 is dephosphorylated, dimerized, and activated in a PIDDosome-independent manner. The activated caspase-2 then processes procaspase-8 monomers between the large and small subunits, thereby priming cancer cells for TNF-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis. The processed procaspase-8 that is recruited to death-inducing signaling complex by TRAIL engagement becomes fully activated, and cancer cells undergo apoptosis. PKCK2 activity is low in TRAIL-sensitive cancer cell lines but high in TRAIL-resistant cancer cell lines. Thus, downregulating PKCK2 activity is required for TRAIL-mediated apoptosis to occur in TRAIL-resistant cancer cells. Our data provide novel insights into the regulation, mode of activation, and function of caspase-2 in TRAIL-mediated apoptosis.
Keywords: caspase-2, caspase-8, PKCK2, priming, TRAIL
Introduction
Caspases are cysteine–aspartate proteases that play critical roles in the initiation and execution of apoptosis. Caspases exist as zymogens that contain a prodomain and a protease domain (Nicholson, 1999). They can be divided into initiator and effector caspases based on the presence of a large prodomain at their N-terminal region (Salvesen and Dixit, 1999). Initiator caspases that have long prodomains are activated with the help of adaptor molecules that bring these procaspases into close proximity, permitting autoprocessing (Fesik, 2000). Caspases with short prodomains are downstream effector caspases. Full processing and activation of these caspases depend on the upstream initiator caspases (Nicholson, 1999).
Caspase-2, one of the initiator caspases, has not been extensively studied, perhaps because caspase-2-deficient mice showed only subtle phenotypes (Bergeron et al, 1998; O'Reilly et al, 2002). Previous reports demonstrated that caspase-2 is required in stress-mediated apoptosis (Lassus et al, 2002) or in death receptor-mediated apoptosis (Duan and Dixit, 1997; Droin et al, 2001; Wagner et al, 2004). However, the exact function, mode of activation, and regulation of caspase-2 remain unknown.
TNF-related apoptosis-inducing ligand (TRAIL) is a promising cancer-specific therapeutic agent since it induces apoptosis in cancer cells but not in most normal cells or tissues (Ashkenazi et al, 1999; Walczak et al, 1999). One potential obstacle that may limit the clinical efficacy of TRAIL is the development of a TRAIL resistance phenotype in many cancer cells (Zhang et al, 1999; Kim et al, 2000). Specific protein kinase CK2 (PKCK2, formerly know as casein kinase 2) inhibitors have been successful at sensitizing TRAIL-resistant cancer cells to TRAIL, but the mechanism of sensitization remains controversial (Ravi and Bedi, 2002; Izeradjene et al, 2004, 2005). PKCK2 is a constitutively active, growth factor-independent serine/threonine protein kinase (Litchfield et al, 1994) composed of two catalytic α (and/or α′) subunits and two regulatory β subunits (Xu et al, 1999). It plays a key role in cell cycle control, cellular differentiation, and proliferation and also participates in the regulation of apoptosis by phosphorylating some apoptosis-related factors (Luscher et al, 1989; Allende and Allende, 1995; McElhinny et al, 1996; Wang et al, 2000; Keller et al, 2001; Li et al, 2002).
In an effort to elucidate the mechanism by which PKCK2 inhibition sensitizes TRAIL-resistant cancer cells to TRAIL, we found that procaspase-2 is activated by PKCK2 inhibition. The present study was therefore undertaken to examine whether the activation of procaspase-2 is regulated by PKCK2-mediated phosphorylation, to test whether caspase-2 is required to sensitize cancer cells to TRAIL, and to investigate the function of activated caspase-2 in TRAIL-mediated apoptosis. We show here that PKCK2 activity is high in TRAIL-resistant cancer cell lines but low in TRAIL-sensitive cancer cell lines and that PKCK2 phosphorylates procaspase-2 at serine-157. When PKCK2 is downregulated, procaspase-2 is dephosphorylated at serine-157. This causes procaspase-2 to dimerize and become activated in a PIDDosome-independent manner. The activated caspase-2 then processes procaspase-8 monomers between the large and small subunits. Once the processed procaspase-8 is present in the cell, the cancer cells are primed for TRAIL-mediated apoptosis.
Results
PKCK2 determines TRAIL sensitivity
Because specific PKCK2 inhibitors sensitized TRAIL-resistant cancer cells to TRAIL (Ravi and Bedi, 2002; Izeradjene et al, 2004), we hypothesized that TRAIL sensitivity of cancer cells is determined by intracellular PKCK2 activity and we analyzed intracellular PKCK2 activity in cancer cell lines. Consistent with our assumption, PKCK2 activity is low in the TRAIL-sensitive cancer cell lines but high in the TRAIL-resistant cell lines (Figure 1A and B and Supplementary Figure 1). 5,6-Dichloro-1-β-D-ribofuranosyl benzimidazole (DRB), a specific PKCK2 inhibitor, sensitized all the TRAIL-resistant cancer cell lines to TRAIL (Figure 1A). To further confirm our hypothesis, siRNA SMART Pool® was used to silence endogenous PKCK2α in TRAIL-resistant HCE4. When PKCK2α was silenced (Figure 1C, lanes 1–4, and Supplementary Figure 2), the cells were killed by TRAIL alone (Figure 1C). To test the effect of increased PKCK2 activity on TRAIL sensitivity, PKCK2α was exogenously overexpressed in TRAIL-sensitive TE2 cells (Figure 1D, lanes 3 and 4). This led to an increase in PKCK2 activity, and the cells became resistant to TRAIL (Figure 1D). Therefore, we conclude that the degree of PKCK2 activity determines TRAIL sensitivity.
Figure 1.
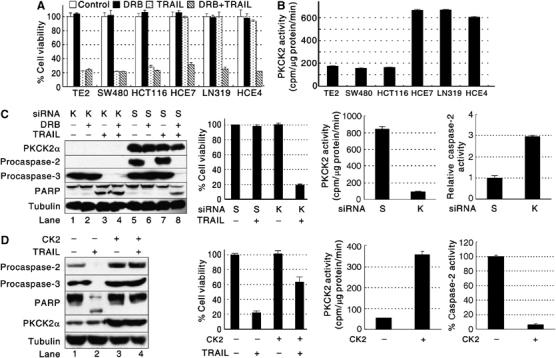
PKCK2 determines TRAIL sensitivity. (A) TRAIL cytotoxicity toward cancer cells. Cell viability was evaluated by the MTT assay using cancer cells incubated with or without DRB for 24 h and then subsequently treated with TRAIL for 2 h. The data are expressed as mean±s.d. for triplicate, and similar results were obtained from two independent experiments. (B) Endogenous PKCK2 activity in cancer cell lines. (C) Silencing of PKCK2α sensitizes cells to TRAIL. TRAIL-resistant HCE4 cells were transfected with scrambled siRNA (S) or PKCK2α SMART Pool® siRNAs (K) for 48 h and subsequently treated with DRB and/or TRAIL. Western blotting and cell viability assay were performed. PKCK2 activity and caspase-2 activity assays were performed using the siRNA-transfected cells. Relative caspase-2 activity, compared to the scrambled siRNA-transfected HCE4 cells, was calculated. (D) Acquisition of TRAIL resistance by exogenous PKCK2α expression. TRAIL-sensitive TE2 cells were transfected with an empty vector (CK2−) or with a PKCK2α expression vector (CK2+) for 24 h and subsequently incubated with TRAIL for 2 h. Western blotting and cell viability assay were performed. PKCK2 activity and caspase-2 activity assays were performed using TE2 cells transfected with an empty vector (−) or with the PKCK2α expression vector (+) for 24 h.
