

Abstract
It has been proposed that microtubules (MTs) participate in skeletal muscle cell differentiation. However, it is still unclear how this happens. To examine whether MTs could participate directly in the organization of thick and thin filaments into sarcomeres, we observed the concomitant reorganization and dynamics of MTs with the behavior of sarcomeric actin and myosin by time-lapse confocal microscopy. Using green fluorescent protein (GFP)-EB1 protein to label MT plus ends, we determined that MTs become organized into antiparallel arrays along fusing myotubes. Their dynamics and orientation was found to be different across the thickness of the myotubes. We observed fast movements of Dsred-myosin along GFP-MTs. Comparison of GFP-EB1 and Dsred-myosin dynamics revealed that myosin moved toward MT plus ends. Immuno-electron microscopy experiments confirmed that myosin was actually associated with MTs in myotubes. Finally, we confirmed that MTs were required for the stabilization of myosin-containing elements prior to incorporation into mature sarcomeres. Collectively, our results strongly suggest that MTs become organized into a scaffold that provides directional cues for the positioning and organization of myosin filaments during sarcomere formation.
Keywords: microtubules, muscle differentiation, sarcomeric myosin, transport
Introduction
Skeletal myogenic differentiation involves extensive changes in cell morphology and subcellular architecture. Upon differentiation, myoblasts fuse to form multinucleated syncitia that eventually develop into terminally differentiated muscle cells, the myofibers. Each myofiber is essentially a bundle of myofibrils that provide contractile properties to the whole muscle. A myofibril is built of repeats of contractile units, the sarcomeres. Assembly of contractile proteins into sarcomeres is a complex process that requires polymerization of sarcomeric actin and myosin into thin and thick filaments, remodeling of each polymer into filaments of precise length, and association and alignment of the two filament systems (Craig, 1994). The striking regularity of myosin and actin filaments within the sarcomere is not the result of self-assembly properties of their major constituting proteins, but requires the synthesis and the regulation of numerous muscle proteins expressed during myogenic differentiation, some of them displaying specific interactions with the cytoskeleton lattice (Fürst et al, 1989; Small et al, 1992; Seiler et al, 1996; Ehler et al, 1999). In spite of numerous earlier observations, an important and still unsolved question in muscle research concerns the assembly of myosin filaments with opposite polarity into arrays of actin filaments within the sarcomere. Based on the effect of microtubule (MT)-directed drugs, several observations suggest that MTs could participate in the organization of thin and thick filament complexes and in the formation of sarcomeres (Warren, 1968; Goldstein and Entman, 1979; Holtzer et al, 1985; Guo et al, 1986; Saitoh et al, 1988). Moreover, MURF2, a specific myogenic protein, could function as a transient adaptator between Titin, MTs and sarcomeric myosin (Pizon et al, 2002). In addition to sarcomere formation, a striking characteristic of myogenic differentiation is the extensive morphologic change occurring during the myoblast to myotube transition. Changes in myogenic cell morphology are related to MT network reorganization (Tassin et al, 1985; Musa et al, 2003; Bugnard et al, 2005). In proliferating myoblasts, a single juxtanuclear centrosome, also called MT organizing center (MTOC) nucleates MTs. In myotubes, the centrosome is eliminated and some MTs that form linear arrays parrallel to the long axis of the cell become detyrosinated, a sign of their increased stability (Gundersen et al, 1989). The importance of MTs for proper myogenesis has been inferred from studies in which myotube formation was aberrant because MTs were either disrupted or stabilized during differentiation (Warren, 1968; Holtzer et al, 1985; Saitoh et al, 1988). Therefore, regulation of MT dynamics seems to be of critical importance for myoblast differentiation. In spite of all these observations, little progress has been made on a potential causal relationship between MT organization and dynamics and formation of the sarcomeres. Although descriptive data report on MT network reorganization, no thorough analysis of MT dynamics has been performed so far in living muscle cells. Moreover, research devoted to investigating how muscle-specific proteins are assembled into sarcomeres has been performed mainly with immunofluorescence and electron microscopy experiments. Since myogenic differentiation is an asynchronous event leading to various phenotypes within various differentiating cells as well as within an individual cell, the data obtained could not reflect the ordered spatiotemporal phenomena leading to myofiber formation. In this study, we used fluorescent protein technology and time-lapse dual-wavelength spinning-disk confocal microscopy to analyze the dynamics and organization of MTs and sarcomeric myosin in living myotubes during their differentiation. This analysis has revealed that MTs are organized in highly dynamic antiparallel bundles that serve as scaffolding structures to direct the transport and organization of nascent myosin structures in early myotubes. Furthermore, the observation of large numbers of movies showed that, within a myotube, MTs displayed specific compartmentalization, reflected in different dynamics and orientation.
Results
Spatial distribution of sarcomeric actin and myosin in relation to the MT network during myotube morphogenesis
To investigate whether MTs could represent a scaffolding element during myofibrillogenesis, we examined simultaneously the location of MTs, and endogenous sarcomeric actin and myosin heavy chain (MHC) in the mouse C57 cell line (Pizon et al, 2002). Previous reports indicated that the number of MT diminishes during myogenic differentiation (Fischman, 1967; Cartwright and Goldstein, 1982; Saitoh et al, 1988), suggesting that MTs could participate in sarcomere organization during early myofibrillogenesis. Here, using triple immunofluorescence and confocal microscopy, we report observations on early stages of cell differentiation. At 24 h after differentiation, myotubes already synthesized sarcomeric actin and myosin, and displayed longitudinally oriented MTs (Figure 1A and B). As observed previously (Van Der Ven et al, 1999), sarcomeric actin formed a filamentous network in close proximity to the sarcolemma (Figure 1A, arrows). In the central portion of the cell, the paucity of distribution of actin staining contrasted strongly with the presence of numerous myosin-containing elements. In the perinuclear region myosin accumulated as a filamentous meshwork, while myosin appeared as dots and short disordered filaments within the sarcoplasm of the myotube. These myosin-containing elements ranged from 0.2 to 2 μm in length, and could represent folding and intermediate myosin structures preceding myosin fiber formation (Lin et al, 1994; Rudy et al, 2001; Srikakulam and Winkelmann, 2004); therefore, we refer to these structures as ‘nascent myosin structures'. Close examination of nascent myosin structures showed that they distributed along MTs in the central region of the cell (Figure 1B, arrowheads). At this stage of differentiation, no obvious colocalization of actin with either myosin or MTs could be observed. As myofibrillogenesis proceeded (48 h differentiation), unstriated actin filaments became densely packed and accumulated predominantly under the plasma membrane of the myotubes (Figure 1C, arrow). In these cells, staining with anti-MHC antibodies revealed slender filaments aligned with MTs within the central portion of the cytoplasm (Figure 1C, arrowhead). Observation of three successive x–y stacks located 1.2 μm apart within the depth (z-direction) of the same myotube revealed various patterns of organization for MTs, actin and myosin. From the cell interior toward the cell membrane, we successively observed bundles of MTs (Figure 1D), colocalization of myosin filaments and MTs (Figure 1E), and then alignments of myosin and actin filaments (Figure 1F). As reported previously (Cartwright and Goldstein, 1982), after 72 h of differentiation we observed (Figure 1G) that myosin filaments became more numerous and multilayered along the longitudinal axis of the myotube. They formed fibers and MT density decreased within the central region of the myotube. No colocalization of actin and MTs could be detected at this stage of differentiation.
Figure 1.
