

Abstract
Upon ligand binding at the subunit interfaces, the extracellular domain of the nicotinic acetylcholine receptor undergoes conformational changes, and agonist binding allosterically triggers opening of the ion channel. The soluble acetylcholine-binding protein (AChBP) from snail has been shown to be a structural and functional surrogate of the ligand-binding domain (LBD) of the receptor. Yet, individual AChBP species display disparate affinities for nicotinic ligands. The crystal structure of AChBP from Aplysia californica in the apo form reveals a more open loop C and distinctive positions for other surface loops, compared with previous structures. Analysis of Aplysia AChBP complexes with nicotinic ligands shows that loop C, which does not significantly change conformation upon binding of the antagonist, methyllycaconitine, further opens to accommodate the peptidic antagonist, α-conotoxin ImI, but wraps around the agonists lobeline and epibatidine. The structures also reveal extended and nonoverlapping interaction surfaces for the two antagonists, outside the binding loci for agonists. This comprehensive set of structures reflects a dynamic template for delineating further conformational changes of the LBD of the nicotinic receptor.
Keywords: acetylcholine-binding protein, α-conotoxin, conformational flexibility, crystal structure, nicotinic acetylcholine receptor
Introduction
Nicotinic acetylcholine receptors (nAChRs) are well-characterized transmembrane allosteric proteins involved in rapid gating of ions elicited by acetylcholine. They belong to the ‘Cys-loop' superfamily of ligand-gated ion channels (LGIC) that includes GABA-A and -C, 5-HT3, and glycine receptors (Grutter and Changeux, 2001; Karlin, 2002; Lester et al, 2004). The nAChRs are homo- or heteromeric pentamers of structurally related subunits that encompass an extracellular N-terminal ligand-binding domain (LBD), four transmembrane-spanning regions that form the ion channel, and an extended intracellular region between spans M3 and M4. They exist in at least three conformational states with distinctive sensitivities to the nicotinic ligands that dictate channel gating and function: basal or resting (closed, but rapidly activatable), activated (open), and desensitized (closed). Indeed, ligand binding triggers conformational changes that are transmitted to the transmembrane-spanning region, leading to gating and changes in membrane potential. Owing to their functional importance and structural and functional differences in numerous pathologies, the nAChRs have been thoroughly investigated at the pharmacological, biochemical, and structural levels. However, structural analyses of the nAChR are impaired by the large size, the transmembrane spans, and, in most species, the low abundance and heteropentameric assembly.
The recent pharmacological and structural characterization of the soluble acetylcholine-binding protein (AChBP) from the freshwater snail, Lymnaea stagnalis (L-AChBP) (Brejc et al, 2001; Smit et al, 2001), has considerably increased our knowledge of the structure and recognition determinants of the nAChR. L-AChBP shows limited sequence identity with the nAChR LBD (Figure 1A), but it assembles as a stable homopentamer and displays ligand recognition properties similar to those of the neuronal homopentameric α7 receptor subtype (Smit et al, 2001). The refined 4 Å resolution electron microscopy structure of the heteropentameric muscle-type, (α1)2βγδ nAChR has elegantly illustrated considerable structural similarity of L-AChBP with the nAChR LBD (Unwin, 2005). Moreover, coupling of L-AChBP, where three membrane-facing loops have been modified, with the transmembrane domain of the 5-HT3A receptor yielded a chimeric receptor with lower affinity for acetylcholine, as expected for an activatable receptor, and the capacity to trigger opening of the ion channel (Bouzat et al, 2004). Therefore, L-AChBP is now considered a structural and functional surrogate of the nAChRs.
Figure 1.
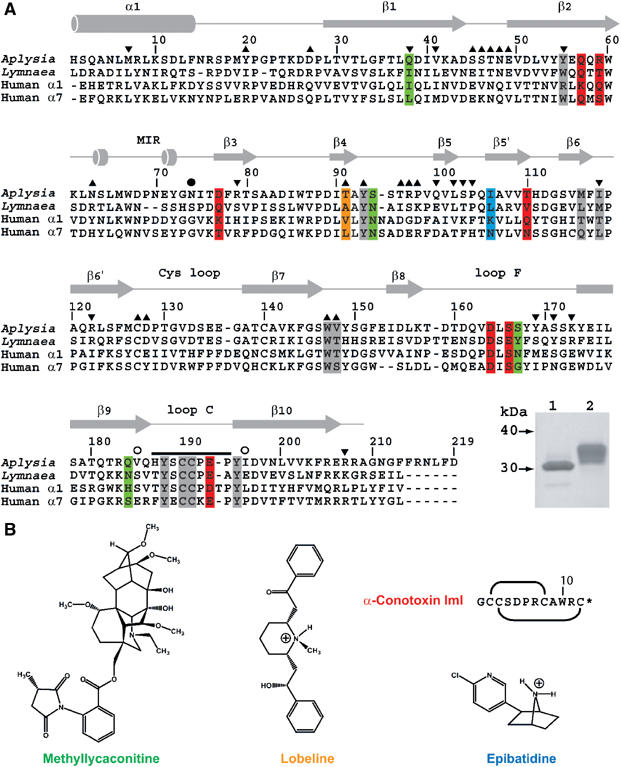
Sequence alignment of A-AChBP. (A) Structural alignment of the subunit sequences of A-AChBP (Hansen et al, 2004) and L-AChBP (Smit et al, 2001) with those of the human α1 and α7 LBDs (LGIC database). The A-AChBP sequence reported in Celie et al (2005) differs by Val substitutions at positions 43 and 138 and a N-terminal two-residue deletion. Secondary structure elements are indicated. The bar and open circles above the A-AChBP sequence indicate the loop C tip and hinge regions, respectively. The solid circle denotes the glycosylated Asn74. Tip up and down triangles denote A-AChBP residues from the (+) and (−) faces that are within a 3.5 Å radius of interaction at the subunit interface in the apo conformation. A-AChBP residues whose side chains interact within 4.5 Å with all four ligands are on a gray background. Residues specific for the nicotinic antagonists, α-conotoxin ImI and MLA, are on a red and green background and those specific for the nicotinic agonists, LOB and EPI, on an orange and a blue background, respectively. Inset: SDS–PAGE analysis (16% gel) of A-AChBP expressed from GnTI− (lane 1) and standard HEK cells (lane 2). The protein expressed in GnTI− cells migrates faster and as a thinner band, indicative of a smaller size and greater homogeneity in oligosaccharide structure. (B) Schematic view of the organic ligands, MLA (the lycoctonine ring is at the top and the N-ethylpiperidine ring at the bottom), LOB and EPI. Top right: sequence and disulfide bonding of α-conotoxin ImI; the star denotes C-terminal amidation.
The initial structures of L-AChBP, in a Hepes-bound form and as two complexes with the small nicotinic agonists, (−)-nicotine and carbamylcholine, bound in the ligand-binding site, show minimal conformational differences (Brejc et al, 2001; Celie et al, 2004) and have been suggested to reflect a desensitized state of the nAChR (Grutter and Changeux, 2001). In turn, the structure of L-AChBP in complex with the large peptidic toxin and nicotinic antagonist α-cobratoxin (Cbtx), tightly inserted at the subunit interface, has revealed concerted large movements of loops C and F that line the binding interface, and has provided a template resembling a resting state conformation of the nAChR (Bourne et al, 2005).
The recently characterized A-AChBP from the saltwater mollusk, Aplysia californica, shares only 33% amino-acid identity with L-AChBP, but it also assembles as a soluble homopentamer and possesses all the functional residues identified in L-AChBP (Hansen et al, 2004; Celie et al, 2005), including those that form the aromatic nest characteristic of the nAChR LBD (Figure 1A). Of the five aromatic residues present in the L-AChBP-binding pocket, only the conservative Trp55Tyr substitution is found in A-AChBP. However, A-AChBP, similar to the nAChR, lacks the sixth aromatic residue present as Tyr164 in L-AChBP. It also displays an Ala194Pro substitution within loop C on the principal face (the (+) face) of the interface and an Arg108Val substitution on the complementary face (the (−) face), which are both conserved in the human α1 and α7 subunits. Accordingly, A-AChBP displays distinctive binding affinities and specificities for nicotinic agonists and antagonists compared with L-AChBP, exemplified by its lower affinity for acetylcholine but higher affinity for the small α-conotoxin peptides, the natural ImI (Hansen et al, 2004) (Table I) and the PnIA Ala10Leu variant (Celie et al, 2005), two α7-specific antagonists.