PKCK2 inhibits procaspase-2 activation by phosphorylation
To examine whether the activity of caspases is changed by treatment with DRB and/or TRAIL, colorimetric caspase activity assays were performed using HCE4 cells. Caspase-2, -3, -8, and -9 were activated in the presence of both DRB and TRAIL, but none of them were activated by TRAIL treatment alone (Figure 2A, left). It was noted that DRB alone induced caspase-2 activation without inducing cell death (Figure 2A, left, and Figure 1A). This result was confirmed using in vivo labeling of active caspases with biotin-VAD-fmk (Figure 2A, right). These results suggest that PKCK2-mediated phosphorylation may inhibit the activation of procaspase-2. To confirm this, HCE4 cells were treated with DRB in the presence or absence of okadaic acid (OA), a protein phosphatase PP-1 and PP-2A inhibitor. Procaspase-2 was phosphorylated at serine residue(s) (Figure 2B, bottom, lane 1), and DRB caused it to become dephosphorylated (Figure 2B, bottom, lane 2) indicating that PKCK2 is the kinase for procaspase-2. Dephosphorylation of procaspase-2 was not observed when the cells had been pretreated with OA (Figure 2B, bottom, lane 4 versus 2), suggesting the involvement of OA-sensitive phosphatase(s) for dephosphorylation of procaspase-2. When dephosphorylated, procaspase-2 is cleaved and activated; however, when OA pretreatment is used to maintain phosphorylation, procaspase-2 activation is prevented (Figure 2B, top). In addition, when PKCK2α was silenced, procaspase-2 was processed and activated even in the absence of both DRB and TRAIL in TRAIL-resistant HCE4 cells (Figure 1C). When PKCK2α was overexpressed, procaspase-2 was not processed and activated, even in the presence of TRAIL in TRAIL-sensitive TE2 cells (Figure 1D). Consistent with this, there was an inverse correlation between the intracellular PKCK2 activity and the caspase-2 activity in the cancer cell lines (Figure 2C versus Figure 1B). To test whether PKCK2 and procaspase-2 interact directly, HCE4 cells were transfected with an empty vector or HA-tagged wild-type procaspase-2 and their lysates were mixed with active human recombinant PKCK2. Western blotting revealed that PKCK2 co-immunoprecipitated with procaspase-2 in vitro (Figure 2D, top) and the interaction was confirmed in vivo (Figure 2D, bottom). Taken together, these results indicate that PKCK2 inhibits procaspase-2 activation by direct phosphorylation.
Figure 2.
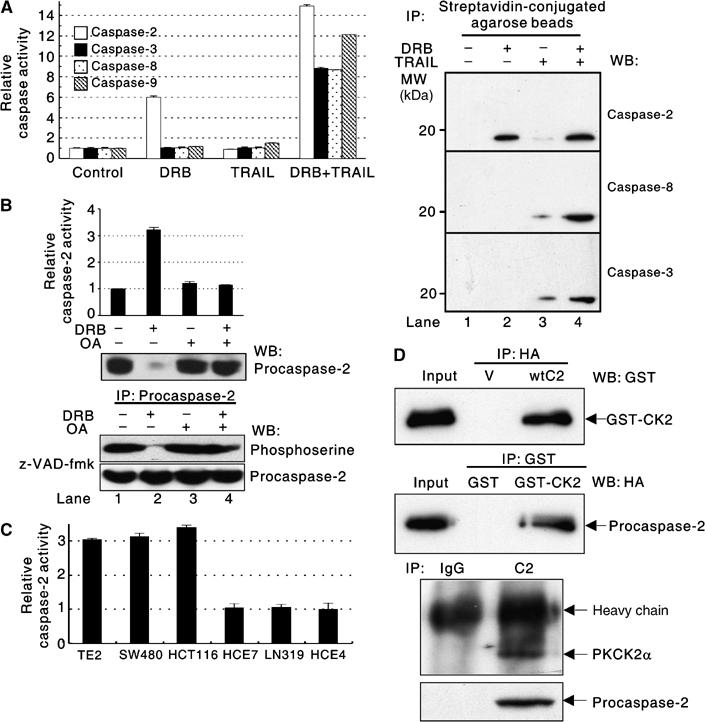
PKCK2 inhibits procaspase-2 activation by direct phosphorylation. (A) Changes in the activity of caspases in DRB- and/or TRAIL-treated HCE4 cells. (Left) Colorimetric caspase-2, -3, -8, and -9 activity assays were performed using cell extracts of HCE4 cells treated as in Figure 1A. (Right) In vivo peptide affinity labeling of active caspases was performed as described in Materials and methods. The activation of caspase-2, -8, or -3 was assessed by Western blotting. (B) PKCK2 inhibition induces dephosphorylation and activation of procaspase-2. Caspase-2 activity assays were performed using HCE4 cells that had been treated or untreated (control) with DRB (40 μM) for 24 h in the presence or absence of OA pretreatment (1.5 h, 30 nM). The data are presented as relative activity compared to the control and are expressed as mean±s.d. for triplicate, and similar results were obtained from two independent experiments. Immunoblotting for endogenous procaspase-2 using the same lysates is given below (top). HCE4 cells were treated with z-VAD-fmk (20 μM) and then incubated with DRB (40 μM) for 24 h in the presence or absence of OA (1.5 h pretreatment, 30 nM). Immunoprecipitation (IP) using anti-procaspase-2 antibody (Ab) was followed by Western blot analysis using anti-phosphoserine Ab. The same blot was reprobed with anti-procaspase-2 Ab (bottom). (C) Endogenous caspase-2 activity in cancer cell lines. Relative caspase-2 activity compared to HCE4 was calculated. (D) (Top) HCE4 cells were transfected with empty vectors (V) or HA-tagged wild-type procaspase-2 (wtC2); cell lysates were mixed with active human recombinant PKCK2 (GST-CK2). IP with a GST- or HA-specific Ab was followed by Western blotting with the indicated Abs. (Bottom) The lysates from the HCE4 cells were immunoprecipitated with anti-procaspase-2 Ab (C2) or nonimmune IgG (IgG) followed by Western blotting using a PKCK2α-specific Ab. The same blot was reprobed with anti-procaspase-2 Ab.