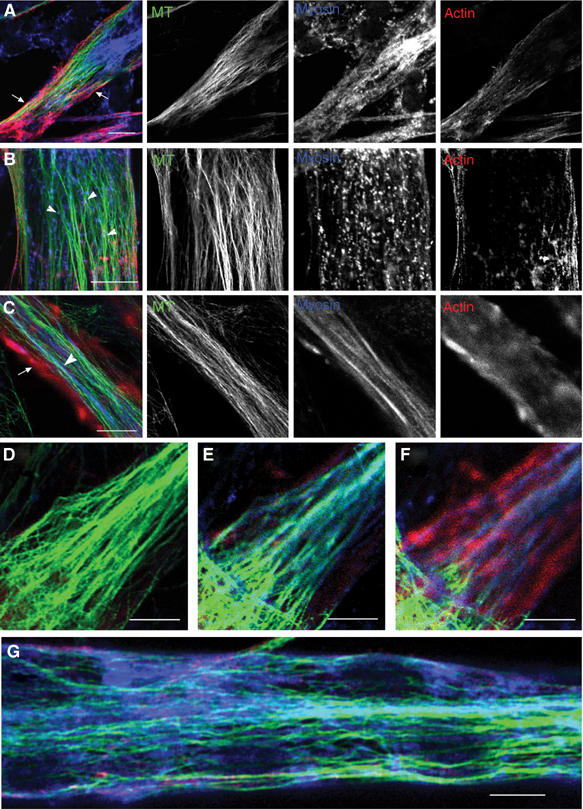
Location of endogenous sarcomeric actin, myosin and MT during myogenic differentiation. Cells differentiated for 24 h (A, B), 48 h (C–F) and 72 h (G) were stained with antibodies directed against sarcomeric actin (red), sarcomeric myosin (blue) and MT (green). Bar=10 μm (B, D–G), 20 μm (A, C).
These results show that, prior to sarcomere formation, sarcomeric actin and myosin display specific intracellular location and that nascent myosin structures and myosin fibers localize along the MT network.
Antiparallel organization of MTs in differentiating myotubes
The finding that early myosin structures are aligned along MTs raised the possibility that MTs could participate in the transport and organization of myosin filaments during myogenesis. We therefore decided to examine the three-dimensional organization and dynamics of MTs in differentiating myotubes. Using myogenic cells expressing green fluorescent protein (GFP)-tubulin, lifetime experiments were conducted with a time-lapse dual-wavelength spinning-disk confocal microscope to examine MT organization. Figure 2 shows representative frames of movies obtained with myoblasts (Figure 2A) and myotubes (Figure 2B). As commonly observed in various animal cells, in myoblasts, MTs emanated from the MTOC associated with centrioles (Figure 2A, arrowhead) and displayed a radial organization extending towards the cell periphery. Following fusion, as MTs became organized into bundles running parallel to the long axis of the syncytium, no clear origin could be discriminated anymore (Figure 2B). The difficulty in clearly identifying individual extremities of MTs within myotubes prompted us to use an alternative approach to study MT dynamics. EB1 protein is a plus-end tracking protein specifically associated with the growing end of MTs, also named MT plus end. Fused to GFP, the fluorescent protein (GFP-EB1) has been used as a marker of MT plus-end growth events (Mimori-Kiyosue et al, 2000; Morrison et al, 2002; Musa et al, 2003). Transfection of skeletal muscle cells with GFP-EB1 yielded comet-like structures in undifferentiated (data not shown) and differentiated cells expressing low amounts of the protein. Many moving fluorescent comets were observed in various regions of myotubes (Figure 2C and D and Supplementary Movie 2). To reveal MT growth direction, EB1 was monitored in one confocal optical section in living myotubes after 48 h of differentiation. Still images extracted from movie 2 show a representative myotube domain localized between two nuclei (Figure 2C, N1 and N2). We observed longitudinal antiparallel GFP-EB1 movements throughout the elongated cell body. In this specific cell, trajectory analyses indicated that 70% of the comets moved from nucleus N2 to N1 (Figure 2C, arrowheads), the other comets moving in the opposite direction (Figure 2C, arrows). Almost all the fluorescent comet trajectories were aligned with the major axis of the cell and could be followed over distances of up to 3.37 μm. In order to determine if MT dynamics was uniform all over an individual myotube, we recorded EB1 excursions at various depths within various myotubes after 48 h of differentiation. To avoid the effect of specific regulation processes due to the interaction of MTs with the cell cortex (Rodriguez et al, 2003), GFP-EB1 behavior was monitored, in two or three stacks 1 μm apart in the z-direction at the level of myotube nuclei within the elongated central portion of the cell. Identical time-lapse recording conditions were used to monitor EB1 in the various stacks of a given myotube. Life history of EB1 movements could be revealed by a projection analysis generated by merging successive frames obtained within an individual stack. Figures 2E and F show the projections obtained, respectively, from movies recorded in two individual levels of cell no. 7 (Table I). According to the position within the cell depth, various patterns of EB1 fluorescence were observed. While EB1 trajectories appeared as long continuous tracks in stack 1 (Figure 2E), the image obtained from stack 2 showed alignments of small tracks (Figure 2F). When the average speed and length of EB1 excursions were determined at each level, we realized that these trajectories reflected distinct MT dynamic behaviors (Table I). MT dynamic properties were analyzed from 18 movies taken in eight different young myotubes. Using a customized recognition program that automatically recorded many EB1 trajectories, thus providing good statistical data (see Materials and methods), we calculated the average rate and the ‘mean tracking distance' (MTD) of EB1 displacement. We defined MTD as the mean distance over which an EB1 comet could be followed before its fluorescence disappeared. The threshold (minimal fluorescence) for EB1 comet identification was set to 4 pixels. Speed analyses revealed that, in myotubes, EB1 comets move with an average speed of 21±6.7 μm/min. This analysis also showed that EB1 rates varied from cell to cell between 14.18±5.2 (Table I, cell 7, stack 2) and 29.40±15.63 μm/min (Table I, cell 2, stack 1). However, rates were fairly homogeneous throughout a given cell (Table I, histogram A). When we analyzed the MTD of EB1 comets, we observed that it varied from 0.88±0.44 (Table I, cell 7, stack 2) to 2.06±1.82 μm (Table I, cell 4, stack 1). Surprisingly, we found that the MTD of EB1 comets varied according to location within the cell depth (Table I, histogram B). This variation did not seem to be due to differences in our ability to track EB1 comets over different distances, but rather reflected the time over which a plus end continued to grow in different domains of the cytoplasm. This suggested that, although growth rates were fairly uniform, MT plus-end dynamics were different in different cytoplasmic domains of a given cell. In contrast to mononucleate cells in which anterograde and retrograde movements referred to the nucleus position, no reproducible specific origin could be assigned to compare EB1 comet behavior between various living multinucleated myotubes. Therefore, we arbitrarily chose to name EB1+ the longitudinal movements occurring from left to right, and EB1− the opposite movement between two successive nuclei. Quantification of EB1+ and EB1– directed movements, in several cells, revealed an asymmetrical distribution of GFP-EB1 directional movements (Table I). In cells no. 1 and 3, we found about 70% of EB1+ and 30% of EB1− comets. In these cells, the main orientation of EB1 movements was similar throughout the cytoplasm. In cell no. 4, where the majority of EB1 tracking was directed in the – direction, we determined that the percentage of the + and – displacements changed according to the stack position, EB1− comets varying from 93 to 71.5%. In cell no. 2 EB1 directionality of movement was asymmetric in stack 1, whereas in the two successive ones (2 and 3) EB1 comets moved symmetrically. These data showed that the antiparallel organization of MTs varied according to position within the myotube.
Figure 2.