Table 1.
Ligand dissociation constants (Kda, nM) for the two AChBP species
| Ligand | A-AChBP | L-AChBP | A/L Kd ratio |
|---|---|---|---|
| Ach | 30 000b | 890c | 34 |
| (−)-Nicotine | 245b | 86c | 2.8 |
| (+)-Epibatidine (EPI) | 14b | 0.16c | 88 |
| α-Lobeline (LOB) | 0.3 | 30 | 0.01 |
| Methyllycaconitine (MLA) | 2.8b | 0.41c | 6.8 |
| α-Bungarotoxin (Bgtx) | 250b | 1.8c | 139 |
| α-Cobratoxin (Cbtx) | 191 | 3.2b | 60 |
| α-Conotoxin ImI | 0.88b | 14 000b | 0.00006 |
| α-Conotoxin MI |
1000 |
2800 |
0.36 |
| aValues are average (n=2) or means (n>2) of individual data that differ by less than 20%. | |||
| bFrom Hansen et al (2004). | |||
| cFrom Hansen et al (2002). | |||
We report the crystal structures of A-AChBP in the apo form and as four complexes with two nicotinic antagonists, the peptidic α-conotoxin ImI and the alkaloid methyllycaconitine (MLA), and two alkaloid nicotinic agonists, α-lobeline (LOB) and (+)-epibatidine (EPI) (Figure 1B and Table II). Compared to the Hepes-bound L- and A-AChBP structures, that of apo A-AChBP, devoid of a bound amine buffer, reveals a distinctive conformation. Structural analysis of the four A-AChBP complexes uncovers, at the subunit interface and outside of the primary competitive binding site, unpredicted anchoring surfaces that contribute nonoverlapping binding loci for the antagonists, and enables one to visualize distinctive conformational changes associated with agonist versus antagonist binding. Hence, these structures provide distinct templates for predicting ligand selectivity for the individual nAChR subtypes and for delineating further the conformational changes associated with channel gating.
Table 2.
Data collection and refinement statistics
| Apo | ImI | MLA | LOB | EPI | |
|---|---|---|---|---|---|
| Data collection | |||||
| Beamline | APS/19-ID | ALS/8.2.1 | ESRF/ID29 | ALS/8.2.1 | ESRF/ID14-EH3 |
| Wavelength (Å) | 0.97934 | 0.97623 | 0.97563 | 0.97623 | 0.931 |
| Space group | C2221 | I222 | P21 | P1 | I23 |
| a, b, c (Å) | 144.1, 146.8, 143.3 | 130.3, 140.0, 153.4 | 67.3, 126.8, 147.3 | 75.8, 85.7, 117.3 | 200.9, 200.9, 200.9 |
| α/β/γ (°) | −/99.5/− | 89.9/97.3/106.6 | |||
| Pentamer/asymmetric unit | 1 | 1 | 2 | 2 | 1 |
| Resolution rangea | 50–2.02 (2.09–2.02) | 50–2.07 (2.14–2.07) | 72–2.45 (2.51–2.45) | 58.3–2.05 (2.16–2.05) | 30–3.4 (3.49–3.4) |
| Rmerge (%)a,b | 5.8 (47.4) | 8.5 (54.8) | 7.9 (48.4) | 6.2 (26.5) | 19.8 (59.4) |
| Observations | 603 776 | 433 003 | 356 687 | 333 038 | 64 112 |
| Unique reflections | 97 846 | 86 601 | 95 739 | 168 545 | 18 257 |
| Completeness (%)a | 98.5 (90.5) | 99.9 (99.8) | 100.0 (100.0) | 95.8 (95.4) | 97.6 (97.6) |
| Redundancya | 6.2 (5.7) | 5.1 (3.8) | 3.7 (3.8) | 2.0 (2.0) | 3.5 (3.5) |
| 〈I/σI〉a | 13.6 (3.8) | 29.5 (2.2) | 11.3 (2.5) | 13.9 (2.5) | 8.3 (2.0) |
| B-factor from Wilson plot (Å2) | 32.4 | 33.5 | 45.4 | 25.6 | 42.3 |
| Refinement | |||||
| Resolution range (Å) | 40–2.02 (2.07–2.02) | 20–2.07 (2.12–2.07) | 20–2.45 (2.51–2.45) | 48–2.05 (2.1–2.05) | 30–3.4 (3.49–3.4) |
| Protein atoms | 8515 | 8411 | 16762 | 16324 | 8521 |
| Solvent/ligand atoms | 882/82 | 1043/469 | 560/490 | 1561/250 | −/70 |
| Rcryst (%)/Rfree (%) | 16.8 (20.8)/20.2 (25.2) | 17.3 (23.1)/21.4 (28.2) | 19.3 (27.7)/23.2 (33.8) | 21.6 (30.8)/25.8 (33.7) | 18.3 (24.9)/25.4 (33.1) |
| Free reflections | 2980 | 1700 | 1916 | 5025 | 929 |
| R.m.s. 1–2 bond distances (Å) | 0.013 | 0.013 | 0.008 | 0.008 | 0.01 |
| R.m.s. 1–3 bond angles (deg) | 1.41 | 1.42 | 1.37 | 1.16 | 1.5 |
| Mean main/side chain B (Å2) | 39.3/41.9 | 39.29/41.03 | 45.3/46.4 | 39.6/40.6 | 45.4/46.6 |
| Mean B solvent/ligand (Å2) | 43.8/62.6 | 43.56/35.26 | 36.8/40.4 | 36.6/31.9 | −/38.58 |
| Main/side chain ΔB for bonded atoms (Å2) | 0.99/1.66 | 1.03/1.60 | 1.14/1.14 | 0.89/1.08 | 1.11/1.35 |
| PDB accession code |
2BYN |
2BYP |
2BYR |
2BYS |
2BYQ |
| aValues in parentheses are for the highest resolution shell. | |||||
| bRmerge=∑hkl∑i∣Ihkli−〈Ihkli〉∣/∑hkl∑i〈Ihkli〉; Rcryst=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo. | |||||
Results and discussion
Determination and quality of the structures
Attempts to solve a structure from crystals obtained from the highly N-glycosylated and nonhomogeneous A-AChBP expressed from a standard HEK cell line (Hansen et al, 2004) were unsuccessful. However, expression from a glycosylation-deficient cell line (Reeves et al, 2002) resulted in a homogenous protein with a shorter glycan chain as assessed by electrophoresis (Figure 1A) and mass spectrometry analyses, yielding crystals suitable for structural studies.
The structures of the homopentameric A-AChBP in the apo form (Figure 2) and as complexes with the nicotinic antagonists α-conotoxin ImI and MLA and agonists EPI and LOB (Figures 3 and 4) were solved in the 1.96–3.4 Å resolution range from crystals grown at different pH's and in distinct space groups, leading to different packing geometries (cf. Materials and methods; Table II). Yet, each of the four complex structures, including the lower-resolution EPI complex, shows well-defined electron density indicative of a high occupancy and a single binding orientation for each of the five ligand molecules bound at the subunit interfaces, along with unambiguous positioning of all side chains in the pentameric complex. Hence, these structures, and their comparison with previous structures of AChBP bound to Hepes (Brejc et al, 2001; Celie et al, 2005), to the small agonists nicotine and carbamoylcholine (Celie et al, 2004), and to the large antagonist Cbtx (Bourne et al, 2005), reveal structural determinants unique to A-AChBP along with a variety of conformations associated with ligand binding at the subunit interfaces.
Figure 2.