PKCK2 phosphorylates procaspase-2 at serine-157
Two potential serine phosphorylation sites were identified in a tryptic digest of procaspase-2 (Figure 3A, top). To determine which serine is phosphorylated, procaspase-2 expression plasmids with various combinations of serine to alanine mutations were constructed (Figure 3A, top, mtC2-1 to mtC2-3) and used for transfection. Metabolic labeling and autoradiography revealed that serine-157 is the phosphorylation site (Figure 3A, middle; the numbering of residues is according to Kumar et al, 1997). To examine how dephosphorylated procaspase-2 is activated, HCE4 cells were transfected with either a C320A (catalytic cysteine-320 replaced with alanine) or a nonphosphorylatable S157A/C320A (serine-157 and cysteine-320 replaced with alanine) mutant procaspase-2 gene. While the C320A mutant formed a dimer only when the cells were treated with DRB (Figure 3B, top, lane 1 versus 2), the nonphosphorylatable S157A/C320A mutant dimerized constitutively (Figure 3B, top, lanes 3 and 4). These results were confirmed by cotransfecting HCE4 cells with the following pairs of mutant procaspase-2 genes: an HA-tagged C320A and V5-tagged C320A, an HA-tagged S157A/C320A and V5-tagged S157A/C320A, or an HA-tagged C320A and V5-tagged S157A/C320A. Western blot analysis following IP showed that only dephosphorylated procaspase-2 monomers were able to interact with each other (Figure 3B, bottom). A recent study revealed that the activation of caspase-2 occurs in the PIDDosome, a complex containing the death domain-bearing protein PIDD (p53-induced protein with a death domain) and the adapter protein RAIDD (Tinel and Tschopp, 2004). To test whether the dephosphorylation-mediated activation of procaspase-2 is dependent on the PIDDosome, siRNA was used to silence the expression of PIDD in HCE4 cells. Procaspase-2 was processed by PKCK2 inhibition even in the absence of PIDD (Figure 3C). Therefore, we conclude that procaspase-2 is activated by dimerization between dephosphorylated procaspase-2 monomers in a PIDDosome-independent manner.
Figure 3.
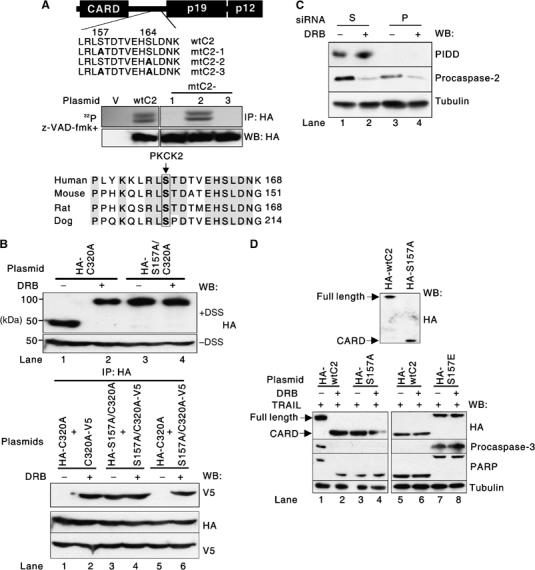
Procaspase-2 is phosphorylated at serine-157. (A) Procaspase-2 is phosphorylated at serine-157. (Top) Schematic representation of procaspase-2. CARD, caspase recruitment domain; p19 and p12, the large and small subunits that form mature caspase-2. The mutations introduced into procaspase-2 are highlighted in bold face. (Middle) Extracts of z-VAD-fmk-pretreated HCE4 cells transfected with either an empty pCMV-HA vector (V), a wild-type procaspase-2 (wtC2) gene, or a mutant procaspase-2 (mtC2-1 to mtC2-3) gene for 24 h and metabolically labeled for 6 h in the presence of orthophosphate (1 mCi/ml) were immunoprecipitated with anti-HA Ab and subjected to SDS–PAGE and autoradiography (32P). (Bottom) Comparison of the human procaspase-2 amino-acid sequence (residues 149–168) with mouse, rat, and dog procaspase-2, showing conservation of serine-157 (boxed). (B) Dimerization of dephosphorylated procaspase-2. (Top) Western blotting was performed using crosslinked (+DSS, disuccinimidyl suberate) or non-crosslinked (−DSS) extracts of HCE4 cells that had been transfected with the indicated plasmids for 24 h and subsequently incubated with or without DRB for 24 h. (Bottom) HCE4 cells were cotransfected with the indicated plasmids for 24 h and subsequently incubated with or without DRB for 24 h. IP was then performed using anti-HA Ab, followed by Western blot analysis with anti-V5 Ab. Western blots of whole-cell lysates confirmed the presence of both HA- and V5-tagged procaspase-2 in the lysates. (C) PIDDosome-independent activation of procaspase-2. Western blotting was performed using extracts of HCE4 cells transfected with either scrambled siRNA (S) or PIDD siRNA for 48 h and subsequently treated with or without DRB for 24 h. (D) Effect of phosphorylation site mutant procaspase-2 on TRAIL-mediated apoptosis. Western blotting was performed using extracts of TRAIL-resistant HCE4 cells that had been transfected with the indicated plasmids for 24 h (top). Western blotting was performed using extracts of HCE4 cells (bottom left) or TRAIL-sensitive TE2 cells (bottom right) that had been transfected with the indicated plasmids for 24 h and subsequently treated with TRAIL in the presence or absence of DRB. ‘Full length' and ‘CARD' indicate the full-length procaspase-2 and the HA-tagged CARD that is generated after activation of procaspase-2, respectively.
We then examined the effect of a phosphorylation site mutant procaspase-2 on TRAIL-mediated apoptosis. When TRAIL-resistant HCE4 cells were transfected with the nonphosphorylatable S157A mutant procaspase-2 gene, the exogenous procaspase-2 was processed and activated even in the absence of TRAIL (Figure 3D, top), and the cells were killed by TRAIL in the absence of DRB (Figure 3D, bottom left, lane 3 versus 1). When TRAIL-sensitive TE2 cells were transfected with an S157E mutant procaspase-2 gene (serine-157 replaced with glutamic acid to mimic constitutive phosphorylation), procaspase-2 was not processed and activated (Figure 3D, bottom right, lanes 7 and 8), and the cells became resistant to TRAIL even in the presence of DRB (Figure 3D, bottom right, lane 8). These results indicate that the S157A and S157E procaspase-2 mutants act as a dominant active and a dominant negative procaspase-2 in TRAIL-mediated apoptosis, respectively. Collectively, the activation of procaspase-2 is negatively regulated by PKCK2-mediated phosphorylation at serine-157.