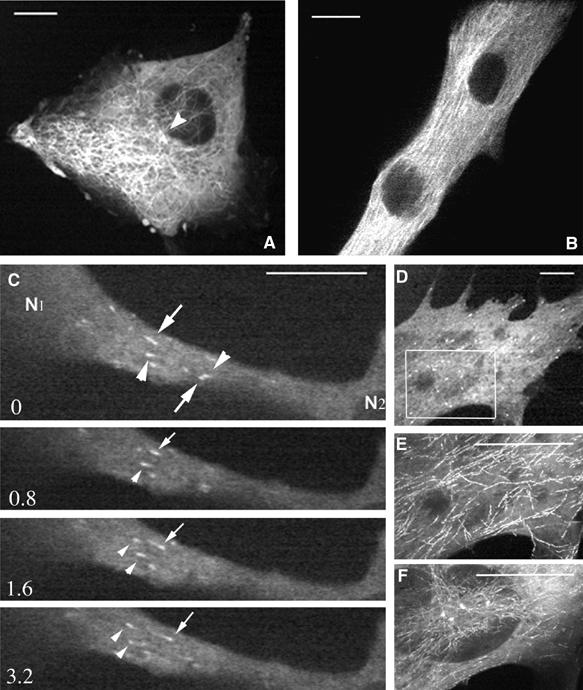
MT network organization during myogenic differentiation. Myogenic cells expressing GFP-tubulin (A, B) or GFP-EB1(C–F) were imaged every second during 12 (A) or 10 min (B); every 0.4 s for 4 min (C) and every 0.6 s for 3 min (D–F). Images A and B, selected from movies, show that MTs emanate from the MTOC in myoblasts (A, arrowhead) and display longitudinal organization in myotubes (B). (C) shows time-lapse sequence of GFP-EB1 movements (movie 2). In a myotube region located between two successive nuclei (N1, N2), we observed that some GFP-EB1 comets moved from nucleus N1 to N2 (arrows) and others in the opposite direction (arrowheads). EB1 comets were followed at two depths (E, F) of the same cell area (D, rectangle). Long linear tracks (E) and dots alignments (F) of EB1 movements were observed by projection analysis generated by merging the images of movies 5S5 and 5S6, respectively. Time is in seconds, bar=10 μm.
Table 1.
Velocity and tracking of GFP-EB1 comets in myotubes
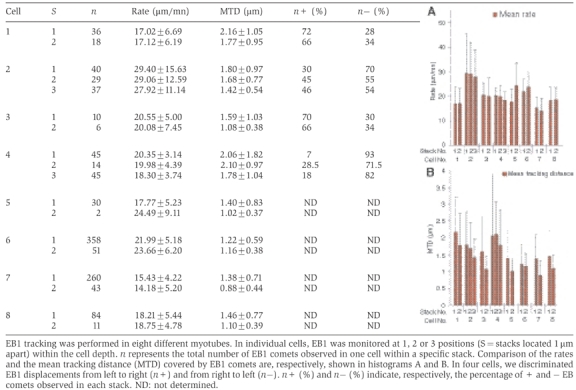 |
These results showed that in myotubes the MT network is composed of antiparallel MTs. MT growth rate is homogeneous throughout the cytoplasm, whereas MT plus-end dynamics (at least the stability of MT plus-end growth phase) vary according to the position within the cell. The variability of antiparallel organization of MTs and their variable stability probably reflects the fact that we are observing myotubes evolving through a morphogenetic process.
Sarcomeric myosin migrates on MT towards their plus ends
Our initial experiments on fixed cells indicated that, at early stages of differentiation, nascent myosin structures seemed to undergo migratory processes along the elongated MTs located in the central region of myotubes (Figure 1). In order to determine whether myosin elements were indeed moving along MTs, in vivo experiments were performed. Thus, α-tubulin tagged with the GFP-tubulin was used to visualize MTs, and a DsredII-tagged myosin light chain 3f (Dsred-myosin) expression vector was developed to visualize myosin. We verified by immunofluorescence experiments that exogenous proteins displayed the same location as their endogenous counterparts (data not shown). Myosin and MTs were then observed simultaneously in living cells, using time-lapse dual-wavelength spinning-disk confocal microscopy. At 24 h after double transfection, myoblasts were induced to differentiate for various times. A representative frame from movies obtained in myotubes differentiated for 24 h showed that the exogenous fluorescent proteins labeled MTs uniformly, and that bright fluorescent myosin structures distributed from the perinuclear region to the cell periphery (Figure 3A). After 72 h of differentiation, MTs aligned parallel to the long axis of the myotubes, nascent myosin structures scattered into the cell body, and myosin filaments organized into nascent myofibrils registered with MTs along the lateral cell borders (Figure 3B, arrowheads). As previously observed with GFP-myosin (Auerbach et al, 1997), integration of Dsred-myosin into mature sarcomeres did not inhibit contraction (Supplementary Movie 5_2). The dynamics of nascent myosin structures was then determined in myotubes after 24 h of differentiation. Figures 3C–E show representative frames from Supplementary Movie 3, in which the fluorescent proteins were imaged at 25-s intervals during 52 min. We monitored nascent myosin structures with sustained translocation covering 2 to 18 μm. Time-lapse fluorescent confocal microscopy revealed that myosin structures displayed many changes in shape, including coalescence of small aggregates into larger ones during their movements. Trajectory analyses revealed that, within the central region of the myotube, myosin moved along MTs and that the overall direction of myosin was oriented from the cell center toward the cell tip (Figure 3C, arrowheads, and 3F). However, myosin excursions were not always simple: some myosin structures could remain still for long periods of time, while others displayed short pauses. We also observed myosin structures changing direction: moving first in one orientation (Figure 3E, arrow), then in the opposite one (Figure 3E, arrowhead). Neighboring nascent myosin structures displaying antiparallel translocation were also observed (Figure 3D, arrowhead, arrow). Nevertheless, when these structures underwent multidirectional trajectories, displacements seemed always to take place along MT tracks, as revealed by life history analysis of myosin displacements (Figure 3F). Velocity analyses performed on 17 particles detected in four different myotubes indicated that the average mobility of myosin was independent of direction and could vary from 0.52±0.26 to 3.39±1.28 μm/min. These variable rates are due to the fact that individual myosin structures showed a combination of rapid and slow movements and also underwent pauses. This did not seem to be related to the distance covered by the structures. These data suggested that MTs could direct the organization and sorting of nascent myosin structures at the beginning of myofibrillogenesis. Since the rate of myosin particles was much slower than the growing rates of MT plus ends measured previously, myosin movements could not be due to myosin molecules anchored at the plus ends of growing MTs. In order to determine the directionality of myosin movements along MTs, GFP-EB1 protein was used as a reporter to label the plus end of MTs. Myotubes expressing GFP-EB1 and Dsred-myosin were observed after 24 h of differentiation. Figure 4 shows representative frames of a 12-min movie in which images were captured every 2 s. Close examination of the cell region displaying movements of myosin particles (Figure 4A) showed fast yellow EB1 dots running regularly through the entire length of the red myosin structures (Supplementary Movie 4_1). Still images showed colocalization of myosin structures with bright EB1 fluorescence (Figure 4B, arrow). The average myosin displacement (Figure 4B, merge) was revealed by merging myosin recording extracted from movie 4_1 captured at time 0 (red), 165 (green) and 345 s (blue). Since EB1 velocity was 5–10 times faster than myosin displacements, movements of both proteins could not be recorded simultaneously. Therefore, in order to compare EB1 and myosin directions, four successive images of the GFP-EB1 recording were extracted from movie 4_1 (Figure 4C and Supplementary Movie 4_2). In the cell region where myosin displacements were previously observed, robust EB1 labelings associated with MT plus ends displayed similar overall trajectories (Figure 4C, arrow and arrowhead). When the life history of EB1 displacement, obtained by merging images isolated from movie 4_2 at 2 (red), 4 (green) and 8 s (blue) (Figure 4C, merge), was compared to the life history of myosin movement (Figure 4B, merge), we observed that MT plus ends and myosin rods moved in the same direction. These data strongly suggested that, at the beginning of differentiation, small myosin structures displayed polarized transport and moved toward the plus end of MTs.
Figure 3.