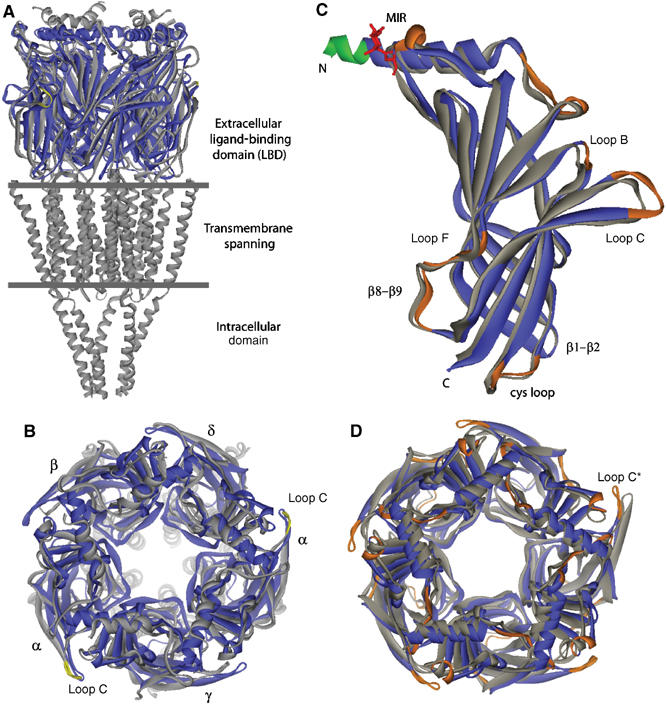
Overall view of the apo A-AChBP structure and structural comparisons. (A) Side view and (B) apical view of apo A-AChBP (blue) overlaid with the muscle-type nAChR (gray) (Unwin, 2005). The tip of loop C in the nAChR α subunits is colored yellow. Loop C in apo A-AChBP overlays most closely with loop C in the α subunit. (C) Side view and (D) top view of the apo A-AChBP subunit and pentamer (blue) overlaid with the Hepes-bound A-AChBP subunit and pentamer (gray) using residues 20–200 from a single and all five subunits, respectively. In apo A-AChBP, the Asn74-linked glycan is in red and the FLAG epitope that prolongs helix α1 in green. The ‘MIR' and loops where there is a large departure in position or conformation are shown in orange. In four of the five Hepes-bound A-AChBP subunits, loop C is more closed than in apo A-AChBP, whereas in the fifth subunit (star), devoid of a bound buffer, it is slightly more extended.
Figure 3.
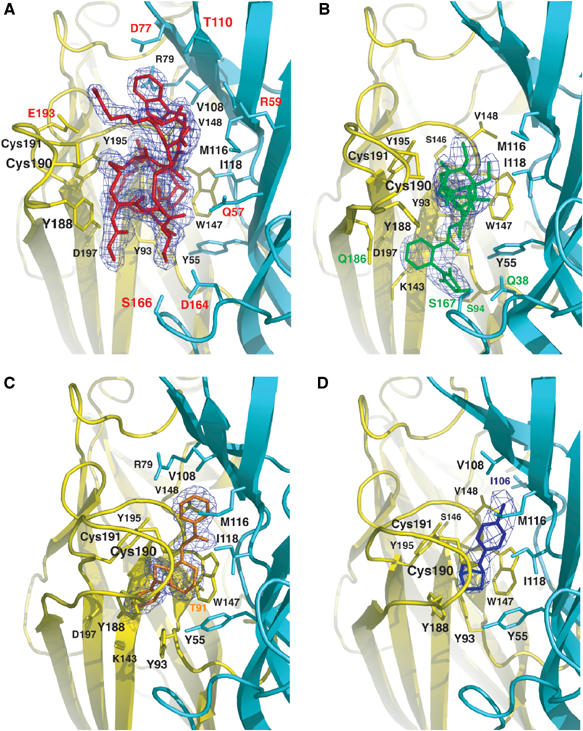
The A-AChBP subunit interface in the antagonist and agonist complexes. Side views of the bound antagonists (A) ImI, buried under loop C, and (B) MLA (same orientation), and of the bound agonists (C) LOB and (D) EPI in similar orientations. The 2.0–3.4 Å resolution omits 2FO–Fc electron density maps contoured at 1.4σ are shown in blue. The main and side chains from the (+) and (−) faces of the subunit interface are in yellow and cyan, respectively. Those side chains that interact specifically with ImI, MLA, LOB, and EPI are labeled red, green, orange, and blue, respectively.
Figure 4.
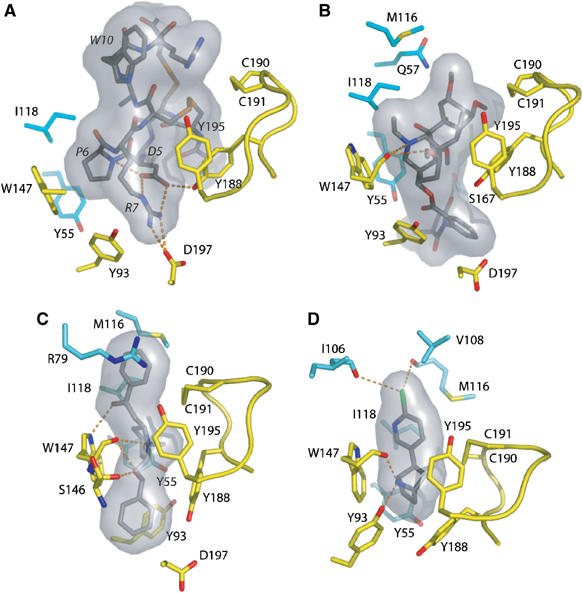
Expanded views of the bound ligands. Hydrogen bonding of key residues for the bound (A) ImI, (B) MLA, (C) LOB, and (D) EPI, viewed from inside of the ion channel vestibule looking in a radial direction. Ligands are bound between the Cys190–Cys191 disulfide on the left and Trp148 on the right. Labels for the ImI residues are italicized. The molecular surfaces of the ligands are in gray. Subunit coloring is identical to that in Figure 3.
Structure of A-AChBP in the apo form
The structure of apo A-AChBP shows the same homopentameric assembly of tightly associated subunits as found in structures of the Hepes-bound AChBPs (Brejc et al, 2001; Celie et al, 2005) and in microscopy images of the muscle-type, (α1)2βγδ nAChR from Torpedo (Unwin, 2005) (Figure 2). Each A-AChBP subunit consists of an N-terminal, 20-residue α-helix that includes the FLAG epitope, two short α310 helices, and a 10-strand β-sandwich core made of an inner β-sheet of six strands (β1, β2, β3, β5, β6, β8) and an outer β-sheet of four strands (β4, β7, β9, β10). Apart from flexible surface loop regions, residue positions in the five subunits within the pentamer are very similar (r.m.s.d. values in the 0.3–0.35 Å range for 211 Cα atoms). A second apo structure, solved from a different crystal form grown at pH 8.5 instead of 5.6, was virtually identical (not shown).
The subunit interface, which is made of six loops (loops α1–β1, β1–β2, β3–β4, β4–β5 (or A), β6′–β7 (Cys loop), and β7–β8 (B)) from the (+) face and of secondary structure elements (helix α1 and strands β2, β3, β5, and β6) from the (−) face (Figure 1A), buries, to a 1.4 Å probe radius, a surface area of ∼1300 Å2 on each subunit, in agreement with the Hepes-bound L-AChBP structure (Brejc et al, 2001). The interface, which is dominated by apolar residues, encompasses several direct polar contacts involving residues Asn48, Glu49, Arg97, and Glu153 from the (+) face and Gln3, Gln38, Tyr55, Arg79, Arg122, and Lys173 from the (−) face, which are weakly conserved. Only nine solvent molecules mediate direct contacts between residues from each subunit, an observation that emphasizes the requirement of a high surface complementarity between subunits in the nAChR to maintain a functional pentameric assembly. Of the several loops that emerge from the β-sandwich core, five (loops A, B, β9–β10 (or C), β8–β9 (F), and the conserved Cys-loop) have been shown to be critical for nAChR function, with several of their residues being involved in both subunit assembly and ligand binding (Karlin, 2002).