Procaspase-2 activation is a prerequisite for TRAIL-mediated apoptosis
To determine whether the activation of procaspase-2 by PKCK2 inhibition is required for TRAIL-mediated apoptosis, siRNA was used to silence the expression of endogenous procaspase-2. When procaspase-2 was silenced, DRB could not sensitize HCE4 cells (Figure 4A, top, lane 4 versus 8) and LN319 cells (data not shown) to TRAIL. TRAIL-sensitive SW480 cells became resistant to TRAIL by the silencing of procaspase-2 as well (Figure 4A, bottom, lane 3 versus 7 and 4 versus 8). To test whether TRAIL-mediated apoptosis indeed required caspase-2, we attempted to restore sensitivity to TRAIL by expressing caspase-2 ectopically (Figure 4B). To prevent the destruction of the ectopic caspase-2 mRNA by the caspase-2 siRNA, we introduced two silent mutations into the region of the caspase-2 cDNA that is complementary to the siRNA (C2sil). Expression of C2sil restored the sensitivity of TRAIL-resistant HCE4 cells to TRAIL in the presence of DRB (Figure 4B, lane 2 versus 6), indicating that the effect of the siRNA (Figure 4A) could be explained by the silencing of caspase-2. When PKCK2α and procaspase-2 were co-silenced in HCE4 cells, the cells were not killed by TRAIL (Figure 4C). To examine whether PKCK2 inhibition also sensitizes normal cells to TRAIL, human primary normal oral keratinocytes and HCE4 cells were treated with TRAIL in the presence or absence of DRB pretreatment. Although procaspase-2 was processed and activated by DRB in both cells (Figure 4D, lanes 3 and 7), only HCE4 cells underwent TRAIL-induced apoptosis (Figure 4D, lane 4 versus 8). In addition, the siRNA-mediated ablation of caspase-2 expression blocked the ability of TNF-α (Figure 4E, lane 2 versus 4) or an agonistic anti-FAS Ab (Figure 4E, lane 6 versus 8) to induce cell death in the presence of DRB. Therefore, we conclude that PKCK2 acts upstream of procaspase-2 and that the activation of procaspase-2 is a prerequisite for death receptor-mediated apoptosis to occur in cancer cells but not in normal cells.
Figure 4.
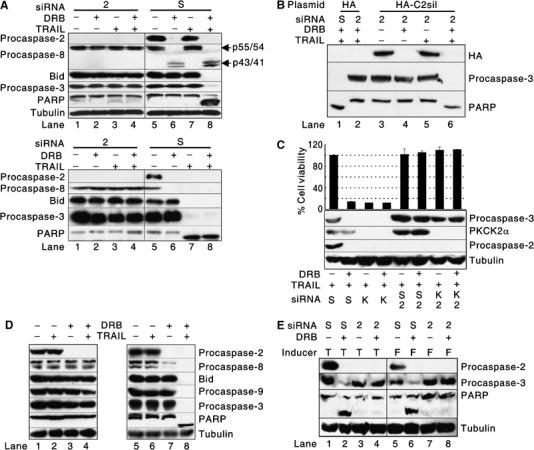
Caspase-2 activation is a prerequisite for TRAIL-mediated apoptosis. (A) Effect of procaspase-2 silencing on TRAIL-mediated apoptosis. Western blotting was performed using extracts of TRAIL-resistant HCE4 (top) or TRAIL-sensitive SW480 (bottom) cells that had been transfected with either a scrambled siRNA (S) or procaspase-2 siRNA (2) for 48 h and subsequently treated with TRAIL and/or DRB. (B) Restoration of sensitivity of HCE4 cells to TRAIL-mediated apoptosis after ectopic expression of caspase-2. Western blotting was performed using extracts of HCE4 cells cotransfected with either scrambled siRNA (S) and empty vectors (HA), caspase-2 siRNA (2) and empty vectors, or caspase-2 siRNA and C2sil (HA-C2sil) for 48 h. The cells were subsequently treated with DRB for 24 h and TRAIL (100 ng/ml) for 2 h. (C) Co-silencing of PKCK2α and procaspase-2. HCE4 cells were transfected with PKCK2α SMART Pool® siRNA (K) and/or procaspase-2 siRNA (2) and then treated with TRAIL and/or DRB. Western blotting (bottom) and cell viability (top) assays were performed. (D) Difference in the mechanism of TRAIL resistance between human primary normal cells and TRAIL-resistant cancer cells. Human primary normal oral keratinocytes (left) and HCE4 cells (right) were treated or untreated with DRB for 24 h and subsequently treated with TRAIL for 2 h. Western blotting was performed using indicated Abs. (E) Caspase-2 is also required in apoptosis that is induced by apoptosis-inducing ligands other than TRAIL. HCE4 cells were transfected with either scrambled siRNA (S) or caspase-2 siRNA (2) and then treated with TNF-α (Inducer, T) or an agonistic anti-FAS Ab (Inducer, F) for 2 h in the presence or absence of DRB pretreatment. Western blotting was performed using indicated Abs.
Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8
It was previously reported that full activation of caspase-8 requires two sequential cleavage events: the first one separates the large and small subunits producing the p43/41 form and the second one separates the large subunit and the prodomain (Medema et al, 1997; Chang et al, 2003). When procaspase-2 was activated by DRB, procaspase-8 was processed to its p43/41 form even in the absence of TRAIL engagement. However, when procaspase-2 was silenced, the processing was not observed (Figure 4A, lane 6 versus 2), indicating that caspase-2 processes the procaspase-8 monomer between the large and small subunits. Consistent with this observation, the processed form of procaspase-8 was present in TRAIL-sensitive cancer cell lines (Figure 5A, Procaspase-8, z-VAD-fmk−), in which a notable fraction of the endogenous procaspase-2 pool was activated due to low intracellular PKCK2 activity (Figure 5A, Procaspase-2, z-VAD-fmk−). These results suggest that caspase-2-mediated processing of procaspase-8 may prime cancer cells for TRAIL-mediated apoptosis. To examine whether short-term inhibition of caspase-2 alters the responsiveness of the primed cancer cells to TRAIL, TE2 cells were pretreated with z-VDVAD-fmk, a caspase-2 inhibitor, for 0.5 h before addition of TRAIL. The processed form of procaspase-8 was present in the cells at the time of TRAIL engagement (Figure 5B, bottom, lane 1) and the cells were killed by TRAIL (Figure 5B). In contrast, in the cells pretreated with z-VDVAD-fmk for 6 h, the processed form of procaspase-8 was not present (Figure 5B, bottom, lane 3) and the cells became resistant to TRAIL (Figure 5B). These results suggest that the processed procaspase-8 in the primed cancer cells may be rapidly eliminated possibly through the ubiquitin–proteasome pathway (Kim et al, 2004a). Indeed, when the cells were coincubated with proteasome inhibitor MG-132 and z-VDVAD-fmk for 6 h, the processed form of procaspase-8 was detected (Figure 5B, bottom, lane 5 versus 3) and the cells were killed by TRAIL (Figure 5B). To examine the status of procaspase-8 in the death-inducing signaling complex (DISC), HCE4 cells were primed by pretreating with DRB and then subsequently incubated with z-VDVAD-fmk for 0.5 h or 6 h. Only the processed form of procaspase-8 was found in the DISC when the cells were primed by DRB (Figure 5C, top, lane 2) or when the primed cells were pretreated with z-VDVAD-fmk for 0.5 h (Figure 5C, top, lane 3). In contrast, when caspase-2 was inhibited for 6 h in the primed cells, only the unprocessed procaspase-8 was present in the DISC (Figure 5C, top, lane 4) as observed in unprimed control HCE4 cells (Figure 5C, top, lane 1) and the cells became resistant to TRAIL (Figure 5C, bottom). These results indicate that TRAIL-mediated apoptosis can occur only in the primed cells in which the processed form of procaspase-8 is present at the moment of TRAIL engagement. Therefore, we conclude that the processing of procaspase-8 between the large and small subunits by caspase-2 primes cancer cells for TRAIL-mediated apoptosis, and we define caspase-2 as a ‘priming caspase' in TRAIL-mediated apoptosis.