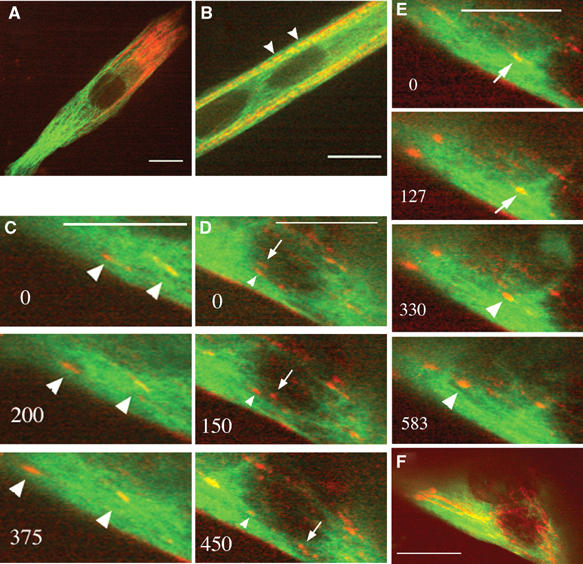
Living myogenic cells expressing GFP-tubulin (green) and Dsred-myosin (red) were analyzed by dual-wavelength spinning disk confocal microscopy. Myotubes are shown after 24 h (A, C–E) and 72 h (B) of differentiation, when nascent myofibrils start to organize at the cell membrane (B, arrowheads). (C–E) contain selected images of movie 3. The images were acquired every 25 s during 52 min. Numbers in the left corners indicate time in seconds. Time 0 indicates the initial location of the selected myosin structures we followed. Arrowheads indicate myosin displacements from right to left, arrows reveal movements from left to right. During the observations, myosin structures moved in the same direction (C, arrowheads), in opposite direction (D, arrows and arrowheads) or changed direction (E, arrows and arrowheads). Long linear tracks (F) of myosin particle movements aligned along MTs were observed by projection analysis generated by merging the images of movie 3. Bar=10 μm.
Figure 4.
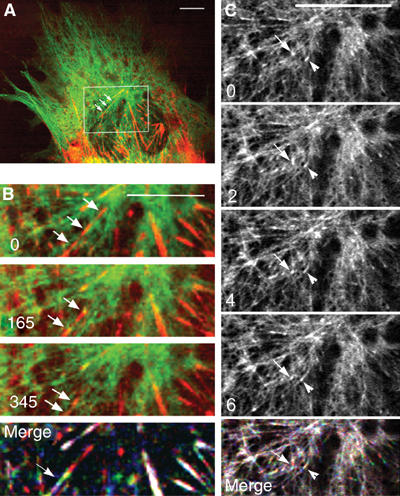
Myosin intermediates tracks MT plus end at the beginning of differentiation. A live myotube (24 h of differentiation) expressing GFP-EB1 (green) and Dsred-myosin (red) was analyzed by confocal microscopy. This figure contains images from movie 4_1. The rectangle in (A) represents the area magnified in (B, C) (movies 4_2 and 4_3, respectively). In (B), images of EB1 and myosin recording show that myosin structures (arrows) displayed red and yellow labeling during their excursion. Life history of myosin displacement (merge) was visualized by merging myosin images, from movie 4_2, captured at time 0 (red), 165 (green) and 345 s (blue). (C) shows successive images of the EB1 recording. Two distinct EB1 comets (arrow, arrowhead) move in the same direction as myosin rods. In (B, C), time 0 indicates the initial location of selected fluorescent myosin or EB1 that was followed. Life history of EB1 displacement (merge in C) was obtained by merging images, from movie 4_3, at time 2 (red), 4 (green) and 6 s (blue). Bar=10 μm.
MT direct location and movement of nascent myosin structures and sarcomere assembly
Although the fluorescence movies were strongly suggestive of myosin movement along MTs, we wanted to confirm this observation by independent experiments. Therefore, we first examined the behavior of myosin in cells in which MTs were disrupted by nocodazole (Supplementary Movie 5_1). Still images extracted from movie 5_1 show that, after 24 h of differentiation and before drug addition, myotubes expressing GFP-tubulin and Dsred-myosin displayed nascent myosin structures in the central region of the cell and myosin filaments packed along the plasma membrane (Figure 5A). Higher magnification showed that myosin structures colocalized with MTs (Figure 5B–D, arrow). Shortly after addition of 15 μM nocodazole (105 s), some MTs persisted, although most of them appeared blurred (Figure 5E–G), and cytoplasmic myosin structures were barely detectable (Figure 5E, F and H). Since nocodazole-resistant MTs have been described as stable MTs, nascent myosin structures should be associated to dynamic MTs (Gundersen et al, 1989). Myosin fibers localized nearby the plasma membrane were not disturbed (Figures 5F and H, arrowhead), as expected for myosin associated with actin fibers accumulating at the plasma membrane. Visualization of myosin at the cell periphery ruled out the possibility that disappearance of the cytoplasmic myosin structures reflected an out-of-focus imaging induced by cell shape changes. Since it has been suggested that MTs could participate in sarcomere formation (Antin et al, 1981; Toyama et al, 1982), we examined Dsred-fluorescent myosin stability in sarcomeres. Movie 5_2 shows that, 92 h after transfection, the contractile apparatus organized and started to be functional. Myosin was incorporated into mature contracting sarcomeres (Figure 5I, arrowhead) and nascent myofibrils (Figure 5I, arrow). Upon nocodazole addition (1 min) nascent myofibrils started to disorganize (Figure 5J, arrow; Supplemental Movie 5_3), while mature sarcomeres continued to exhibit spontaneous contractions (Figure 5J, arrowhead). After 35 min nascent myofibrils displayed low fluorescence (Figure 5K, arrow), while mature sarcomeres remained functional (Figure 5K, arrowhead, and Supplementary Movie 5_3).
Figure 5.
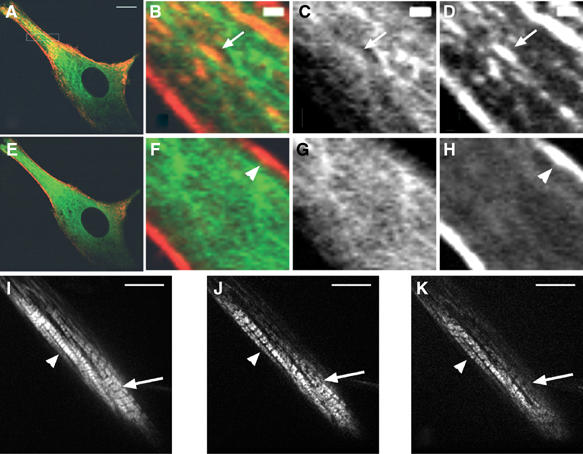
MTs are needed for myosin stability and sarcomere formation. Before nocodazole addition, young myotubes (A) displayed myosin structures colocalized with MTs. Images (B–D, F–H) are magnifications of the rectangle in (A). (B, F) are merged images of MTs (C, G) and myosin (D, H). At 105 s after drug addition (E), myosin remained only at the cell membrane (arrowhead in (F) and (H), movie 5_1). During myofibril formation (I, movie 5_2), Dsred-myosin incorporated into sarcomeres (arrowhead) and into nascent myofibrils (arrow). Nocodazole addition destabilized nascent myofibrils after 1 min (J, arrow; movie 5_3), but did not affect mature sarcomeres after 35 min of treatment (K, arrowhead). Bar=10 μm (A, I–K). Bar=1 μm (B–D).
Due to high MT concentration and low amount and small size of the nascent myosin structures observed at the beginning of differentiation, visualizing the colocation of both structures could be difficult even using confocal microscopy. Furthermore, one can imagine that nocodazole could indirectly affect myosin associated with cellular structures dependent on MTs. Therefore, we relied on immunoelectron microscopy to investigate whether myosin structures were directly associated with MTs. Out of seven myotubes differentiated for 24 h and examined randomly, six displayed myosin immunoreactivity detected on MTs. Out of 101 MTs examined 32 displayed at least one gold particle, whereas in a control cell, out of 29 MTs observed, only one MT displayed one gold particle. Electron micrographs of immunogold labeling of myosin are shown in two regions of a myotube (Figure 6A, rectangles 1 and 2). Enlarged views of region 1 (Figure 6B and D and region 2 (Figure 6C) revealed well-defined longitudinally oriented MTs (mt). Gold particles of 10 nm (arrowheads) were detected directly on MTs (Figure 6C) or within amorphous cytoplasmic material associated with MTs that resisted extraction (Figure 6B and D). Since these gold particles localized within a distance of 21 nm, equal to the sum of lengths of the primary and secondary antibody molecules, they should decorate myosin associated with MTs (Verkade et al, 1997). Filaments adjacent to MTs were devoid of immunogold particles (Figure 6B and D) and extremely rare gold particules were detected in fixed control cells prepared in the absence of primary antibody, indicating the specificity of our results.