The architecture of the ligand-binding pocket, with the aromatic nest made of Tyr93, Trp147, Tyr188, and Tyr195 from the (+) face of the interface and Tyr55 from the (−) face, is highly conserved in the AChBPs. Yet, in A-AChBP, compared to L-AChBP, major rearrangements are observed within the (−) face that may arise from replacement of Trp53 by Tyr55, as often found in the GABA-A and -C LBDs, and of the bulky Arg104 by Val108 (Figure 1A). In fact, residue Arg79, which originates from a different region in A-AChBP, positions its guanidinium group close to that of Arg104 in L-AChBP. Near the pocket entry, two small A-AChBP residues, Thr36 and Ser167, replace the bulkier L-AChBP Lys34 and Tyr164. The largest difference between the apo A-AChBP and Hepes-bound AChBP subunits occurs in the loop C segment Val183–Tyr193, with deviation up to 6.5 Å at position Cys190 (Figure 2C and D). This large opening movement of loop C, seen at each subunit interface, markedly enhances ligand access and reduces the loop's proximity to the subunit interface.
Apo A-AChBP versus the muscle-type α1 subunit
Structural overlay of apo A-AChBP subunit with the muscle-type α1 subunit (Unwin, 2005) shows large positional differences (r.m.s.d. values of 1.7 Å for only 130 Cα atoms), but reveals a similar orientation of the ‘untwisted' loop C, with positional differences of only 0.7 and 1.6 Å for the two sulfur atoms from the vicinal Cys190–Cys191 residues (Figure 2A and B). Loop β1–β2, which in the muscle-type nAChR is thought to control the displacement of the transmembrane, pore-lining M2 segment, also adopts in apo A-AChBP a conformation similar to that in the α1 subunit. Although the curvature of the inner β-sheet is roughly conserved between the α1 and apo A-AChBP subunits, the outer β-sheet in the α1 subunit is rotated by 11–14° compared to its position in the A-AChBP subunit.
In the muscle-type nAChR, the main immunogenic region (MIR) is a reactive epitope in autoimmune Myasthenia gravis (Richman and Agius, 2003). This region, localized within loop β2–β3 on the apical surface of the α1 subunit, is separated from the N-terminal helix by a cleft that contributes to the major antibody-binding site (Unwin, 2005). In the AChBPs, the corresponding region, encompassing residues Trp67–Glu71, is weakly conserved (Figure 1A). In A-AChBP, compared to L-AChBP, loop β2–β3 is two residues longer and displays a large positional change that exposes the side chains of Asp68 and Glu71 to the solvent. These residues correspond to the immunoreactive Asn67 and Asp71 in the α1 subunit. However, while in the α1 subunit the N-terminal helix is solvent-exposed (Unwin, 2005), in A-AChBP it packs tightly against the β-sandwich core in a conformation that may restrict antibody access.
Antagonist binding
The ImI–AChBP complex. Certain conotoxin peptides, from the venom of marine snails of Conus sp, are among the most selective nAChR antagonists. The largest family of α-conotoxins has been divided into the structural α3/5, α4/3, α4/6, and α4/7 subfamilies based on the number of residues between the second and third Cys residues (loop I) and the third and fourth Cys residues (loop II). In the α4/3 family, the dodecapeptides ImI (Figure 1B) and ImII are the smallest α-conotoxins yet identified. They are α7 and α3β2 nAChR selective, with lower affinity for ImII (Ellison et al, 2004). These two peptides, that differ by three residues (Gly1Ala, Pro6Arg, and Ala9Arg), appear to bind nonoverlapping sites on the α7 nAChR (Ellison et al, 2003). Moreover, ImI, with a 14 000-fold preference for A-AChBP compared to L-AChBP as found by direct binding (Table I), is the most selective of the nAChR ligands.
The ImI–A-AChBP complex structure (Figures 3A and 4A) shows that bound ImI retains the rigid scaffold found in solution. The peptide folds around two compact loops, made of an α310-helical region followed by a β-turn, that are defined by the two disulfide bridges, Cys2–Cys8 (loop I) and Cys3–Cys12 (loop II), and are separated by a deep cleft (Maslennikov et al, 1999; Rogers et al, 1999). However, the conformations of the two disulfide bonds markedly differ from those for the free peptide, indicating subtle conformational rearrangement upon binding. The resulting r.m.s.d. value (0.87 Å for 12 Cα atoms) is significantly greater than the averaged value (0.15 Å) between the best 20 energy-minimized conformers of the free ImI structure (Maslennikov et al, 1999).
ImI, inserted under A-AChBP loop C, entirely fills the ligand-binding pocket, where it buries 71% of its solvent-accessible surface area (Figures 3A and 4A). Only residues Gly1, Ser4, and Arg11 and the C-terminal amine group remain solvent-exposed. At the (+) face of the interface, the Asp5–Pro6–Arg7 tripeptide in ImI loop I is deeply anchored on the ‘membrane' side of the pocket. This tripeptide represents a major binding determinant, consistent with earlier mutagenesis data showing dramatic affinity decreases (Quiram and Sine, 1998; Servent et al, 1998). Indeed, ImI Asp5, as a key residue, stabilizes the loop I conformation by establishing contacts with the ImI Arg7 guanidinium group and backbone nitrogen, and with A-AChBP Tyr188 in loop C. ImI Pro6 is located deep in the pocket and in an edge-to-face aromatic interaction with the 4.1 Å distant A-AChBP Trp147. The ImI Arg7 guanidinium group establishes a salt bridge with the invariant A-AChBP Asp197 in loop C and contacts the Tyr93 hydroxyl and the Ile196 carbonyl oxygen.
Moreover, the ImI Cys2–Cys8 disulfide bridge faces the A-AChBP Cys190–Cys191 disulfide 4 Å away and interacts with Tyr188 and Tyr195 in loop C. At the (−) face, ImI Ala9 contributes van der Waals contacts with A-AChBP Met116 and Ile118, and ImI Trp10 establishes extensive interactions with A-AChBP Arg79, Val108, Met116, and Arg59, consistent with its key role for ImI binding to the α7 nAChR (Quiram and Sine, 1998; Servent et al, 1998) (Figures 3A and 4A). At the complex interface, only eight ordered solvent molecules are found, indicating a high degree of surface complementarity that is consistent with the nM dissociation constant of ImI for A-AChBP (Hansen et al, 2004) (Table I). Overall, the bound ImI buries, to a 1.4 Å probe radius, equivalent surface areas of 348 and 331 A2 on the (+) and (−) faces of the A-AChBP subunit interface. The number of residues involved in the resulting interfacial area is consistent with the ImI large preference for A-AChBP, compared to L-AChBP (Table I). Compared to ImI, the PnIA Ala10Leu/Asp14Lys variant, of the α4/7 family, is bound ∼1 Å deeper in the ligand-binding pocket of A-AChBP (Celie et al, 2005), due to the Pro residue that replaces the bulky Arg7 at the tip of ImI loop I. Yet, the conformation of loop I in the two peptides is similar, whereas large deviations occur in loop II, which in PnIA is longer (by four residues) and larger (in size) than in ImI. This argues for loop I being the major determinant conferring nAChR selectivity.
The methyllycaconitine–AChBP complex. The diterpene alkaloid MLA, the principal toxic component of seeds from the Aconitum and Delphinium generi, is the most potent, nonpeptidic nAChR antagonist known. MLA is highly selective for the neuronal α7 receptor (Ward et al, 1990). Yet, it displays similar affinities for the A- and L-AChBP species (Table I). MLA consists of a bulky, oxygen-rich lycoctonine moiety lacking nicotinic potency and linked, via a carbonyl ester, to an N-phenyl succinimide (2-methylsuccinimidobenzoyl) side chain (Figure 1B). Ester hydrolysis to produce lycoctonine markedly diminishes MLA affinity on rat brain preparations, suggesting that the N-phenyl succinimide moiety of MLA is a major determinant for α7 selectivity.