Figure 5.
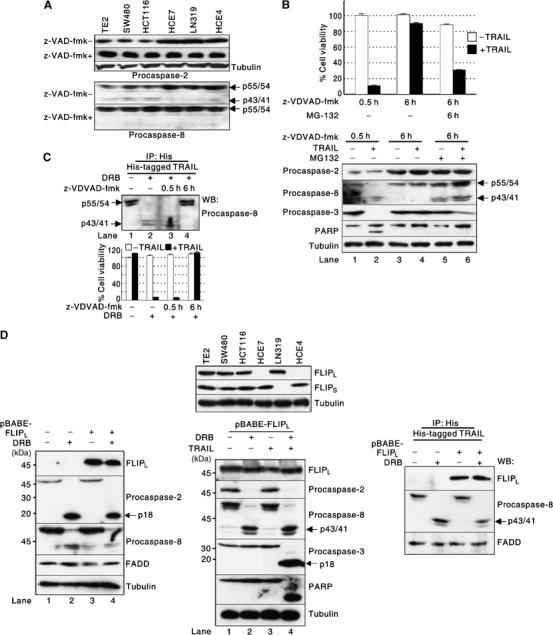
Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. (A) Expression level and status of endogenous procaspase-2 and procaspase-8 in the cancer cell lines. Western blotting was performed using extracts of the cancer cell lines treated or untreated with z-VAD-fmk. (B) Priming by the presence of processed form of procaspase-8. TRAIL-sensitive TE2 cells were incubated in the presence or absence of MG-132 (10 μM) with z-VDVAD-fmk for 6 h or incubated with z-VDVAD-fmk for 0.5 h and then treated with TRAIL for an additional 2 h. Cell viability assay (top) and Western blotting (bottom) were performed. (C) Procaspase-8 in the DISC. HCE4 cells were treated or untreated with DRB for 24 h. The cells were further incubated with z-VDVAD-fmk for the indicated times. DISC IP was performed followed by Western blotting (top). A cell viability assay was performed in the presence or absence of TRAIL (bottom). (D) Effect of c-FLIP overexpression on DRB-mediated sensitization of cancer cells to TRAIL. Western blotting was performed using extracts of the cancer cell lines with indicated Abs (top). HCE4 cells were transduced with c-FLIPL-expressing retroviruses and the cells were treated with or without DRB for 24 h. Western blotting was performed using indicated Abs (bottom left). HCE4 cells were transduced with c-FLIPL-expressing retroviruses and the cells were treated with TRAIL for 2 h in the presence or absence of DRB pretreatment. Western blotting was performed using indicated Abs (bottom middle). HCE4 cells were transduced with c-FLIPL-expressing retroviruses and the cells were treated with or without DRB for 24 h. DISC IP was performed followed by Western blotting using indicated Abs (bottom right).
Primed cancer cells undergo apoptosis even in the presence of FLIP overexpression
It has been known that FLICE inhibitory protein (FLIP) regulates apoptosis by precluding recruitment of caspase-8 to the DISC and thereby prevents its proteolytic cleavage (Krueger et al, 2001; Rippo et al, 2004). To evaluate the correlation between FLIP expression and TRAIL sensitivity, we first checked the expression level of c-FLIP short (c-FLIPS) and long form (c-FLIPL) in the cell lines. c-FLIPL was expressed in all the TRAIL-sensitive cell lines and one TRAIL-resistant cell line (Figure 5D, top, FLIPL). In the case of c-FLIPS, it was expressed in all of the TRAIL-sensitive cell lines and two TRAIL-resistant cell lines (Figure 5D, FLIPS). These results indicate that endogenous FLIP may not be a determinant of TRAIL sensitivity. We then examined whether the overexpression of c-FLIPL alters the responsiveness of the primed cancer cells to TRAIL. When c-FLIPL was overexpressed, the activation of procaspase-2 (Figure 5D, bottom left, lane 4; bottom middle, lane 2) and the processing of procaspase-8 (Figure 5D, bottom, lane 2 versus 4) were still observed by PKCK2 inhibition, and the cells were also sensitized to TRAIL (Figure 5D, bottom middle, lane 4). To examine whether there is a difference in the amounts of recruited procaspase-8 between the presence and the absence of c-FLIPL overexpression, DISC IP was performed. We found that there was no difference in the amount of recruited procaspase-8 in the DISC, even in the presence of overexpressed c-FLIPL (Figure 5D, bottom right). These results suggest that overexpressed FLIP may not act as a negative modulator of the processed procaspase-8 activation in TRAIL-induced DISC.
Discussion
Although previous reports showed that caspase-2 is required in stress-mediated apoptosis (Lassus et al, 2002) or in death receptor-mediated apoptosis (Duan and Dixit, 1997; Droin et al, 2001), little is known about the exact function, mode of activation, and regulation of caspase-2. Our data show that the activation of procaspase-2 is regulated by PKCK2-mediated phosphorylation at serine-157, that dephosphorylated procaspase-2 is activated by dimerization, and that active caspase-2 cleaves procaspase-8 monomer between the large and small subunits, thereby priming TRAIL-resistant cancer cells for TRAIL-mediated apoptosis. Therefore, we propose a novel mechanism by which TRAIL-mediated apoptosis is regulated (Figure 6).
Figure 6.