Figure 6.
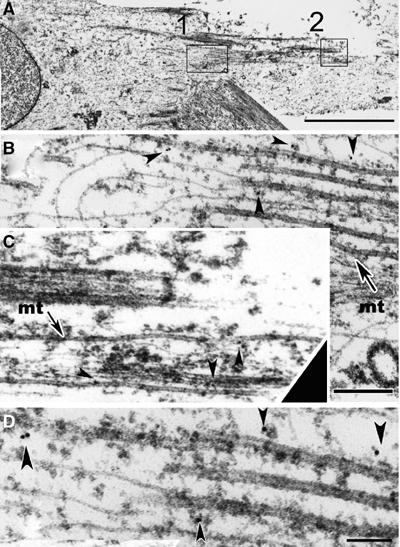
Electron micrographs of immunogold labeling of myosin in a myotube. Two regions of a myotube differentiated for 24 h are marked (A, rectangles 1 and 2). Two magnifications of region 1 are reported (B, D), while an enlarged view of region 2 is shown in (C) (same scale as B). Myosin labeled with 10 nm gold particles (arrowheads) is in close association with MTs (B and C, mt). Adjacent filaments are devoid of immunogold particles. Bar=3 μm (A). Bar=200 nm (B). Bar=100 nm (D).
Taken together, immunoelectron microscopy pictures showed at the ultrastructural level that nascent myosin structures were associated with MTs, and that an intact MT network is required for the stability of nascent myofibrils and of myosin fibers during sarcomere formation.
Actin fibers are not required for myosin movements
Although sarcomeric actin was not observed in the cytoplasm of young myotubes (Figure 1), it remained possible that undetectable microfilaments were involved in myosin movements along MTs. To address this point, we examined the effect of the barbed-end F-actin assembly inhibitor, cytochalasin D (Cooper, 1987), on the organization of nascent myosin structures. Myotubes expressing GFP-α tubulin and Dsred myosin were used for in vivo experiments after 24 h of differentiation. As exemplified in Figure 7A, before drug addition, myotubes displayed cytoplasmic myosin particles (small arrows) and myosin filaments aligned under the plasma membrane (large arrow). At 10 min after 1 μg/ml cytochalasin D addition to the medium, due to actin disorganization, the myotube shape was modified and few myosin filaments were observed nearby the plasma membrane (Figure 7B, large arrow, and Supplementary Movie 7). In contrast, numerous nascent myosin structures remained associated with MTs (Figure 7B, small arrows). In a series of pictures isolated from movie 7, we observed that, despite cytochalasin D, myosin structures could still display short movements along MTs and could fuse (Figure 7C, arrows). At 45 min after drug addition, the cell displayed numerous myosin patches (Figure 7D and E, arrowheads). At the cell margin, many growing MTs appeared to bend away from the edge of the cell and continued to elongate (Figure 7E, arrows). Myosin structures decorated some of these MTs and remained tightly bound to them (Figure 7E, hollow arrowhead). We also determined that myosin continued to fuse and to form large aggregates (Figure 7E, arrowheads). No myosin fiber elongation could be revealed all along the time course of the experiment.
Figure 7.
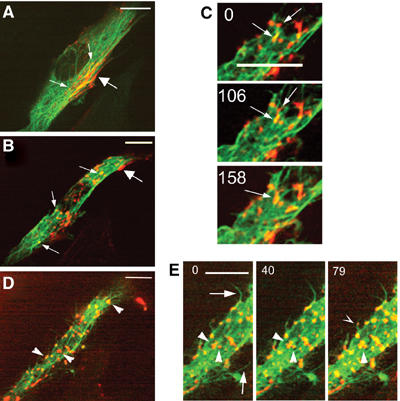
Myosin organization in cytochalasin D-treated myotubes. Myotubes differentiated for 36 h and expressing GFP-tubulin and Dsred-myosin were imaged at 13-s intervals by spinning disk confocal microscopy for 48 min. Isolated images of movie 6_1 show a myotube displaying myosin localized in the cytoplasm (A, small arrows) and under the plasma membrane (A, large arrow). At 10 min after cytochalasin D addition (B, movie 6_2) myosin fibers disappeared from the plasma membrane (B, large arrow), while myosin structures remained on MTs (B, small arrows) and fused as exemplified in (C) (arrows). At 45 min after drug addition, myotubes dispayed large myosin aggregates (D, arrowheads). Formation of these structures was followed (E, arrowheads), as well as myosin structures (E, hollow arrowhead) localized on growing MT (E, arrows). Time 0 indicates the initial location of selected myosin structures. Time is in seconds, bar=10 μm.
These results indicate that disorganization of the actin network did not impede movements of nascent myosin structures along MTs. However, it could impair some steps of myosin filament maturation, since no fiber elongation could be revealed during the time course of the experiment.
Discussion
In this study, we have examined the interaction between the acto-myosin system and MTs during myotube formation. In particular, we wanted to know whether MTs could represent an intermediate scaffolding and organizing element during myofiber development.
Myogenic differentiation is asynchronous and different levels of organization can be observed at the same time within a single cell. Although earlier observations already reported the temporal pattern of myofibrillogenesis in various myogenic systems, none of them described the simultaneous behavior of MTs, of sarcomeric actin and of sarcomeric myosin during myofibrillogenesis (Sanger et al, 2000, 2002). Therefore, we conducted triple immunofluorescence experiments to describe the organizational pattern of these cytoskeletal components during differentiation of the C57 cell line. Over a time window of 24–36 h after the onset of differentiation, MTs aligned parallel to the cell axis of fusing myoblasts. During this period, actin and myosin fibers were mainly found in the perinuclear region of the myotube, and myosin dots and rods were observed along MTs in the central region of the myotube. According to their size, location and time of occurrence, these myosin structures could be the myosin particles described previously as precursors of myosin fibers (Lin et al, 1994; Rudy et al, 2001; Srikakulam and Winkelmann, 2004). Therefore, we called these structures ‘nascent myosin structures'. Between 48 and 72 h of differentiation, upon actin and myosin accumulation, fibers started to organize in nascent myofibrils beneath the myotube membrane, while myosin fibers elongated on MTs in the central region of the cell. Beyond 72 h of differentiation, sarcomeres and nascent myofibrils represented the major components of the myotube. As previously observed, MTs were nearly undetectable in muscle fibers displaying spontaneous beating (Fischman, 1967; Saitoh et al, 1988). In agreement with various models of sarcomere formation (Rhee et al, 1994; Gregorio, 1997; Holtzer et al, 1997; Ojima et al, 1999), our observations showed that thin and thick filaments formed independently and that only sarcomeric myosin was distributed along MTs during myofibrillogenesis.
MTs are polar filaments, which can undergo alternative phases of growth, pause and shortening, a behavior termed dynamic instability (Desai and Mitchison, 1997). In living cells, a large number of proteins regulate dynamic instability, thus determining the shape of MT networks in different phases of the cell cycle and in different cell regions. Several immunofluorescence studies showed that, during the myoblast/myotube transition, MTs reorganized, leading to the formation of an aligned MT network parallel to the long cell axis (Tassin et al, 1985; Gundersen et al, 1989; Franzini-Armstrong and Fischman, 1994; Musa et al, 2003; Bugnard et al, 2005). However, no detailed kinetic analysis of MT behavior had been performed in skeletal muscle cells. To address this question, we performed in vivo time-lapse experiments using GFP-EB1 protein. EB1 is an MT-associated protein that binds specifically to the plus end of growing MTs and that detaches from the tip of still or depolymerizing MTs. Thus, GFP-EB1 has been used in various cells as a reporter to label the plus end of growing MTs (Mimori-Kiyosue et al, 2000; Morrison et al, 2002; Musa et al, 2003). Our first observations during myofribrillogenesis showed that the complete reorganization of the MT network involved the formation of antiparallel arrays of MTs aligned with the long axis of myotubes. Speed analysis revealed that EB1 fluorescent comets moved in myotubes at a rate similar to the one reported in COS-1 cells (Stepanova et al, 2003) and that this rate did not vary remarkably according to location in each individual cell. Owing to EB1 interaction specificity with MT plus end, the distance measured for EB1-GFP comets (MTD) is not strictly identical to the distance covered by growing MTs. However, the two values are related and MTD can be used as an indirect evaluation of the time spent by MTs in a growing phase. We determined that MTD of EB1 comets depends on the location of MTs within the cell, which suggests that local regulations of MT dynamic instability occur in myotubes. Specific regional activities of MT-associated proteins and MT-based motors could affect MT organization and dynamics (Ginkel and Wordeman, 2000).