In the MLA–A-AChBP complex (Figures 3B and 4B), the long axis of the bulky and rigid lycoctonine skeleton, that resembles a 7 × 4 Å cylinder, is located at the subunit interface and abuts against A-AChBP residues Tyr188 and Tyr195 and the Cys190–Cys191 disulfide from the (+) face, and against Met116 and Ile188 from the (−) face. The key interaction is an edge-to-face stacking of the N-ethylpiperidine ring, in a chair conformation, with Trp147 at the ‘membrane' side of the binding site pocket. This conformation ideally positions the lycoctonine tertiary amine within hydrogen bond distance to the Trp147 carbonyl oxygen, whereas the ethyl group makes van der Waals contacts with Trp147 and Val148 on the (+) face and with Ile118 on the (−) face. Yet, the lycoctonine ring makes limited polar contact with A-AChBP, consistent with retention of antagonist potency by simplified MLA derivatives that contain only an N-ethylpiperidine moiety linked to the succinimidoylanthranilate ester (Bergmeier et al, 2004). Owing to the flexibility of the ester linkage, the carbonyl oxygen is ideally positioned to be hydrogen bonded to Tyr55 on the (−) face. The well-ordered succinimidoylanthranilate moiety partially occupies a cavity lined by Ser94, Met126, Lys143, Gln186, and Asp197 on the (+) face and Gln38 and Ser167 on the (−) face, with the methyl group at position 3 of the imide ring contributing van der Waals contacts. The three hinge regions in MLA are essential to complement the shape of the binding pocket, resulting in a near-perpendicular orientation of the lycoctonine cylinder axis relative to the direction of the ester linkage, while within the succinimidoylanthranilate ester the succinimide and phenyl rings are twisted by 60°.
The structurally related alkaloid aconitine, a voltage-gated Na+ channel activator, differs from MLA in the oxygen pattern and stereochemistry and by the presence of bulky benzoyl and acetyl functional groups. Moreover, the N-phenyl succinimide group linked to the lycoctonine portion of MLA is replaced by an O-methyl ether. The MLA–A-AChBP complex structure reveals that all aconitine substituents could be easily accommodated in A-AChBP, consistent with observation that addition of an N-phenyl succinimide moiety to aconitine produces a compound with nicotinic potency comparable to that of MLA, but lacking Na+ channel activation properties (Hardick et al, 1995).
Agonist binding
The LOB–AChBP complex. The lipophilic alkaloid LOB, isolated from the Indian tobacco plant, Lobelia inflate, has been described as a mixed agonist/antagonist (Terry et al, 1998). Comparison of the large, three-ring LOB (Figure 1B) with the chemically related but smaller nicotine and cytisine has suggested that the phenyl 2-keto-ethyl moiety, more than the phenyl 2-hydroxyethyl group, may be the primary functional determinant for LOB binding to the nAChR (Barlow and Johnson, 1989). Its 100-fold preference for A-AChBP over L-AChBP indicates its selectivity among the nicotinic agonists (Table I).
In the LOB–A-AChBP complex (Figures 3C and 4C), the LOB molecule adopts an extended conformation devoid of internal hydrogen bonding that differs from the highly flexible conformation observed by NMR (Maslennikov et al, 1999), but favors its interaction with both faces of the subunit interface. The central piperidine ring exhibits a stacking interaction with A-AChBP Trp147 that favors hydrogen bonding between the tertiary amine and the Trp carbonyl oxygen, while the methyl group contributes van der Waals contacts with Cys190, Tyr188, and Tyr195. Within the binding pocket, the LOB carbonyl oxygen is bound to the Trp47 indole nitrogen and establishes a water-mediated contact with the Ile106 carbonyl and Ile118 nitrogen. At the ‘membrane' side of the binding pocket, the LOB hydroxyl is bound to the Ser146 and Trp147 carbonyl oxygens, consistent with its hydrogen-bonding potential. The phenyl 2-keto-ethyl moiety is enclosed at the binding pocket entrance, where it interacts with the Cys190–Cys191 disulfide on the (+) face and with Arg79, Met116, and Ile118 on the (−) face. In contrast, the phenyl 2-hydroxyethyl moiety, distant from the other phenyl moiety by ∼11 Å, is buried at the ‘membrane' side of the binding pocket, where it is exclusively lined by residues Tyr93, Lys143, Gly145, Trp147, and Asp197 from the (+) face. Consistent with the requirement of retaining both arms for optimal affinity (Flammia et al, 1999), the high flexibility of the two arms appears essential for the tight fit of the large LOB molecule to the shape of the binding pocket with a closed loop C conformation. In the complex, this results in the two distal phenyl rings being twisted and bent by ∼15° from colinear planarity. Given the expanded surface area that LOB occupies relative to smaller nicotinic agonists, multiple residues contribute to its preference for A-AChBP over L-AChBP (Table I).
The EPI–AChBP complex The chlorine-containing alkaloid EPI, from the skin of the Ecuadoran frog, Epipedobates tricolor, displays high potency and efficacy for several neuronal nAChR subtypes (Badio and Daly, 1994; Gerzanich et al, 1995). EPI has 90-fold lower affinity for A-AChBP than for L-AChBP (Table I). However, substitution of A-AChBP Tyr55 for a Trp, as found in the L-AChBP and human α7 subunits (Figure 1A), restores the higher affinity (SB Hansen, unpublished results), suggesting that the nature or size of the aromatic side chain at this position is critical for EPI binding.
The mode of binding of EPI to A-AChBP closely resembles that of nicotine to L-AChBP (Celie et al, 2004) (Figures 3D and 4D). EPI is sandwiched between the Trp147 side chain and the Cys190–Cys191 disulfide positioned at the midpoint of the two ring systems. The pyridine ring in the bound EPI is oriented similar to the pyridine ring in the bound nicotine, while the nitrogen-containing alicyclic skeleton (bridge ring) in EPI, which abuts on the Tyr55 ring, coincides with the slightly smaller pyrolidine ring in nicotine, resulting in similar intra-nitrogen distances (4.5 Å in A-AChBP versus 4.4 Å in L-AChBP). As a result, a similar network of hydrogen bonds is observed in the two complexes, with the pyridine amine bound to Ile118 and Trp147 via a solvent molecule, while the bridge ring amine targets the Trp147 carbonyl oxygen. In three A-AChBP subunits in the pentamer, a second hydrogen bond is seen between the bridge ring amine and Tyr93. The bulky bridge ring in EPI favors additional aromatic interactions with Tyr188, and the aromatic chloride ideally contributes polar contacts with the Ile106 and Val108 carbonyl oxygens from the (−) face of the interface in the apical region of the binding pocket. The mode of binding of EPI, with the chloride contribution and the large bridge ring that compensates for the A-AChBP smaller Tyr ring, compared to the L-AChBP Trp, is consistent with the mutagenesis data and provides an explanation for EPI's higher affinity than nicotine for A-AChBP (Table I).
Conformational fit for nicotinic antagonists and agonists
The availability of an apo A-AChBP structure along with two antagonist and two agonist complexes permits a direct comparative analysis of the conformational changes induced differentially by antagonist and agonist binding. Structural comparisons of the five structures (r.m.s.d. values in the 0.55–0.65 Å range for ∼1000 Cα atoms) and of the subunits from each pentamer (r.m.s.d. values in the 0.35–0.55 Å for 210 Cα atoms) indicate that the large conformational changes are localized to loops C and F, with smaller changes in helix α1 and surrounding loop regions on the apical surface of the subunit. Superimposition of the apo structure and the MLA and LOB complexes through their subunit (+) faces does not reveal significant variations in the relative orientations of the subunits that contribute the (−) faces. In contrast, for the EPI and ImI complexes this superimposition reveals rigid body movements, of ∼1.5° and in opposite directions, of the (−) faces in the complexes compared to their position in the apo structure. This movement, which differs from the 3–4° movement of subunits observed between the Hepes- and PnIA-bound AChBP structures (Celie et al, 2005), indicates that ligand binding may induce selective influences on the orientation of the subunits within the pentamer. Associated with the C loop enveloping the agonist is a small, diaphragm-like contraction at the cross-sectional level of the Cys and β1–β2 loops. This would serve to reduce the diameter of the pentamer near the extracellular membrane face by ∼1 Å. Minimal conformational change in the vicinity of the linking region might be expected in the AChR, since the transitions between the open channel and resting states are rapid and require small differences in energy states (Sakmann, 1992). Nevertheless, it is unclear whether such conformational differences are part of the linkage in the gating mechanism, since AChBP lacks the complement of residues necessary in the functioning receptor to couple to channel gating (Bouzat et al, 2004).