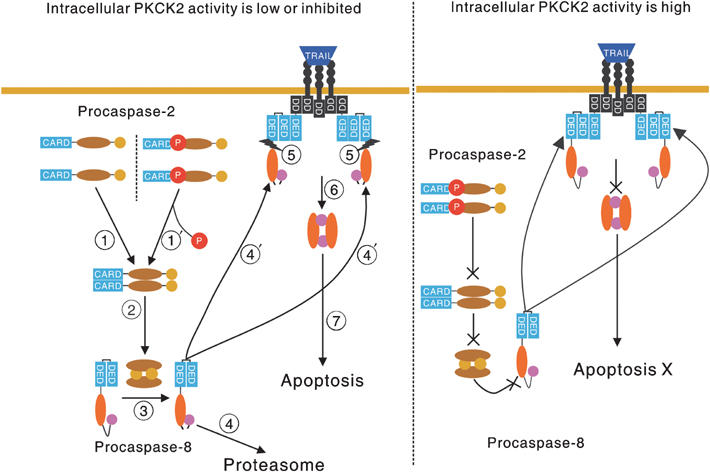
Proposed mechanism by which TRAIL-mediated apoptosis is regulated. (Left) When intracellular PKCK2 activity is low ( ) or when high intracellular PKCK2 activity is downregulated by a specific inhibitor (
) or when high intracellular PKCK2 activity is downregulated by a specific inhibitor ( ), dephosphorylated procaspase-2 monomers form dimers. Procaspase-2 is then activated through the dimerization (
), dephosphorylated procaspase-2 monomers form dimers. Procaspase-2 is then activated through the dimerization ( ), and activated caspase-2 cleaves procaspase-8 monomer between the large and small subunits (
), and activated caspase-2 cleaves procaspase-8 monomer between the large and small subunits ( ). At this point, the cancer cells are ‘primed' for TRAIL-mediated apoptosis. If TRAIL is not bound to the TRAIL-death receptor(s), the cleaved procaspase-8 goes to the proteasome for degradation (
). At this point, the cancer cells are ‘primed' for TRAIL-mediated apoptosis. If TRAIL is not bound to the TRAIL-death receptor(s), the cleaved procaspase-8 goes to the proteasome for degradation ( ). On the other hand, if TRAIL is bound to its receptor, the cleaved procaspase-8 is recruited to the TRAIL death receptor(s), thereby forming DISC (
). On the other hand, if TRAIL is bound to its receptor, the cleaved procaspase-8 is recruited to the TRAIL death receptor(s), thereby forming DISC ( ). The second cleavage between the prodomain and large subunit could occur efficiently by DISC-mediated dimerization of the cleaved procaspase-8 (
). The second cleavage between the prodomain and large subunit could occur efficiently by DISC-mediated dimerization of the cleaved procaspase-8 ( ), which leads to the activation of procaspase-8 (
), which leads to the activation of procaspase-8 ( ), and TRAIL-mediated apoptosis (
), and TRAIL-mediated apoptosis ( ). (Right) When intracellular PKCK2 activity is high, procaspase-2 cannot be activated; thus, procaspase-8 cannot be processed. Even with the engagement of TRAIL, procaspase-8 in the DISC cannot be fully activated, and thus, TRAIL-mediated apoptosis cannot occur.
). (Right) When intracellular PKCK2 activity is high, procaspase-2 cannot be activated; thus, procaspase-8 cannot be processed. Even with the engagement of TRAIL, procaspase-8 in the DISC cannot be fully activated, and thus, TRAIL-mediated apoptosis cannot occur.
Recent studies have shown that formation of a stable dimeric intermediate induces procaspase-8 activation by intramolecular processing between the large and small subunits. This is the first step in the cleavage of procaspase-8 after dimer formation and renders the region between the large subunit and prodomain susceptible to further cleavage (Chen et al, 2002; Boatright et al, 2003; Chang et al, 2003). However, caspase-8 activation is inefficient in the dimer when intramolecular processing between the large and small subunits does not occur (Chen et al, 2002; Chang et al, 2003). Consistent with this, procaspase-8 was not activated by DISC-mediated dimerization in TRAIL-resistant cancer cells, in which PKCK2 activity is high (Figure 5C, top, lane 1 versus 2).
A recent study revealed that activation of procaspase-2 occurs in the PIDDosome (Tinel and Tschopp, 2004). However, we found that the dephosphorylation-mediated dimerization and activation of procaspase-2 can occur in the absence of PIDD (Figure 3C). It is possible that PIDDosome-dependent caspase-2 activation may operate only in p53-dependent genotoxic stress-mediated apoptosis.
It is notable that the sequence around serine-157 is highly conserved between human, mouse, rat, and dog procaspase-2 (Figure 3A, bottom). However, this conservation is not observed in chicken (data not shown), suggesting that the regulatory mechanism of procaspase-2 activation by PKCK2 may operate only in mammals.
PKCK2 activity is invariably elevated in tumors but not in normal tissues (Gapany et al, 1995; Faust et al, 1996; Ahmed et al, 2000), and normal cells are resistant to TRAIL (Ashkenazi et al, 1999; Walczak et al, 1999). When we examined whether normal primary epithelial cells can be sensitized to TRAIL by DRB, we found that procaspase-2 was activated in both cells, but the processing of procaspase-8 and apoptotic cell death were observed only in TRAIL-resistant cancer cells (Figure 4D). Accordingly, we speculate that primary normal cells or normal cells in tissues are resistant to TRAIL because they may develop a mechanism(s) by which activated caspase-2 cannot recognize and cleave procaspase-8. This could be the reason(s) why caspase-2-deficient mice have only a few abnormalities (Bergeron et al, 1998). It is possible that the overt phenotypes of caspase-2 deficiency can be observed only in certain pathologic condition(s), such as cancer.
PKCK2 inhibition by DRB also sensitized cancer cells to TNF-α or agonistic anti-FAS Ab-mediated apoptosis, but the silencing of procaspase-2 expression blocked the ability of TNF-α or an agonistic anti-FAS Ab to induce cell death even in the presence of DRB (Figure 4E). These results suggest that the priming of procaspase-8 by caspase-2-mediated processing is a general mechanism for death receptor-mediated apoptosis. In addition, it has been reported that the combined use of a DNA-damaging cancer chemotherapeutic and TRAIL synergistically kill TRAIL-resistant cancer cells (Lacour et al, 2001), and that cytotoxic stress such as DNA damage induces cell death through procaspase-2 activation (Lassus et al, 2002). When we checked PKCK2 activity after cytotoxic stress, PKCK2 activity was downregulated and procaspase-2 was activated by etoposide or cisplatin treatment (unpublished observation). Therefore, PKCK2-regulated procaspase-2 activation plays a central role in both death receptor-mediated and stress-mediated apoptosis.
In summary, we have shown the novel mechanism by which caspase-2 activation is regulated, and the role of caspase-2 in TRAIL-mediated apoptosis.
Materials and methods
Cell lines and culture conditions
The human esophageal cancer cell lines (TE2, HCE4, and HCE7) and human colon cancer cell lines (SW480 and HCT116) were grown as previously described (Kim et al, 2000). The human malignant glioma cell line LN319 was cultured at 37°C in DMEM containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. Human primary normal oral keratinocytes were kindly provided by Dr Jongin Yook (Yonsei University College of Dentistry). The keratinocytes were grown in Keratinocyte-SFM medium (Gibco BRL, Rockville, MD) containing bovine pituitary extract (40 μg/ml) and epidermal growth factor (1 ng/ml). The cells were treated for 2 h with either 100 ng/ml TRAIL (Kim et al, 2004b), 35 ng/ml TNF-α (Sigma-Aldrich Co., St Louis, MO), or 50 ng/ml of an agonistic anti-FAS Ab (clone CH11; Upstate Biotechnology, Lake Placid, NY). OA, a protein phosphatase PP-1 and PP-2A inhibitor, was purchased from Sigma-Aldrich. Cells were pretreated with OA for 1.5 h at a final concentration of 30 nM. Inhibition of PKCK2 was achieved by incubating the cells with DRB (Calbiochem, La Jolla, CA) for 24 h at a final concentration of 40 μM (Zandomeni, 1989). The irreversible pan-caspase inhibitor, z-VAD-fmk (R&D Systems, Minneapolis, MN), was used at a final concentration of 20 μM. Biotin-VAD-fmk was purchased from Calbiochem. The caspase-2 inhibitor, z-VDVAD-fmk, was purchased from R&D Systems and used at a 20 μM concentration. MG-132, a proteasome inhibitor, was purchased from Calbiochem. DSS (5 mM; Pierce, Rockford, IL) was used to crosslink interacting proteins and to maintain the crosslinks under the reducing conditions of SDS–PAGE.
siRNAs
siRNAs were purchased from Dharmacon Inc. (Option A4; Lafayette, CO). The target sequences for caspase-2 (Lassus et al, 2002) and PIDD (Tinel and Tschopp, 2004) were previously described. Dharmacon SMART Pool® siRNAs were used for silencing PKCK2α. A scrambled siRNA was used as a control.