Assembly of myosin in striated muscles entails events at three levels: the folding of the molecule (Lin et al, 1994; Rudy et al, 2001; Srikakulam and Winkelmann, 2004), the assembly and the incorporation of thick filaments of specific length into sarcomeres (Epstein and Fischman, 1991). Our direct observations of living myotubes expressing fluorescent MTs and sarcomeric myosin reveal that, at the beginning of differentiation, nascent myosin structures localize and move along MTs. Comparison of the trajectories of nascent myosin structures and of EB1 protein shows that myosin exhibits polarized excursion toward MT plus ends. Speed analysis indicated that the rate of myosin movements corresponded to the speed of various MT motors. Therefore, it is possible that nascent myosin structures are transported by kinesin-like plus-end-directed motors along MTs to reach the distal region of the cell (Schnapp, 2003). Myosin occasionally changed direction during its movement. Although we cannot exclude that nascent myosin structures could reverse direction by using minus-end MT motors, changes in myosin movements could also be explained by MT network architecture. Actually, in myotubes a plus-end motor loaded with myosin molecules could display various directions, traveling successively on adjacent MTs showing opposite polarities.
Nocodazole experiments and immunoelectron microscopy confirm that nascent myosin structures are physically associated with MTs. In addition, treatment with nocodazole suggests that myosin is associated with dynamic MTs. Indeed, stable MTs found in myotubes upon differentiation are resistant to nocodazole (Gundersen et al, 1989). Electron microscopy revealed that nascent myosin structures are often embedded in an amorphous matrix, which could correspond to protein complexes. These complexes could be constituted of MT motors and various myosin-binding proteins. Good candidates for interactions with nascent myosin structures could be: chaperones mediating folding and assembly of myosin (Price et al, 2002; Srikakulam and Winkelmann, 2004), the myosin-binding protein C, which favors myosin polymerization (Davis, 1988; Clark et al, 2002) and MURF2, a transient adaptator between MTs, titin and myosin (Pizon et al, 2002). Differences in MT polarity have been used to explain polarized axonal transport (Burak et al, 2000). Our results suggest that, within myotubes, irregular antiparallel MT bundles could provide local asymmetry, producing specifically oriented kinetic gradients used by MT-directed motors to transport nascent sarcomeric structures within myotubes (Figure 8). In addition to myosin, these motors could guide other molecules to the zones of myotubes where sarcomeres assemble. Therefore, it is possible that antiparallel MT arrays could play a role in the insertion of the self-assembled antiparallel myosin rods (Huxley, 1963) into the A-bands of sarcomeres. The ability of muscle cells to generate movement is related to the mechanical interaction between actin and myosin. To determine whether myosin tracking along MTs was actin dependent, myotubes expressing fluorescent MTs and myosin were treated with cytochalasin D. Upon actin depolymerization, myosin filaments previously located beneath the cell membrane disappeared, indicating that they were already assembled with actin in unstriated myofibrils. In contrast, nascent myosin structures remained attached to MTs, continued to travel over short distances and eventually fused to form large aggregates. Therefore, actin did not interfere with nascent myosin structures movement along MTs. In Dictyostelium cells, previous data showed that myosin can move independently of its own motor activity (Yumura and Uyeda, 1997). Although more experiments will be needed to determine if this is the case for myosin displacements observed in myotubes, various results support this hypothesis. Actually: (i) MTs have never been shown to stimulate muscle myosin by direct association, (ii) myosin movement is dependent on MTs and independent of actin, (iii) myosin displacements fit well with the direction and rate of active kinesin-like transport.
Figure 8.
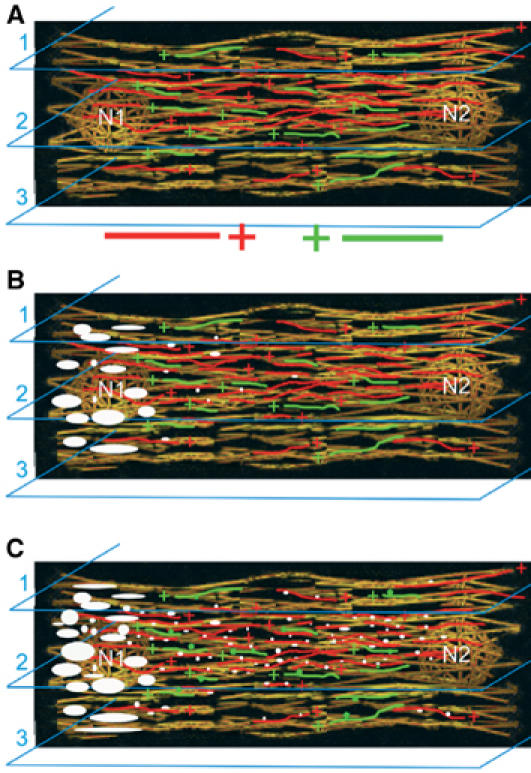
A model summarizing movements and organization of sarcomeric myosin in relation with MT network during myogenic differentiation. After myoblast fusion (A), MTs align and organize in a nonhomogenous longitudinal antiparallel network. MTs display opposite polarities (+ indicates the growing end of MTs) and various dynamic properties. Upon cell differentiation, stable MTs appear in myotubes (shown in yellow). Dynamic MTs elongating from nucleus N1 to N2 are shown in red, while MTs growing from nucleus N2 to N1 are in green. The amount and the organization of dynamic MTs vary according to cell layers (1, 2 and 3). Between 24 and 48 h of differentiation (B), sarcomeric myosin is synthetized and accumulates (large white spots) near an active nucleus (N1). Few nascent myosin structures (small white dots) associated with kinesin-like motor move along MTs toward the distal region of the cell. After 48 h of differentiation (C), due to the specific organization of the dynamic MTs, the majority of the nascent myosin structures (small white dots) move from N1 to N2 and mainly localize within the internal region of myotubes (cell layer 2). By traveling along opposite MTs (green), some nascent myosin structures (small green dots) travel from N2 to N1.
In conclusion, myofibrillogenesis is a complex process involving dramatic changes in gene expression as well as complex dynamic changes of the cytoskeleton. Obviously, differentiation requires cellular mechanisms capable of regulating and coordinating cell shape and the distribution of specific sarcomeric proteins from their site of synthesis to the site of sarcomere assembly. Using real-time observations, we have determined that MT organization and dynamics are temporally and spatially regulated within the differentiating cell and that the MT network is required for sarcomeric myosin movement and organization during myofibrillogenesis.
Materials and methods
Cell cultures, transfections and plasmids
C57 myoblastic cells were grown and induced to differentiate as described previously (Pizon et al, 2002). For transient transfections, cells were exposed to vectors (1–5 μg/ml of DNA) and jetPEI reagent from Qbiogene Molecular Biology (Illkirch, France). The Dsred-myosin vector was constructed from the eGFP-tagged myosin vector (light chain isoform MLC 3f) kindly provided by Dr Perriard (Auerbach et al, 1997). The EcoR1–Sal1 myosin light chain 3 cDNA insert was cloned at the same sites in the pDsRed2-N1 Clontech vector. This construct was restricted with Not1 and treated with Klenow enzyme to get a blunt end and was then digested with HindIII. The isolated myosin-Dsred insert was then cloned at the HindIII–Sma1 sites of vector pJ3 behind SV40 promoter (Mulligan and Berg, 1981). The EB1-GFP vector was a generous gift from Dr Mimori-Kiyosue. DsredII and GFP-α tubulin vectors were from Clontech (Palo Alto, CA).