The largest differences arise from the rigid-body motion of loop C, which swings as much as 11 Å between the two extreme positions observed in the ImI and EPI complexes (Figure 5A and B), around hinge regions defined within dipeptides Gln184–Val185 and Tyr195–Ile196, respectively, located in strands β9 and β10 (Figure 1A). In fact, the loop C positions throughout the five structures cluster into three groups that contain: (i) the peptide antagonist-bound ‘open' conformations (ImI complex); (ii) the apo and organic antagonist-bound ‘intermediate' conformations (apo structure and MLA complex); (iii) the agonist-bound ‘closed' conformations (LOB and EPI complexes) (Figure 5B). Indeed, A-AChBP in the apo conformation lies between the peptide antagonist- and the agonist-bound conformations. Loop C in the Cbtx–L-AChBP complex adopts a similar intermediate conformation as seen in the apo structure and the MLA–A-AChBP complex, but it is tangentially displaced by ∼4 Å toward loop β7–β8 (Bourne et al, 2005), suggesting either a common mechanism for peptide toxin antagonism or that the Cbtx fingers lock loop C in a unique position. The slower ligand association rates observed for the peptide toxins (Hansen et al, 2002, 2004), being well below the diffusion limitation, may be a consequence of the transition to the extended (open) loop C conformation.
Figure 5.
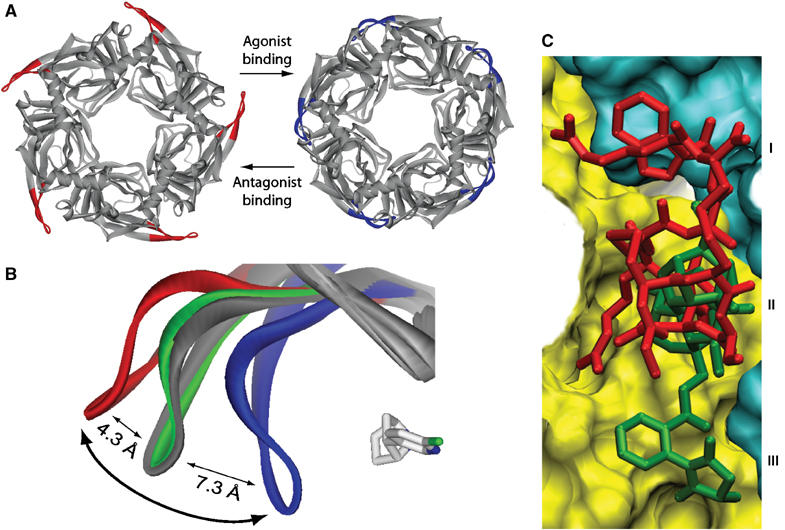
Conformational fit mechanism and ligand selectivity. (A) Top views of the ImI-bound (red loops C) and EPI-bound A-AChBP pentamers (blue loops C) showing the distinctive conformations for the antagonist and agonist complexes. (B) Overlay of loop C in apo A-AChBP (gray) and the ImI (red), MLA (green), and EPI complexes (blue); the bound EPI molecule is shown in light gray. The curved arrow denotes opening and closure of loop C upon antagonist and agonist binding. (C) Overlay of the A-AChBP-bound antagonists ImI (red) and MLA (green). The surfaces of the (+) and (−) faces of the subunit interface are shown in yellow and cyan, respectively. The common competitive binding site is at the center (labeled II); the two peripheral nonoverlapping sites are distinguished by ImI Trp10 (I) and the MLA N-phenyl succinimide moiety (III).
Residues at the tip of loop C contribute to the subunit interface and wrap around the bound agonist, with Ser196 bound to Asp164 in loop F from the (−) face as seen for Hepes-bound L-AChBP (Brejc et al, 2001). Hence, a series of local conformational changes specific to each ligand is observed, and loop C behaves as an induced-fit ‘sensor' adapting its configuration to the structural characteristics of the ligand entering the binding pocket (Figure 5B). This motif may not only govern ligand specificity but also whether the ligand elicits channel-opening events, that is, the efficacy of the ligand as an agonist. These findings are consistent with early studies where the vicinal Cys residues on loop C of the nAChR were reduced and labeled with quaternary sulfhydryl reactive reagents. Those reagents with less bulk and shorter distances between the quaternary nitrogen and reactive electrophile produced a depolarization, while the more bulky ligands with larger intersite distances maintained the receptor in an inactive state (Karlin, 1969).
In the apo A-AChBP structure, the architecture of the binding pocket is reminiscent of a resting state of the nAChR (Unwin, 2005) with low affinity for the nicotinic ligands. In contrast, the agonist-elicited closure of loop C is associated with substantial reorganization of the recognition determinants, for example, Gln186, Tyr188, Glu193, and Tyr195; this results in a binding pocket configuration that perhaps reflects a ligand-induced open channel or desensitized state of the nAChR. At two of the five-subunit interfaces in the ImI–A-AChBP complex, loop F adopts two alternative conformations, indicating that this region also displays significant conformational flexibility when antagonists are bound.
Ligand selectivity
The aromatic nest rich in Tyr and Trp residues that characterizes the ligand-binding site of AChBP and the nAChR emphasizes the importance of cation–π interactions to stabilize the cation of the nicotinic ligands (Lester et al, 2004). Although several cation–π interactions are observed in the A-AChBP and L-AChBP complexes, the secondary amine in EPI and tertiary amines in LOB and MLA are located within ∼1.0 Å of the Trp147 carbonyl oxygen; this association contributes charge compensation to the cation (Figures 3B–D and 4B–D). In the ImI complex, the Arg7 backbone amide, distant by only 2 Å from the amine centroid position in the alkaloid ligands, serves as the cation (Figures 3A and 4A). Hence, the multiple ligand-bound A-AChBP structures largely support the previous assumption that a partial negative charge of the Trp147 carbonyl oxygen, mediated via the invariant Asp85, may favor interaction with the common cationic ammonium of the nicotinic ligands (Celie et al, 2004).
This comparative analysis also permits identification of the structural determinants required to accommodate specifically these chemically diverse ligands. It has been suggested that the ligand may access the binding pocket from either the apical side or the ‘membrane' side of loop C (Brejc et al, 2001). The structure of the MLA–A-AChBP complex reveals an unpredicted binding pocket, well removed from Trp147 at the ‘membrane' side of the agonist-binding site and loop C. This pocket accommodates the MLA N-phenyl succinimide moiety (Figures 3B, 4B, and 5C). In turn, the structure of the ImI–A-AChBP complex identifies a second binding region, located on the apical side of loop C, which accommodates ImI Trp10 and neighboring residues (Figures 3A, 4A, and 5C). The residues that line these two distant functional sites are weakly conserved within the nAChRs and may confer subtype selectivity. In apo A-AChBP with its open loop C, these pockets at the subunit interface are freely accessible from the outside (Figure 2), whereas in the agonist complexes (closed loop C) access is impaired by the close proximity of the invariant Tyr188 and Lys143 residues (located on the ‘membrane' side) and the tight packing of the Cys190–Cys191 disulfide (apical side) that act as barriers that isolate the competitive binding site (Figures 3C, D and 4C, D). In the LOB complex, subtle reorientations of side chains near the binding pocket, as seen for Tyr93, displaced by one of the LOB phenyl moieties, are required to accommodate a specific ligand (Figures 3C and 4C).
The mode of binding of ImI reveals that the critical Asp5–Pro6–Arg-7 triad, rather than Trp10 (Maslennikov et al, 1999), is the primary region that targets A-AChBP (Figures 3A and 4A), a finding consistent with the low conformational flexibility of these residues in solution. We would anticipate that substitution of ImI Pro6 or Ala9 by a residue with a larger side chain as found in ImII would drastically impair ImI binding, whereas the binding contribution of Trp10 would be less sensitive to mutation. Based on these structural criteria, α-conotoxin EpI should bind A-AChBP; this would be consistent with data on binding to the α7 receptor (Nicke et al, 2004). To some extent, this rationale could apply to α-conotoxin PnIB, whereas α-conotoxin MII should not be a potent blocker, as found for MI (Table I). Overall, the crystalline ImI–A-AChBP complex provides a suitable template for designing synthetic peptides for further characterization of nAChR subtypes, and highlights the absolute requirement of a α310 helical region within loop I of the α-conotoxin to confer nAChR antagonism.