Site-directed mutagenesis
To introduce silent mutations into the regions of the procaspase-2 cDNA that are cognate to the corresponding siRNA, the nucleotide pair GC in positions 161–162 of the procaspase-2 gene was mutated into AT (C2sil) (Lassus et al, 2002) using a Quick change site-directed mutagenesis kit (Stratagene, La Jolla, CA). Potential serine phosphorylation sites in wild-type procaspase-2 were also mutagenized into alanine as shown in Figure 3A.
cDNAs
Wild-type or mutant procaspase-2 cDNAs were cloned into either pCMV-HA (Clonetech, Palo Alto, CA) or pcDNA3.1/V5-His (Invitrogen, Carlsbad, CA). PIDD and PKCK2α cDNAs were cloned into pcDNA3.1/V5-His.
Transfection
Cells were transfected with the siRNAs and/or plasmids using either Lipofectamine 2000 (Invitrogen) or Nucleofector (AMAXA, Gaithersburg, MD, Program A-28) according to the manufacturers' protocol.
Cell viability assay
Cell viability was measured using the MTT assay (Kim et al, 2000). The data are expressed as mean±s.d. for triplicate, and similar results were obtained from two independent experiments.
Western blot immunostaining
Western blot analysis was performed as previously described (Kim et al, 2000). The cell lysates were prepared by directly adding 1 × SDS sample loading buffer to the cells followed by 15 bursts of sonication (cycle 0.5, amplitude 80%) and boiling at 100°C for 5 min. The blotted membranes were immunostained with Abs specific for the following antigens: phosphoserine (Zymed Laboratories Inc., South San Francisco, CA), caspase-2 (BD Transduction Laboratories, San Jose, CA and Delta Biolabs, Campbell, CA), caspase-3 (Santa Cruz Biotechnology Inc., Santa Cruz, CA), caspase-8 (Cell Signaling Technology Inc., Beverly, MA), Bid (Cell Signaling Technology), Fas-associated death domain protein (FADD; Upstate Biotechnology, Lake Placid, NY), FLICE-like inhibitory protein (FLIPL; Alexis Biochemicals, San Diego, CA), FLIPS (Calbiochem), PIDD (Alexis Biochemicals), poly ADP-ribose polymerase (PARP; Cell Signaling Technology Inc.), PKCK2α (Upstate Biotechnology), HA (Covance, Berkeley, CA), V5 (Abcam Inc., Cambridge, MA), GST (Santa Cruz Biotechnology Inc.), and α-tubulin (Oncogene Science, Cambridge, MA).
Caspase activity assays
Activity of caspase-2, -3, -8, or -9 was detected using colorimetric assay kits (R&D Systems). The data are expressed as mean±s.d. for triplicate, and similar results were obtained from two independent experiments.
PKCK2 activity assay
PKCK2 activity was measured using a Casein Kinase-2 Assay Kit (Upstate Biotechnology). A recombinant full-length human PKCK2 protein that contains an α-subunit N-terminal 6xHis tag and a β-subunit N-terminal GST tag (Upstate Biotechnology) was used as a positive control.
Immunoprecipitation
Cells were collected and lysed in 1 ml IP lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, and 0.5% NP-40) with 1 × protease inhibitor cocktail (Roche Diagnostics). The cell lysates were precleared and then incubated with the indicated Abs for 1 h at 4°C. The complexes were precipitated with Protein A/G-Sepharose beads (Santa Cruz Biotechnology Inc.), washed, and resuspended in 40 μl 1 × SDS sample buffer. Nonimmune mouse IgG (Santa Cruz Biotechnology Inc.) served as a negative control.
Retrovirus production and viral transduction
FLIPL was cloned into pBABE-puro. An amphotropic retroviral packaging cell line, φNX-ampho, was transfected with the plasmid. At 48 h after transfection, the supernatant was collected. The supernatant with polybrene added (800 μg/ml) was used to infect HCE4 cells. At 48 h after infection, the cells were used for experiments.
DISC IP
To perform DISC IP, cells were grown, harvested, and resuspended in 2 ml of DMEM (1.5 × 107 cells/ml). TRAIL (500 ng/ml) was added to the resuspended cells. The cells were treated with TRAIL for 10 min at 37°C in a water bath with intermittent agitation. TRAIL treatment was stopped by adding 5 ml of ice-cold PBS, followed by immediate centrifugation. After washing once more with 2 ml of ice-cold PBS, the cells were lysed with IP lysis buffer with 1 × protease inhibitor cocktail for 30 min on ice. The cell lysates were cleared by centrifugation (15 000 r.p.m. × 15 min × 2) at 4°C. IP was then performed with anti-His Ab (R&D systems).
In vivo labeling of active caspases
To label the active site of caspases, 1 × 107 cells were incubated for 1 h with 10 μM biotin-VAD-fmk following apoptosis induction. Cells were collected and lysed in 1 ml of IP lysis buffer with 1 × protease inhibitor cocktail. The biotinylated proteins were captured using 30 μl of streptavidin-conjugated agarose beads (Calbiochem). After overnight rotation at 4°C, the agarose beads were extensively washed in lysis buffer containing 0.5% Nonidet P-40. The biotinylated proteins were eluted from the beads by the addition of 60 μl of 1 × SDS sample loading buffer and incubation at 95°C for 10 min. The blotted membranes were immunostained with Abs specific for caspases.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Acknowledgments
This study was supported by a grant of the Korea Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea (02-PJ1-PG10-20708-0006) and was partly supported by KOSEF through National Core Research Center for Nanomedical Technology (R15-2004-024-00000-0).