Immunofluorescence
For triple immunofluorescence experiments, cells were briefly rinsed with warm (37°C) BRB80 buffer (8 mM K-Pipes, pH 6.8, 1 mM EGTA and 1 mM MgCl2), then permeabilized with 0.1% Triton X-100 in BRB80 for 45 s before methanol fixation at −20°C for 7 min. Primary antibodies, diluted in PBS containing 0.1% saponin and 5% FCS, were sequentially applied for 1 h at room temperature. Following three washes with 0.1% saponin in PBS, staining was obtained by incubating conjugated secondary antibodies for 30 min. Coverslips were mounted in AF1 solution (Citifluor) and observed with an LSM 510 confocal microscope (Zeiss). Rabbit polyclonal antibody against the ‘KVEGEGEEEGEEY' sequence of rabbit α-tubulin was raised according to established procedures used at the animal house of EMBL (Heidelberg, Germany). The antibody was purified against the same peptide. Mouse monoclonal IgG1 antibody against skeletal myosin (MY-32) and mouse monoclonal IgM antibody against α-skeletal sarcomeric actin (5C5) were purchased from Sigma-Aldrich. We used the following secondary antibodies: CyTM5-conjugated rabbit anti-mouse IgG, CyTM3-conjugated goat anti-mouse IgM from Zymed (San Francisco, CA) and Alexa 488-conjugated goat anti-rabbit from Molecular Probes (Leiden, The Netherlands).
Immunoelectron microscopy
Cells were permeabilized for 1 s with 0.5% Triton X-100 in warm BRB80 (37°C), then fixed successively at 37°C for 15 min in BRB80 buffer containing 0.3% Triton and 0.3% glutaraldehyde, then for 15 min in BRB80 buffer containing 0.3% glutaraldehyde. Following fixation, cells were incubated for 15 min in PBS containing 50 mM NH4Cl and incubated with the primary monoclonal myosin antibody for 1 h. Following three washes with 0.1% saponin in PBS for 15 min, incubation with secondary rabbit anti-mouse antibody was performed for 30 min. After three washes, protein A gold incubation was performed for 30 min. The gold (10 nm)-conjugated protein A was purchased from Utrecht University. Following another round of washes, post-fixation was performed as described by Yi et al (2001). After osmication, cells were dehydrated and flat embedded in Epon resin. After resin polymerization, small pieces were dissected from flat-embedded cultures, mounted in plastic stubs and sectioned. Ultrathin sections were stained with uranyl acetate and lead citrate. Sections were examined in a Philips CM120 electron microscope.
Microscopy
For in vivo observations, cells were kept in the microscope chamber at 37°C, in minimum essential medium eagle (MEM) without phenol red from SIGMA (Saint Louis, MO, USA). To reduce lethality, 30 mM Hepes was added to cell medium. Dual-wavelength time-lapse fluorescent microscopy was performed on a spinning-disk confocal microscope system. Light from a KryptoArgon ion laser was delivered to a Yokogawa spinning disk confocal scan-head (Ultra-View; Perking-Elmer) on an inverted Leica microscope. Images were collected by an × 100 1.4NA Plan-Apo DIC objective and captured with an Orca ER camera (Hamamatsu). Microscope functions were controlled by MetaMorph software (Universal Imaging). Sequential images were collected using 488 nm light for GFP-tagged protein and 568 nm light for Dsred protein. Exposure times for each chromophore varied between 0.3 and 0.9 s and time intervals between each frame were optimized according to the sample and the particle speed. Movies from 5 to 60 min were recorded. Images for presentation were prepared with Adobe PhotoShop (Adobe Systems, Mountain View, CA).
Data analysis and quantifications
Tracking of EB1 trajectories at the tip of MT was automatically performed by a customized software written as a Java plug-in for Image J. High-quality contrast fluorescence images allowed automatic recognition of the EB1 complex visualized as white spots of 4–10 pixels. The algorithm uses both the information of intensity and size to initially detect and track the spots. Once detected, spots are erased from the image stack, thus allowing recursive detection of the remaining trajectories. To avoid false recognition, a filter algorithm rejects records showing jerky motion (i.e. steps with more than 20° angle with the initial trajectory). Blind tests showed that all automatically recorded trajectories are good by comparison with manual detection and that automatic selection of spots does not induce any bias in the statistics. Direction, length, mean speed and RMS are computed from recorded trajectories. This computer analysis allowed recognition of many EB1 trajectories (up to 358 for one single cell stack), thus providing good statistical data.
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Movie 7
Supplementary Movie 8
Legends of Supplementary Movies
Acknowledgments
We are grateful to T Zimmermann, J Rietdorf and R Pepperkok for helpful discussions and expert assistance in teaching to VP how to use confocal microscopes. We thank C Antony for reagents and initial help in EM experiments, T Piolot and C Chamot for assistance in movies mounting and manual tracking experiments, P Bozin for photographic assistance, G Baldacci for critical reading of the manuscript. FG thanks Olivier Cardoso for teaching Java and singing Brassens songs. This work was supported in part by the European Molecular Biology Laboratory (Heidelberg, Germany) and in part by CNRS, Paris 7 and Paris 6 Universities and INSERM (Paris, France). VP was the recipient of a European Advanced Light Microscopy Facility Fellowship in 2001 and of an EMBO short-term fellowship in 2003. This work was also partially funded by a grant from the Association Française de Lutte contre les Myopathies (AFM) to VP.
References
- Antin P, Forrey-Shaudies S, Tapscott S, Holtzer S (1981) Taxol induces postmitotic myoblasts to assemble interdigitating microtubules–myosin arrays that exclude actin filaments. J Cell Biol 90: 300–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach D, Rothen-Ruthishauser B, Bantle S, Leu M, Ehler E, Hefman D, Perriard J-C (1997) Molecular mechanisms of myofibril assembly in heart. Cell Struct Funct 22: 139–146 [DOI] [PubMed] [Google Scholar]
- Bugnard E, Zaal K, Ralston E (2005) Reorganization of microtubule nucleation during muscle differentiation. Cell Motil Cytoskelet 60: 1–13 [DOI] [PubMed] [Google Scholar]
- Burak M, Silverman M, Banker G (2000) The role of selective transport in neuronal protein sorting. Neuron 26: 465–472 [DOI] [PubMed] [Google Scholar]
- Cartwright J, Goldstein M (1982) Microtubules in soleus muscles of postnatal and adult rat. J Ultrastruct Res 79: 74–84 [DOI] [PubMed] [Google Scholar]
- Clark K, McElhinny A, Beckerle M, Gregorio C (2002) Striated muscle cytoarchitecture: an intricate web of form and function. Annu Rev Cell Dev Biol 18: 637–706 [DOI] [PubMed] [Google Scholar]
- Cooper J (1987) Effects of cytochalasin and phalloidin on actin. J Cell Biol 105: 1473–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R (1994) The structure of the contractile filaments. In Myology, Engel AG, Franzini-Armstrong C (eds), pp 134–175. New York: McGraw-Hill [Google Scholar]
- Davis J (1988) Interaction of C-protein with pH 8.0 synthetic thick filaments prepared from the myosin of vertebrate skeletal muscle. J Muscle Res Cell Motil 9: 174–183 [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison T (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13: 83–117 [DOI] [PubMed] [Google Scholar]
- Ehler E, Rothen B, Hammerle S, Komiyama M, Perriard JC (1999) Myofibrillogenesis in the developing chicken heart: assembly of Z-disk, M-line and the thick filaments. J Cell Biol 112: 1529–1539 [DOI] [PubMed] [Google Scholar]
- Epstein H, Fischman D (1991) Molecular analysis of protein assembly in muscle development. Science 251: 1039–1044 [DOI] [PubMed] [Google Scholar]