Previous structural analysis of the Cbtx–L-AChBP complex along with unpublished mutagenesis data led us to examine whether the lower affinity of Cbtx for A-AChBP (Table I) could be enhanced, to approach that found for L-AChBP, by a His197Phe substitution that may eliminate electrostatic repulsion on the external face of loop C (Bourne et al, 2005). In contrast to the large and flat Cbtx molecule, the smaller and globular ImI molecule binds deeper in the binding pocket, and residues that line the pocket are primary candidates for mutation. Docking of ImI onto L-AChBP, by superimposing the L-AChBP and A-AChBP coordinates from their respective Cbtx and ImI complexes, does not reveal steric occlusion except for a small overlap between ImI Trp10 and the side chain of L-AChBP Gln78. It therefore appears that ligand selectivity arises from subtle differences between L-AChBP and A-AChBP such as electrostatic repulsion, most particularly with residues in loop F at the (−) face of the subunit interface. Based on their structural similarity, the 310 helical turn in ImI and the tip of loop II in Cbtx have been proposed to exhibit similar spatial organization upon binding to the α7 receptor, with the ImI Trp10/Arg7 and Cbtx Phe29/Arg33 pairs mimicking each other and being deeply anchored within the binding pocket (Tsetlin, 1999). However, comparative analysis of the respective complexes shows that these structural determinants are separated by 7 Å and rotated by 50° from each other. Hence, the toxins may orient differently in targeting distinct nAChR subtypes.
Concluding remarks
The structure of apo A-AChBP reveals several features not evident in those of the Hepes-bound L-AChBP and A-AChBP. First of all, the radial extension of loop C creates a portal for ligand access. Such a conformation in the absence of a bound cationic ligand may better resemble a resting state of the nAChR than the open conformation previously observed for Cbtx-bound L-AChBP. This structure provides a lead template for drug-design studies.
Nicotinic agonists bind within a discrete site, as seen for the LOB- and EPI-A-AChBP and nicotine– and carbamylcholine–L-AChBP complexes. They elicit loop C closure, and this may be an essential feature of a conformational change linked to channel opening in the nAChR. Nicotinic antagonists primarily use distinct nonoverlapping regions of the subunit interface for stabilization of their complexes. Consistent with their disparate structures and modes of association, they either fit to the apo conformation of loop C (MLA–A-AChBP complex) or further distend it radially (ImI–A-AChBP complex) or tangentially (Cbtx–L-AChBP complex), suggesting that loop C extension may be a common feature for most competitive antagonists. Accordingly, loop C extension and closure may be the distinguishing feature between agonist and antagonist complexes with the nAChR.
Materials and methods
Protein expression and purification
A-AChBP, flanked with an N-terminal FLAG epitope numbered (−8)DYKDDDDKL(0), was expressed from chemically synthesized cDNA (Hansen et al, 2004) as a soluble exported protein from stable HEK293S cells lacking the N-acetylglucosaminyltransferase I (GnTI−) gene (Reeves et al, 2002) and selected for G418 resistance. Culture media containing A-AChBP (6–8 mg/l) were collected at 24–36-h intervals and stored at 4°C with 0.02% NaN3. A-AChBP was purified on immobilized anti-FLAG antibody (Hansen et al, 2002), dialyzed against 50 mM Tris, pH 7.4, 150 mM NaCl, 0.02% NaN3, and concentrated to 18 mg/ml by ultrafiltration (YM50 Centricon unit). Mass spectrometry analyses yielded a monoisotopic mass of 27 254 Da, that is, 3.9% higher than the theoretical mass, and indicated the presence of a single heptasaccharide Man5GlcNAc2 linked to Asn74. Gel filtration FPLC on prepacked Superdex-200 (Amersham Biosciences) showed a single peak corresponding to a pentamer of subunits.
Ligand binding
Kd's for the high-affinity ligands (values <100 nM) were determined from the ratio of dissociation to association rates by monitoring intrinsic Trp quenching with stopped-flow spectrofluorometry, while the low-affinity ligands (values >100 nM) were measured by equilibrium fluorescence quenching (Hansen et al, 2004).
Crystallization and data collection
The peptidic α-conotoxin ImI, from Conus imperialis venom, was from the American Peptide Co. The organic ligands MLA and LOB were from Tocris and EPI from Sigma-Aldrich. Formation of the four A-AChBP complexes used a 1.1-fold molar excess of ligand and 1 h incubation at room temperature. Crystallization was achieved by vapor diffusion either at 18°C using a protein-to-well ratio of 1:1 in 1 μl hanging drops, or at 20°C using a protein-to-well ratio of 2:1 in nanoliter sitting drops setup with automated crystallization TECAN Genesis and Cartesian robots (Sulzenbacher et al, 2002). The well solutions were: for apo A-AChBP, 12–14% PEG-4000 (Fluka), 0.1 M sodium citrate, pH 5.6, 20% isopropanol, 5% glycerol; for the ImI complex, 11–14% PEG-4000, 0.1 M Tris, pH 7.5, 0.4 M MgCl2; for the MLA complex, 21–22% PEG-4000, 0.1 M Tris, pH 7.5, 0.4 M MgCl2; for the EPI complex, 18–22% PEG-3350 (Nextal Biotech.), 0.1 M Tris, pH 7.5, 0.2 M sodium citrate, and the crystals were improved by macroseeding; for the LOB complex, 25% PEG-4000, 0.1 M Hepes, pH 7.5. The crystals were flash-cooled in liquid nitrogen after successive short soaks into well solutions supplemented with 18–23% PEG and 3–5% glycerol. Data were processed with HKL2000 (Otwinowski and Minor, 1997) or Mosflm (Leslie, 1992), and all further computing carried out with the CCP4 program suite (CCP4, 1994), unless otherwise stated.
Structure determination and refinement
The five structures were solved by molecular replacement with AMoRe (Navaza, 1994), using, as search model, the structure of Hepes-bound L-AChBP (PDB code 1UX2; Celie et al, 2004) for the apo A-AChBP structure, and the latter for each of the four complexes. The initial electron density maps were improved considerably using the automatic rebuilding procedure as implemented in ARP/wARP (Perrakis et al, 1999) and manual adjustment with the graphics programs Xtalview v4.1 (McRee, 1992) and TURBO-FRODO (Roussel and Cambillau, 1989). All structures were refined with REFMAC (Murshudov et al, 1997) using the maximum likelihood approach and incorporating bulk solvent corrections, anisotropic Fobs versus Fcalc scaling, and TLS refinement with each subunit defining a TLS group. Random sets of reflections were set aside for cross-validation purposes. NCS restraints were applied for refinement of the MLA, EPI, and LOB complex structures. Automated solvent building was performed with ARP/wARP. Data collection and refinement statistics are reported in Table II.
The final apo A-AChBP and MLA, LOB, and EPI complex structures comprise residues His1–Arg207/Arg208 in all five subunits. A partially ordered PEG molecule occupies the binding pocket in all five apo A-AChBP subunits. The final ImI complex structure comprises A-AChBP residues His1–Arg208 and Im1 residues Gly1–Cys12 in all five subunits and bound toxins. In all structures, most of the N-terminal FLAG epitope is clearly visible in most subunits, but weak electron density is associated with surface loop Asn15–Met19. A well-ordered GlcNAc moiety linked to Asn74 is visible in a single subunit in the apo and the ImI complex structures. The stereochemistries of the structures were analyzed with PROCHECK (Laskowski et al, 1993); no residues were found in the disallowed regions of the Ramachandran plot. Figures 2 and 5 were made with ViewerLite (Accelyrs) and Figures 3 and 4 with PyMOL (DeLano Scientific LLC). Atomic coordinates and structure factors have been deposited with the Protein Data Bank (cf. Table II for accession codes).