References
- Ahmed K, Davis AT, Wang H, Faust RA, Yu S, Tawfic S (2000) Significance of protein kinase CK2 nuclear signaling in neoplasia. J Cell Biochem Suppl 35: 130–135 [DOI] [PubMed] [Google Scholar]
- Allende JE, Allende CC (1995) Protein kinases. 4. Protein kinase CK2: an enzyme with multiple substrates and a puzzling regulation. FASEB J 9: 313–323 [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH (1999) Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 104: 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J (1998) Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 12: 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS (2003) A unified model for apical caspase activation. Mol Cell 11: 529–541 [DOI] [PubMed] [Google Scholar]
- Chang DW, Xing Z, Capacio VL, Peter ME, Yang X (2003) Interdimer processing mechanism of procaspase-8 activation. EMBO J 22: 4132–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Orozco A, Spencer DM, Wang J (2002) Activation of initiator caspases through a stable dimeric intermediate. J Biol Chem 277: 50761–50767 [DOI] [PubMed] [Google Scholar]
- Droin N, Bichat F, Rebe C, Wotawa A, Sordet O, Hammann A, Bertrand R, Solary E (2001) Involvement of caspase-2 long isoform in Fas-mediated cell death of human leukemic cells. Blood 97: 1835–1844 [DOI] [PubMed] [Google Scholar]
- Duan H, Dixit VM (1997) RAIDD is a new ‘death' adaptor molecule. Nature 385: 86–89 [DOI] [PubMed] [Google Scholar]
- Faust RA, Gapany M, Tristani P, Davis A, Adams GL, Ahmed K (1996) Elevated protein kinase CK2 activity in chromatin of head and neck tumors: association with malignant transformation. Cancer Lett 101: 31–35 [DOI] [PubMed] [Google Scholar]
- Fesik SW (2000) Insights into programmed cell death through structural biology. Cell 103: 273–282 [DOI] [PubMed] [Google Scholar]
- Gapany M, Faust RA, Tawfic S, Davis A, Adams GL, Ahmed K (1995) Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol Med 1: 659–666 [PMC free article] [PubMed] [Google Scholar]
- Izeradjene K, Douglas L, Delaney A, Houghton JA (2004) Influence of casein kinase II in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human rhabdomyosarcoma cells. Clin Cancer Res 10: 6650–6660 [DOI] [PubMed] [Google Scholar]
- Izeradjene K, Douglas L, Delaney A, Houghton JA (2005) Casein kinase II (CK2) enhances death-inducing signaling complex (DISC) activity in TRAIL-induced apoptosis in human colon carcinoma cell lines. Oncogene 24: 2050–2058 [DOI] [PubMed] [Google Scholar]
- Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, Goodman R, Lozano G, Zhao Y, Lu H (2001) A DNA damage-induced p53 serine 392 kinase complex contains CK2, hSpt16, and SSRP1. Mol Cell 7: 283–292 [DOI] [PubMed] [Google Scholar]
- Kim K, Fisher MJ, Xu SQ, El-Deiry WS (2000) Molecular determinants of response to TRAIL in killing of normal and cancer cells. Clin Cancer Res 6: 335–346 [PubMed] [Google Scholar]
- Kim S, Choi K, Kwon D, Benveniste EN, Choi C (2004a) Ubiquitin–proteasome pathway as a primary defender against TRAIL-mediated cell death. Cell Mol Life Sci 61: 1075–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Kim K, Kwagh JG, Dicker DT, Herlyn M, Rustgi AK, Chen Y, El-Deiry WS (2004b) Death induction by recombinant native TRAIL and its prevention by a caspase 9 inhibitor in primary human esophageal epithelial cells. J Biol Chem 279: 40044–40052 [DOI] [PubMed] [Google Scholar]
- Krueger A, Schmitz I, Baumann S, Krammer PH, Kirchhoff S (2001) Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J Biol Chem 276: 20633–20640 [DOI] [PubMed] [Google Scholar]
- Kumar S, Kinoshita M, Dorstyn L, Noda M (1997) Origin, expression and possible functions of the two alternatively spliced forms of the mouse Nedd2 mRNA. Cell Death Differ 4: 378–387 [Google Scholar]
- Lacour S, Hammann A, Wotawa A, Corcos L, Solary E, Dimanche-Boitrel MT (2001) Anticancer agents sensitize tumor cells to tumor necrosis factor-related apoptosis-inducing ligand-mediated caspase-8 activation and apoptosis. Cancer Res 61: 1645–1651 [PubMed] [Google Scholar]
- Lassus P, Opitz-Araya X, Lazebnik Y (2002) Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297: 1352–1354 [DOI] [PubMed] [Google Scholar]
- Li PF, Li J, Muller EC, Otto A, Dietz R, von Harsdorf R (2002) Phosphorylation by protein kinase CK2: a signaling switch for the caspase-inhibiting protein ARC. Mol Cell 10: 247–258 [DOI] [PubMed] [Google Scholar]
- Litchfield DW, Dobrowolska G, Krebs EG (1994) Regulation of casein kinase II by growth factors: a reevaluation. Cell Mol Biol Res 40: 373–381 [PubMed] [Google Scholar]
- Luscher B, Kuenzel EA, Krebs EG, Eisenman RN (1989) Myc oncoproteins are phosphorylated by casein kinase II. EMBO J 8: 1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinny JA, Trushin SA, Bren GD, Chester N, Paya CV (1996) Casein kinase II phosphorylates I kappa B alpha at S-283, S-289, S-293, and T-291 and is required for its degradation. Mol Cell Biol 16: 899–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J 16: 2794–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson DW (1999) Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ 6: 1028–1042 [DOI] [PubMed] [Google Scholar]
- O'Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL, Hacker G, Magnusson C, Pakusch M, Cecconi F, Kuida K, Strasser A, Huang DC, Kumar S (2002) Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ 9: 832–841 [DOI] [PubMed] [Google Scholar]
- Ravi R, Bedi A (2002) Sensitization of tumor cells to Apo2 ligand/TRAIL-induced apoptosis by inhibition of casein kinase II. Cancer Res 62: 4180–4185 [PubMed] [Google Scholar]
- Rippo MR, Moretti S, Vescovi S, Tomasetti M, Orecchia S, Amici G, Catalano A, Procopio A (2004) FLIP overexpression inhibits death receptor-induced apoptosis in malignant mesothelial cells. Oncogene 23: 7753–7760 [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM (1999) Caspase activation: the induced-proximity model. Proc Natl Acad Sci USA 96: 10964–10967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, Tschopp J (2004) The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304: 843–846 [DOI] [PubMed] [Google Scholar]
- Wagner KW, Engels IH, Deveraux QL (2004) Caspase-2 can function upstream of bid cleavage in the TRAIL apoptosis pathway. J Biol Chem 279: 35047–35052 [DOI] [PubMed] [Google Scholar]
- Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5: 157–163 [DOI] [PubMed] [Google Scholar]
- Wang D, Westerheide SD, Hanson JL, Baldwin AS Jr (2000) Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem 275: 32592–32597 [DOI] [PubMed] [Google Scholar]
- Xu X, Toselli PA, Russell LD, Seldin DC (1999) Globozoospermia in mice lacking the casein kinase II alpha′ catalytic subunit. Nat Genet 23: 118–121 [DOI] [PubMed] [Google Scholar]
- Zandomeni RO (1989) Kinetics of inhibition by 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole on calf thymus casein kinase II. Biochem J 262: 469–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Franco A, Myers K, Gray C, Nguyen T, Hersey P (1999) Relation of TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma. Cancer Res 59: 2747–2753 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2