- Fischman D (1967) An electron microscope study of myofibril formation in embryonic chick skeletal muscle. J Cell Biol 17: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Fischman D (1994) Morphogenesis of skeletal muscle fibers. In Myology, Engel AG, Franzini-Armstrong C (eds), pp 74–96. New York: McGraw-Hill [Google Scholar]
- Fürst D, Osborn M, Weber K (1989) Myogenesis in the mouse embryo: differential onset of expression of myogenic proteins and the involvement of titin in myofibril assembly. J Cell Biol 109: 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginkel L, Wordeman L (2000) Expression and partial characterization of kinesin-related proteins in differentiating and adult skeletal muscle. Mol Biol Cell 11: 4143–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein M, Entman M (1979) Microtubules in mammalian heart muscle. J Cell Biol 80: 183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio C (1997) Models of thin filament assembly in cardiac and skeletal muscle. Cell Struct Funct 22: 191–195 [DOI] [PubMed] [Google Scholar]
- Gundersen G, Khawaja S, Bulinski C (1989) Generation of a stable, posttranslationally modified microtubule array is an early event in myogenic differentiation. J Cell Biol 109: 2275–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Jacobson S, Brown D (1986) Rearrangement of tubulin, actin and myosin in cultured ventricular cardiomyocytes of the adult rat. Cell Motil Cytoskelet 6: 291–304 [DOI] [PubMed] [Google Scholar]
- Holtzer H, Forry-Schaudies S, Dlugosz A, Antin P, Dubyak G (1985) Interactions between IFs, microtubules and myofibrils in fibrogenic and myogenic cells. Ann NY Acad Sci 455: 106–111 [DOI] [PubMed] [Google Scholar]
- Holtzer H, Hijikata T, Zhang Z, Holtzer S, Protasi F, Franzini-Armstrong C, Sweeney H (1997) Independant assembly of 1.6 μm long bipolar MHC filaments and I–Z–I bodies. Cell Struct Funct 22: 83–93 [DOI] [PubMed] [Google Scholar]
- Huxley H (1963) Electron microscope studies on the structure of natural and synthetic protein filaments from striated muscle. J Mol Biol 7: 281–308 [DOI] [PubMed] [Google Scholar]
- Lin Z, Lu M, Schultheiss T, Choi J, Holtzer S, DiLullo C, Fischman D, Holtzer H (1994) Sequential appearance of muscle-specific proteins in myoblasts as a function of time after cell division: evidence for a conserved myoblast differentiation program in skeletal muscle. Cell Motil Cytoskelet 29: 1–19 [DOI] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y, Shiina N, Tsukita S (2000) The dynamic behavior of the APC-binding protein EB1 on the distal ends of microtubules. Curr Biol 10: 865–868 [DOI] [PubMed] [Google Scholar]
- Morrison E, Moncour P, Askham J (2002) EB1 identifies sites of microtubule polymerization during neurite development. Mol Brain Res 98: 145–152 [DOI] [PubMed] [Google Scholar]
- Mulligan R, Berg P (1981) Selection for cells that express the Escherichia coli gene coding for xanthine–guanine phosphoribosyl-transferase. Proc Natl Acad Sci USA 78: 2072–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musa H, Orton C, Morrison E, Peckmam M (2003) Microtubule assembly in cultured myoblasts and myotubes following nocodazole induced microtubule depolymerisation. J Muscle Res Cell Motil 24: 301–308 [PMC free article] [PubMed] [Google Scholar]
- Ojima K, Lin Z, Zhang Z, Hijikata T, Holtzer H, Labeit S, Sweeney H, Holtzer H (1999) Initiation and maturation of I–Z–I bodies in the growth tips of transfected myotubes. J Cell Sci 112: 4101–4112 [DOI] [PubMed] [Google Scholar]
- Pizon V, Iakovenko A, Kelly R, van der Ven P, Fürst D, Karsenti E, Gautel M (2002) Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 Ring-finger protein. J Cell Sci 115: 4469–4482 [DOI] [PubMed] [Google Scholar]
- Price M, Landsverk M, Barral J, Epstein H (2002) Two mammalian UNC-45 isoforms are related to distinct cytoskeletal and muscle-specific functions. J Cell Sci 115: 4013–4023 [DOI] [PubMed] [Google Scholar]
- Rhee D, Sanger JM, Sanger JW (1994) The premyofibril: evidence for its role in myofibrillogenesis. Cell Motil Cytoskelet 28: 1–24 [DOI] [PubMed] [Google Scholar]
- Rodriguez O, Schaefer A, Mandato C, Forsher P, Bement W, Waterman-Storer C (2003) Conserved microtubule–actin interactions in cell movement and morphogenesis. Nat Cell Biol 5: 599–609 [DOI] [PubMed] [Google Scholar]
- Rudy D, Yatskievych T, Antin P, Gregorio C (2001) Assembly of thick, thin and titin filaments in chick precardiac explants. Dev Dyn 221: 61–71 [DOI] [PubMed] [Google Scholar]
- Saitoh O, Arai T, Obinata T (1988) Distribution of microtubules and other cytoskeletal filaments during myotube elongation by fluorescence microscopy. Cell Tiss Res 252: 263–273 [DOI] [PubMed] [Google Scholar]
- Sanger JW, Ayoob J, Chowrashi P, Zurawski D, Sanger JM (2000) Assembly of myofibrils in cardiac muscle cells. In Elastic Filaments of the Cells, Granzier, Pollack (ed), pp 89–102. Dordrecht: Kluwer Academic/Plenum Publishers [DOI] [PubMed] [Google Scholar]
- Sanger JW, Chowrashi P, Shaner N, Spalthoff S, Wang J, Freeman N, Sanger JM (2002) Myofibrillogenesis in skeletal muscle cells. Clin Orthopaed Relat Res 403: 153–162 [DOI] [PubMed] [Google Scholar]
- Schnapp B (2003) Trafficking of signaling modules by kinesin motors. J Cell Sci 116: 2125–2135 [DOI] [PubMed] [Google Scholar]
- Seiler S, Fischman D, Leinwand L (1996) Modulation of myosin filament organization by C-protein family members. Mol Biol Cell 7: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small J, Fürst D, Thornell L (1992) The cytoskeletal lattice of muscle cells. Eur J Biochem 208: 559–572 [DOI] [PubMed] [Google Scholar]
- Srikakulam R, Winkelmann D (2004) Chaperone-mediated folding and assembly of myosin in striated muscle. J Cell Sci 117: 641–652 [DOI] [PubMed] [Google Scholar]
- Stepanova T, Slemmer J, Hoogenraad C, Lansbergen G, Dortland B, DeZeeuw C, Grosveld F, van Cappellen G, Akhmanova A, Galjart N (2003) Visualization of microtubule growth in cultured neurons via use of EB3-GFP (end-binding protein 3-green fluorescent protein). J Neurosci 23: 2655–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassin A-M, Maro B, Bornens M (1985) Fate of microtubule-organizing centers during myogenesis in vitro. J Cell Biol 100: 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama Y, Forrey-Shaudies S, Hoffman B, Holtzer S (1982) Effects of taxol and colcemid on myofibrillogenesis. Proc Natl Acad Sci USA 79: 6556–6560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Ven P, Ehler E, Perriard J-C, Fürst D (1999) Thick filament assembly occurs after the formation of a cytoskeletal scaffold. J Muscle Res Cell Motil 20: 569–579 [DOI] [PubMed] [Google Scholar]
- Verkade P, Schrama L, Verkleij A, Gispen W, Oestreicher A (1997) Ultrastructural colocalization of calmodulin in B50/growth associated protein-43 at the plasma membrane of proximal unmyelinated axon shafts studied in the model of regenerating rat sciatic nerve. Neuroscience 79: 1207–1218 [DOI] [PubMed] [Google Scholar]
- Warren R (1968) The effect of colchicine on myogenesis in vivo in Rana pipiens and Rhodnius prolixus. J Cell Biol 39: 544–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Leunissen J, Shi G, Gutekunst C, Hersch S (2001) A novel procedure for Pre-embedding double Immunogold-Silver labeling at the ultrastructural level. J Histochem Cytochem 49: 279–283 [DOI] [PubMed] [Google Scholar]
- Yumura S, Uyeda T (1997) Transport of myosin II to the equatorial region without its own motor activity in mitotic Dictyostelium cells. Mol Biol Cell 8: 2089–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Movie 6
Supplementary Movie 7
Supplementary Movie 8
Legends of Supplementary Movies