Acknowledgments
We are grateful to the ESRF (Grenoble, France), ALS (Berkeley, CA), and APS (Argonne, IL) staff for assistance in data collection, and to G Ghosh, N Nguyen, C Kim, SS Taylor, TT Talley and Z Radic (UCSD) for support and fruitful discussions. This work was supported by USPHS grants R37-GM18360/GM07752 (to PT) and T32DK007233 (to TH), a Tobacco-Related Disease Research fellowship (to SBH), and by the CNRS during SBH's work in Marseille (to YB and PM).
References
- Badio B, Daly JW (1994) Epibatidine, a potent analgetic and nicotinic agonist. Mol Pharmacol 45: 563–569 [PubMed] [Google Scholar]
- Barlow RB, Johnson O (1989) Relations between structure and nicotine-like activity: X-ray crystal structure analysis of (−)-cytisine and (−)-lobeline hydrochloride and a comparison with (−)-nicotine and other nicotine-like compounds. Br J Pharmacol 98: 799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeier SC, Ismail KA, Arason KM, McKay S, Bryant DL, McKay DB (2004) Structure activity studies of ring E analogues of methyllycaconitine. Part 2: Synthesis of antagonists to the α3β4 nicotinic acetylcholine receptors through modifications to the ester. Bioorg Med Chem Lett 14: 3739–3742 [DOI] [PubMed] [Google Scholar]
- Bourne Y, Talley TT, Hansen S B, Taylor P, Marchot P (2005) Crystal structure of a Cbtx–AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J 24: 1512–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM (2004) Coupling of agonist binding to channel gating in an ACh-binding protein linked to an ion channel. Nature 430: 896–900 [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411: 269–276 [DOI] [PubMed] [Google Scholar]
- CCP4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Celie PH, Kasheverov IE, Mordvintsev DY, Hogg RC, van Nierop P, van Elk R, van Rossum-Fikkert SE, Zhmak MN, Bertrand D, Tsetlin V, Sixma TK, Smit AB (2005) Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an α-conotoxin PnIA variant. Nat Struct Mol Biol 12: 582–588 [DOI] [PubMed] [Google Scholar]
- Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK (2004) Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41: 907–914 [DOI] [PubMed] [Google Scholar]
- Ellison M, Gao F, Wang HL, Sine SM, McIntosh JM, Olivera BM (2004) α-Conotoxins ImI and ImII target distinct regions of the human α7 nicotinic acetylcholine receptor and distinguish human nicotinic receptor subtypes. Biochemistry 43: 16019–16026 [DOI] [PubMed] [Google Scholar]
- Ellison M, McIntosh JM, Olivera BM (2003) α-Conotoxins ImI and ImII. Similar α7 nicotinic receptor antagonists act at different sites. J Biol Chem 278: 757–764 [DOI] [PubMed] [Google Scholar]
- Flammia D, Dukat M, Damaj MI, Martin B, Glennon RA (1999) Lobeline: structure–affinity investigation of nicotinic acetylcholinergic receptor binding. J Med Chem 42: 3726–3731 [DOI] [PubMed] [Google Scholar]
- Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J (1995) Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol 48: 774–782 [PubMed] [Google Scholar]
- Grutter T, Changeux JP (2001) Nicotinic receptors in wonderland. Trends Biochem Sci 26: 459–463 [DOI] [PubMed] [Google Scholar]
- Hansen SB, Radic Z, Talley TT, Molles BE, Deerinck T, Tsigelny I, Taylor P (2002) Tryptophan fluorescence reveals conformational changes in the acetylcholine binding protein. J Biol Chem 277: 41299–41302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Talley TT, Radic Z, Taylor P (2004) Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J Biol Chem 279: 24197–24202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardick DJ, Cooper G, Scott-Ward T, Blagbrough IS, Potter BV, Wonnacott S (1995) Conversion of the sodium channel activator aconitine into a potent α7-selective nicotinic ligand. FEBS Lett 365: 79–82 [DOI] [PubMed] [Google Scholar]
- Karlin A (1969) Chemical modification of the active site of the acetylcholine receptor. J Gen Physiol 54: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A (2002) Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci 3: 102–114 [DOI] [PubMed] [Google Scholar]
- Laskowski R, MacArthur M, Moss D, Thornton J (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Leslie A (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4+ESF-EAMCB Newslett Prot Crystallogr 26 [Google Scholar]
- Lester HA, Dibas MI, Dahan DS, Leite JF, Dougherty DA (2004) Cys-loop receptors: new twists and turns. Trends Neurosci 27: 329–336 [DOI] [PubMed] [Google Scholar]
- Maslennikov IV, Shenkarev ZO, Zhmak MN, Ivanov VT, Methfessel C, Tsetlin VI, Arseniev AS (1999) NMR spatial structure of α-conotoxin ImI reveals a common scaffold in snail and snake toxins recognizing neuronal nicotinic acetylcholine receptors. FEBS Lett 444: 275–280 [DOI] [PubMed] [Google Scholar]
- McRee D (1992) A visual protein crystallographic software system for X11/XView. J Mol Graph 10: 44–46 [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr A 50: 157–163 [Google Scholar]
- Nicke A, Wonnacott S, Lewis RJ (2004) α-Conotoxins as tools for the elucidation of structure and function of neuronal nicotinic acetylcholine receptor subtypes. Eur J Biochem 271: 2305–2319 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Quiram PA, Sine SM (1998) Structural elements in α-conotoxin ImI essential for binding to neuronal α7 receptors. J Biol Chem 273: 11007–11011 [DOI] [PubMed] [Google Scholar]
- Reeves PJ, Callewaert N, Contreras R, Khorana HG (2002) Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci USA 99: 13419–13424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman DP, Agius MA (2003) Treatment of autoimmune Myasthenia gravis. Neurology 61: 1652–1661 [DOI] [PubMed] [Google Scholar]
- Rogers JP, Luginbuhl P, Shen GS, McCabe RT, Stevens RC, Wemmer DE (1999) NMR solution structure of α-conotoxin ImI and comparison to other conotoxins specific for neuronal nicotinic acetylcholine receptors. Biochemistry 38: 3874–3882 [DOI] [PubMed] [Google Scholar]
- Roussel A, Cambillau C (1989) TURBO-FRODO. Mountain View, CA: Silicon Graphics [Google Scholar]
- Sakmann B (1992) Elementary steps in synaptic transmission revealed by currents through single ion channels. Science 256: 503–512 [DOI] [PubMed] [Google Scholar]
- Servent D, Thanh HL, Antil S, Bertrand D, Corringer PJ, Changeux JP, Menez A (1998) Functional determinants by which snake and cone snail toxins block the α7 neuronal nicotinic acetylcholine receptors. J Physiol Paris 92: 107–111 [DOI] [PubMed] [Google Scholar]
- Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP (2001) A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 411: 261–268 [DOI] [PubMed] [Google Scholar]
- Sulzenbacher G, Gruez A, Roig-Zamboni V, Spinelli S, Valencia C, Pagot F, Vincentelli R, Bignon C, Salomoni A, Grisel S, Maurin D, Huyghe C, Johansson K, Grassick A, Roussel A, Bourne Y, Perrier S, Miallau L, Cantau P, Blanc E, Genevois M, Grossi A, Zenatti A, Campanacci V, Cambillau C (2002) A medium-throughput crystallization approach. Acta Crystallogr D 58: 2109–2115 [DOI] [PubMed] [Google Scholar]
- Terry AV Jr, Williamson R, Gattu M, Beach JW, McCurdy CR, Sparks JA, Pauly JR (1998) Lobeline and structurally simplified analogs exhibit differential agonist activity and sensitivity to antagonist blockade when compared to nicotine. Neuropharmacology 37: 93–102 [DOI] [PubMed] [Google Scholar]
- Tsetlin V (1999) Snake venom α-neurotoxins and other ‘three-finger' proteins. Eur J Biochem 264: 281–286 [DOI] [PubMed] [Google Scholar]
- Unwin N (2005) Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J Mol Biol 346: 967–989 [DOI] [PubMed] [Google Scholar]
- Ward JM, Cockcroft VB, Lunt GG, Smillie FS, Wonnacott S (1990) Methyllycaconitine: a selective probe for neuronal α-bungarotoxin binding sites. FEBS Lett 270: 45–48 [DOI] [PubMed] [Google Scholar]